

Abstract
Dual-specific A-kinase-anchoring protein 2 (D-AKAP2/AKAP10), which interacts at its carboxyl terminus with protein kinase A and PDZ domain proteins, contains two tandem regulator of G-protein signaling (RGS) domains for which the binding partners have remained unknown. We show here that these RGS domains interact with Rab11 and GTP-bound Rab4, the first demonstration of RGS domains binding small GTPases. Rab4 and Rab11 help regulate membrane trafficking through the endocytic recycling pathways by recruiting effector proteins to specific membrane domains. Although D-AKAP2 is primarily cytosolic in HeLa cells, a fraction of the protein localizes to endosomes and can be recruited there to a greater extent by overexpression of Rab4 or Rab11. D-AKAP2 also regulates the morphology of the Rab11-containing compartment, with co-expression causing accumulation of both proteins on enlarged endosomes. Knockdown of D-AKAP2 by RNA interference caused a redistribution of both Rab11 and the constitutively recycling transferrin receptor to the periphery of cells. Knockdown also caused an increase in the rate of transferrin recycling, suggesting that D-AKAP2 promotes accumulation of recycling proteins in the Rab4/Rab11-positive endocytic recycling compartment.
Introduction
Dual-specific A-kinase-anchoring protein 2 (D-AKAP2/AKAP10), 5 which was first discovered as a binding partner for both the RI and RII regulatory subunits of protein kinase A (PKA), is a ubiquitous scaffold protein containing two tandem RGS (regulator of G-protein signaling) domains and a carboxyl-terminal PDZ (PSD95/Dlg/ZO1)-binding motif (1, 2). The PDZ-binding motif was subsequently shown to interact with the multi-PDZ domain proteins PDZK1 and NHERF-1, allowing D-AKAP2 to link indirectly to membrane proteins such as the sodium-dependent phosphate co-transporter (NaPi-IIa), which is regulated by trafficking into and out of the plasma membrane (3). D-AKAP2 is considered the sole member of its own subfamily of mammalian RGS proteins and is the only one to contain two RGS domains, referred to as RGS-A and RGS-B (4, 5). RGS proteins contain conserved α-helical domains of around 120 residues, which usually interact with heterotrimeric G protein α-subunits, in many cases acting as GTPase-activating proteins (6, 7). Although deuterium exchange-mass spectrometry experiments suggest that RGS-B folds into the traditional α-helical structure, RGS-A is predicted to have a ∼122 residue insertion (residues 170–291), but it is unknown how RGS-A, which is predicted to have a ∼122-residue insertion (residues 170–291), folds or interacts with RGS-B (8). D-AKAP2 has so far demonstrated no specific interactions with heterotrimeric G proteins, and no ability to activate them, raising the possibility of nontraditional targets.6
In this study, we show that the RGS domains of D-AKAP2 interact not with heterotrimeric G proteins but with the small GTPases Rab4 and Rab11, which regulate endocytic recycling. Endocytic membrane traffic determines the cellular destination of internalized receptors and other membrane proteins, receptor ligands, lipid, and solute molecules. The Rab subfamily of Ras-like GTPases is known to play a key role in regulating such processes as vesicle formation, motility, and docking (9, 10). Rab proteins help to establish the identity of membrane compartments and organize distinct membrane domains, typically by interacting in their GTP-bound active state with downstream effectors, which may be recruited to membranes through combinatorial interactions in a highly specific manner (11).
The transferrin receptor (TfnR) is a commonly studied marker of constitutive recycling. TfnR endocytosed in Rab5-positive vesicles is delivered initially to Rab5/Rab4-positive peripheral tubular-vesicular structures called sorting endosomes or early endosomes (12, 13). Bound transferrin, after releasing its iron in the sorting endosomes, is segregated with its receptor and the vast majority of lipid molecules and membrane proteins into the Rab4 domain, where they are separated from the soluble molecules in the lumen of the sorting endosomes by the pinching off of narrow membrane tubules, a process called “geometry-based sorting” (12). The receptor then recycles to the plasma membrane either directly in the “fast” recycling pathway, which is thought to be regulated by Rab4 (14, 15), or indirectly via the Rab4/Rab11-positive endocytic recycling compartment (ERC) in the “slow” recycling pathway. The ERC is a collection of tubular structures associated with microtubules, which in many cell types is condensed around the microtubule-organizing center but in others is more dispersed throughout the cytoplasm. Rab11 localizes mainly to the ERC and regulates trafficking through this organelle (16, 17). Here, we show that through interactions with Rab4 and Rab11, D-AKAP2 can alter the morphology of the Rab11 compartment and affect the recycling of the transferrin receptor.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The following monoclonal (mAb) and polyclonal (pAb) antibodies were used: AKAP10 mAb (Abnova, Taipei, Taiwan); transferrin receptor mAb (Zymed Laboratories Inc., South San Francisco, CA); LAMP-2 mAb (H4B4, developed by J. T. August and J. E. K. Hildreth, Developmental Studies Hybridoma Bank); giantin pAb (Covance, Richmond, CA); γ-adaptin mAb 100/3, FLAG mAb M2, PMP70 pAb, α-tubulin mAb, and EZview Red anti-FLAG M2 affinity gel (Sigma); Hsp90 mAb and EEA1 mAb (BD Biosciences); histone H2 pAb (Cell Signaling Technology, Danvers, MA); AIF mAb E-1 and GFP pAb (Santa Cruz Biotechnology, Santa Cruz, CA); vimentin mAb and calreticulin pAb (Abcam, Cambridge, MA); Rab11a pAb VU57 (raised against a peptide from the carboxyl-terminal variable domain and described previously (18)); Cy5-labeled donkey anti-mouse IgG antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA); and AlexaFluor 488-labeled donkey anti-mouse IgG and goat anti-rabbit IgG antibodies and AlexaFluor 568-labeled goat anti-rabbit IgG antibodies (Invitrogen). Human holotransferrin, deferroxamine, GDP, and GTPγS were from Sigma. AlexaFluor 488-labeled transferrin was from Invitrogen.
DNA Constructs
Enhanced GFP fusions to canine Rab4b WT, Rab4b Q67L, Rab4b S22N, Rab11a WT, Rab11a S25N, Rab5a WT, Rab5a Q79L, Rab5a S34N, and Rab7a WT were obtained from Dr. Robert Lodge (Centre Hospitalier Universitaire de Québec, Quebec, Canada). Additional mutations were generated using QuikChange (Stratagene, La Jolla, CA). All D-AKAP2 constructs used the human gene with valine at position 646. Mouse PDZK1 DNA was obtained from Dr. Heini Murer (University of Zurich, Switzerland). mCherry DNA was obtained from Dr. Roger Tsien (University of California at San Diego). Generation of truncations and subcloning were performed by standard methods. Vectors for expression of mCherry- and FLAG-tagged proteins were generated by inserting the tag as a BamHI/EcoRI fragment into pcDNA3 with the coding sequence for the protein of interest following as an EcoRI/XhoI fragment.
Cell Culture, Fractionation, and Immunofluorescence Microscopy
HeLa and HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum and 1× GlutaMAX-I (Invitrogen). HeLa cells were fractionated using the Qproteome cell compartment kit (Qiagen) according to the manufacturer's protocol. For microscopy, HeLa cells were grown in 35-mm glass-bottom microwell dishes (MatTek). Cells were transfected with FuGENE 6 (Roche Applied Science) and analyzed roughly 20 h later. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) at room temperature for 20 min. For immunostaining, cells were permeabilized with 0.2% Triton X-100 (Fisher) in PBS for 5 min and blocked with 2% normal donkey serum and 0.5% bovine serum albumin (BSA) in PBS. Wash buffer consisted of PBS plus 0.05% Tween 20, and antibodies were diluted into a 1:5 dilution of block buffer into wash buffer. Images were captured on either a Zeiss Axiovert 200M microscope (Photometrics, Tucson, AZ) or a Radiance 2000 laser-scanning confocal microscope (Bio-Rad). For live imaging, cells were washed with Hanks' balanced salt solution and imaged at room temperature on the Zeiss microscope. Images were processed, merged, and pseudocolored using Adobe Photoshop. To quantify colocalization on endosomes, the background was subtracted from both channels, and the intensity value for the 95th percentile of pixels within the cell was determined. All pixels under this threshold were set to zero, and the Pearson's coefficient for the two channels was calculated using the ImageJ plug-in, JACoP (19).
Co-immunoprecipitation
HEK293 cells in 6-cm dishes were transfected with FuGENE 6 and harvested ∼20 h later. Pelleted cells were lysed on ice for 10 min in 400 μl of 20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 5 mm MgCl2, 1 mm dithiothreitol, protease inhibitor mixture set III (Calbiochem), and 1% Nonidet P-40. After centrifugation for 20 min at 21,000 × g, 330 μl of lysate was added to 20 μl of EZview Red anti-FLAG M2 affinity gel (Sigma) suspended in 670 μl of wash buffer, which was equivalent to the lysis buffer but contained only 0.05% Nonidet P-40. After 1 h of incubation at 4 °C with rotation, the affinity resin was spun and washed four times with 1 ml of wash buffer. Protein was eluted with ImmunoPure IgG elution buffer (Pierce), heated with SDS sample buffer, and analyzed by SDS-PAGE and immunoblotting.
GST-Rab4 Pulldown
Free GST and GST-tagged Rab4 were expressed from the pGEX4T-1 vector in BL21 Escherichia coli cells and purified by standard means using glutathione-Sepharose 4B (GE Healthcare). GST-Rab4 was loaded with either 1 mm GDP or GTPγS in 20 mm Tris, pH 8.0, 150 mm NaCl, and 4 mm EDTA at 30 °C for 1 h after which MgCl2 was added to 20 mm. HEK293 cells transfected with a pcDNA3 vector expressing untagged D-AKAP2 were combined with untransfected cells, lysed in 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm MgCl2, 1 mm dithiothreitol, 0.05% Nonidet P-40, and protease inhibitors, and incubated with 75 μg of each GST protein at 4 °C for 2 h. Proteins were precipitated with glutathione resin, washed, eluted with wash buffer supplemented with 10 mg/ml reduced glutathione (Sigma), and analyzed by SDS-PAGE and immunoblotting.
Yeast Two-hybrid Analysis
Protein interactions were assayed in yeast using a split-ubiquitin two-hybrid approach according to the manufacturer's protocol (Dualsystems Biotech). Bait and prey constructs were transformed into the yeast strain NMY32, plated on −Trp−Leu selection plates, and incubated at 30 °C for 3–4 days. Approximately five medium-size colonies were selected and resuspended in water. After all samples were diluted to equivalent densities, serial dilutions were spotted on selection plates −Trp/−Leu, −Trp/−Leu/−His, and −Trp/−Leu/−His/+10 mm 3-amino-1,2,4 triazole. After 3–4 days of growth at 30 °C, the presence of protein interactions was determined using the most stringent selection. The results given are representative of at least three trials.
Transferrin Recycling Assays
Cells were washed and incubated with DMEM supplemented with 0.5% BSA (DMEM/BSA) for 2 h at 37 °C. Cells were pulsed for 20 min at 37 °C with 50 μg/ml AlexaFluor 488-conjugated transferrin in DMEM/BSA. Cells were washed twice with DMEM/BSA plus the iron chelator deferroxamine (50 μm; Sigma). Cells were chased for 0, 15, or 30 min in DMEM/BSA plus 50 μm deferroxamine and 250 μg/ml unlabeled transferrin (Sigma). Cells were then washed once with 4 °C PBS, once with 4 °C stripping buffer (500 mm NaCl, 0.5% acetic acid, pH 3.0), and twice more with PBS. Cells for imaging were fixed immediately. Cells to be quantified by flow cytometry were removed from the plate by incubating them with 0.526 mm EDTA (Irvine Scientific) for 4 min before chilling and centrifugation. Propidium iodide was added to cells just prior to analysis in order to distinguish broken cells. The amount of total fluorescent transferrin remaining in each cell was quantified on a FACSCalibur flow cytometer (BD Biosciences) by counting 10,000 intact cells from each plate, with three plates per condition. The mean fluorescence intensity ± S.E. is reported after subtracting the background fluorescence of control cells with no added transferrin and then normalizing to the fluorescence of the control cells at 0 min. To determine the statistical significance of differences between control and knockdown plates, we used an independent two-sample, two-tailed t test with a p value for type 1 error. To convert the data to a rate of recycling, individual means for each plate were fit to a single exponential decay, F = Fo e−kt, where F0 represents the fluorescence at time zero, and k represents the rate of recycling. The experiment was performed on three separate occasions with similar results.
RNA Interference
To knock down levels of D-AKAP2 with small interfering RNA (siRNA), 35-mm dishes of HeLa cells were transfected with 3 pmol of siRNA (Sigma-Proligo) based on the D-AKAP2 sequence 5′-GAATCTTCCTCTACACTTA-3′ together with 3 μl of RNAiMax transfection reagent (Invitrogen) in 3 ml (final volume) of medium. The negative control siRNA was from Santa Cruz Biotechnology (sc-37007). Cells were analyzed 2 days after siRNA treatment. Any transfection of plasmid DNA was performed 1 day after siRNA treatment. Scaling up for larger dishes of cells was performed with equivalent final concentrations of siRNA and transfection reagent.
RESULTS
D-AKAP2 Can Localize to the Rab4/Rab11 Endocytic Recycling Compartment
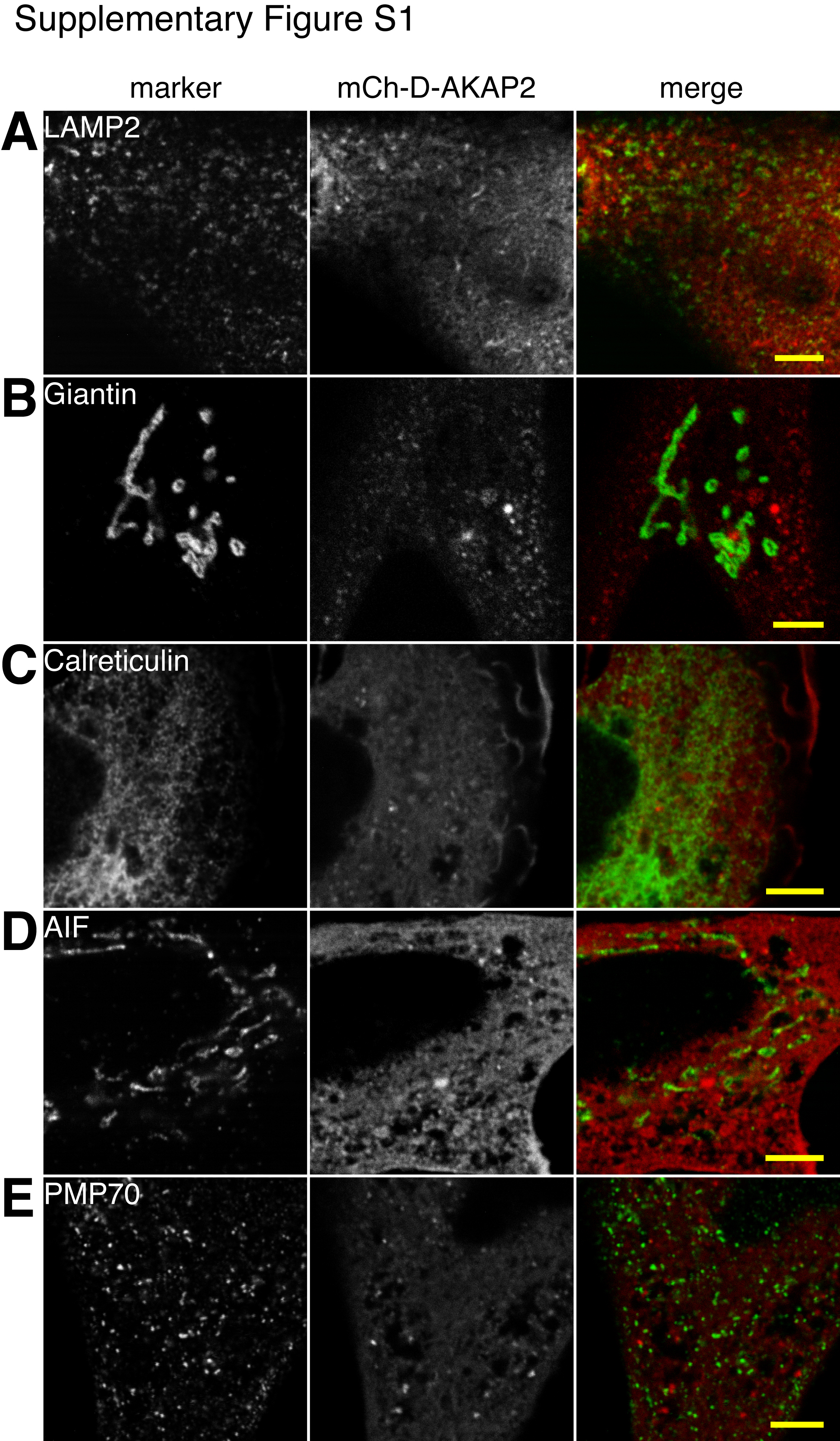
To determine the biological function of D-AKAP2, we first sought to identify its subcellular localization in HeLa cells. Cell fractionation showed that endogenous D-AKAP2 is almost entirely cytosolic (Fig. 1A). Transfection of D-AKAP2 tagged with mCherry (mCh) also showed diffuse cytosolic localization in most cells. However, imaging of live cells revealed a subpopulation of mCh-D-AKAP2 molecules localized to dynamic and motile puncta that was visible against the cytosolic background. D-AKAP2 in these puncta colocalized significantly with internalized fluorescently labeled transferrin, a marker of the endocytic recycling pathway (supplemental Movie 1). Some fixed cells also displayed visible accumulation of mCh-D-AKAP2 on what appeared to be intracellular membranes, and confocal microscopy confirmed that many of these puncta colocalized with internalized transferrin (Fig. 1B). Staining cells for various membrane markers also revealed colocalization with a subset of the endogenous transferrin receptor (Fig. 1C), which was more widely distributed than pulse-chased transferrin. D-AKAP2 showed partial colocalization with the early endosome marker EEA1 but most often appeared to be present on adjacent endosomes or endosomal domains (Fig. 1D). D-AKAP2 showed very little colocalization with markers of the endoplasmic reticulum, the Golgi complex, lysosomes, peroxisomes, or mitochondria (supplemental Fig. S1). D-AKAP2 was suggested previously to localize partially to mitochondria (2). The different result in the previous study may have been because of the use of a polyclonal antibody with a greater degree of cross-reactivity or the use of different cell types.
FIGURE 1.

D-AKAP2 in HeLa cells is primarily cytosolic but can localize to membranes of the transferrin recycling pathway. A, HeLa cells were fractionated into cytosol, membrane, nucleus, and cytoskeleton fractions and probed for endogenous D-AKAP2 and markers. B–D, confocal microscopy of HeLa cells showed some accumulation of overexpressed mCh-D-AKAP2 on membrane structures that colocalized with fluorescently labeled transferrin pulsed for 5 min and chased for 10 min (B) and with the endogenous transferrin receptor (C). The early endosome marker EEA1 showed minimal colocalization with mCh-D-AKAP2, sometimes appearing on adjacent membrane domains (D). Scale bars, 10 μm.
Because Rab GTPases play a large role in determining the identity of particular endosomal compartments, we co-expressed mCh-D-AKAP2 with GFP-tagged Rab proteins as well as their GDP- or GTP-locked mutant forms (Fig. 2). D-AKAP2 not only colocalized with Rab4 and Rab11, two Rab proteins known to regulate endocytic recycling, but also showed increased endosomal localization upon co-expression with the Rab proteins. This was especially true of Rab11; its co-expression with D-AKAP2 caused accumulation of both proteins on enlarged, dynamic endosomes (Fig. 2A and supplemental Movie 2). These enlarged endosomes were still accessible to internalized transferrin (data not shown). Neither the GDP-locked form (dominant negative S25N (16)) nor the GTP-locked form (GTPase-deficient S20V (20)) of Rab11 was as effective as WT in recruiting D-AKAP2 onto endosomes, implying that the cycle of GTP hydrolysis was important for their colocalization (Fig. 2, B and C). Rab11 Q70L also has been used by several groups as a putative GTPase-deficient mutant by analogy to other Rab proteins (21), but it has been found that S20V, and not Q70L, acts as a dominant active mutation (20, 22). Likewise, we found little difference between Rab11 Q70L and the WT with respect to complex formation with D-AKAP2 (data not shown). Unlike Rab11, Rab4 showed extensive colocalization with D-AKAP2 in both the WT and the GTP-locked (Q67L), but not the GDP-locked (S22N), forms (Fig. 2, D–F). Neither Rab5 WT nor the GTP-locked Rab5 Q79L appeared to recruit D-AKAP2 from the cytosol, but like EEA1, they sometimes appeared to reside on an adjacent domain of the same endosome (Fig. 2, G and H), and in live imaging they could be found on some of the same motile puncta (data not shown). GDP-locked Rab5 S34N, as well as the late endosomal Rab7, showed very little colocalization with D-AKAP2 on endosomes (Fig. 2, I and J).
FIGURE 2.

D-AKAP2 is recruited to endosomes by Rab4 and Rab11. HeLa cells were transfected with mCherry-D-AKAP2 together with GFP-tagged Rab proteins or their GDP- or GTP-locked mutant forms: Rab11 WT (A), GTP-locked Rab11 S20V (B), GDP-locked Rab11 S25N (C), Rab4 WT (D), GTP-locked Rab4 Q67L (E), GDP-locked Rab4 S22N (F), Rab5 WT (G), GTP-locked Rab5 Q79L (H), GDP-locked Rab5 S34N (I), Rab7 WT (J), and GFP vector (K). D-AKAP2 on endosomes colocalized extensively with Rab11 (A) and Rab4 (D), the two Rab proteins most involved in regulating the endocytic recycling compartment, but not with Rab5 (G) or Rab7 (J). Co-expression with either Rab4 or Rab11 caused increased localization of D-AKAP2 on endosomes; this was particularly true for Rab11 WT, which tended to accumulate with D-AKAP2 on enlarged endosomes (A). Scale bars, 10 μm. Quantification of endosomal colocalization was determined as described under “Experimental Procedures” as the Pearson's coefficient for the top 5% of pixels within the cell. The value given in the merge panel represents the average ± S.E. for 6–8 confocal images and is depicted in the graph (L).
When mCh-D-AKAP2 was expressed with both GFP-Rab11 and FLAG-Rab4, it generally localized to endosomes positive for both Rab proteins (Fig. 3A). Colocalization of mCh-D-AKAP2 and GFP-Rab11 with the GTP-locked FLAG-Rab4 Q67L was even more pronounced (Fig. 3B). Endogenous D-AKAP2 also clearly colocalized with GFP-Rab4 and mCh-Rab11 on endosomes, with an apparent preference for endosomes positive for both Rab proteins (Fig. 3, C–E). Additionally, D-AKAP2 on endosomes was able to recruit PDZ proteins through its carboxyl-terminal PDZ-binding motif. FLAG-tagged PDZK1, which contains four PDZ domains, localized to GFP-Rab11 endosomes in the presence of mCherry-tagged D-AKAP2 but not D-AKAP2ΔPDZ, which lacks the three residues at the carboxyl terminus required for the PDZ interaction (Fig. 3, F and G). Because other PDZ domains of PDZK1 are known to bind to membrane proteins containing PDZ-binding motifs, D-AKAP2 may serve to link recycling cargo to Rab proteins in a PDZ-dependent fashion.
FIGURE 3.

D-AKAP2 localizes to membrane domains containing both Rab4 and Rab11. A and B, in transfected HeLa cells, mCh-D-AKAP2 localized to endosomes positive for both GFP-Rab11 and FLAG-Rab4 WT (A) or for FLAG-Rab4 Q67L (B). C—E, endogenous D-AKAP2 was recruited to endosomes positive for both GFP-Rab4 and mCh-Rab11 (C) as well as to GFP-Rab4 (D) or mCh-Rab11 (E) expressed separately. F and G, D-AKAP2 can recruit PDZK1 to endosomes through its carboxyl-terminal PDZ-binding motif. FLAG-PDZK1 was recruited onto GFP-Rab11 endosomes when expressed with mCh-D-AKAP2 (F) but not with mCh-D-AKAP2ΔPDZ, in which the carboxyl-terminal three residues of D-AKAP2 (TKL) are truncated to abolish the PDZ-binding motif (G). Endogenous D-AKAP2 and FLAG-tagged proteins were visualized by staining with monoclonal antibodies and Cy5-conjugated secondary antibodies. All images were merged by pseudocoloring the first column green, the second red, and the third blue, with vector expression omitted. A–E represent confocal images. Scale bars, 10 μm.
D-AKAP2 Interacts with Rab4 and Rab11 through Its RGS Domains
To determine whether D-AKAP2 forms protein complexes with Rab4 and Rab11, we co-transfected HEK293 cells with FLAG-tagged D-AKAP2 and GFP-tagged Rab proteins and immunoprecipitated them with anti-FLAG antibodies (Fig. 4A). D-AKAP2 specifically co-immunoprecipitated GFP-tagged Rab4 and Rab11 but not Rab5. Consistent with the colocalization results, D-AKAP2 preferentially pulled down GTP-locked Rab4 (Q67L > WT > S22N) and wild-type Rab11 (WT > S20V = S25N). The observation that neither the GDP- nor the GTP-locked mutant form of Rab11 was precipitated by D-AKAP2 could imply that the interaction was stabilized by additional protein contacts that may be absent with the mutants because of altered localization. A FLAG-tagged truncation of D-AKAP2 containing the RGS domains (residues 1–513) was also able to immunoprecipitate Rab4 Q67L and Rab11 WT. Furthermore, purified GST-tagged Rab4 bound to the nonhydrolyzable nucleotide analog GTPγS was much more able to pull down D-AKAP2 from a HEK293 lysate than GDP-bound GST-Rab4 or GST alone (Fig. 4B). Unfortunately, GST-Rab11 did not form a stable enough complex with D-AKAP2 to be observed in pulldown experiments (data not shown).
FIGURE 4.

D-AKAP2 forms protein complexes with Rab4 and Rab11. A, HEK293 cells were co-transfected with FLAG-tagged D-AKAP2, vector, or a truncation containing the two RGS domains (residues 1–513 (last two lanes)) together with GFP-tagged Rab proteins or mutants. Samples from the lysate and from the elution of the immunoprecipitation (IP) were immunoblotted (IB) with antibodies against either FLAG or GFP. B, free GST or GST-Rab4 protein loaded with GDP or GTPγS was incubated with a HEK293 lysate containing endogenous and overexpressed tagless D-AKAP2, pulled down with glutathione resin, and immunoblotted for co-precipitated D-AKAP2.
To look more closely at the determinants of D-AKAP2 protein complexes with Rab4 and Rab11 on endosomes, we co-expressed mCherry-tagged truncations of D-AKAP2 with Rab4 or Rab11 (Fig. 5, A–K, and supplemental Fig. S2 for the complete panel of truncations). Truncations or mutations of D-AKAP2 that disrupted PKA binding were no longer excluded from the nucleus (Fig. 5, D–F and I–K, and data not shown). This had the effect of lowering the level of cytosolic protein for carboxyl-terminal truncations, which may have led to decreased endosomal localization. Because co-expression with Rab11 often caused substantial recruitment of D-AKAP2 to enlarged Rab11-containing endosomes, it was readily apparent which regions of D-AKAP2 were responsible for this phenotype. The tandem RGS domains (residues 120–513) were necessary and sufficient for this colocalization (Fig. 5F). Truncations missing all or part of one RGS domain (residues 292–662, 1–369, 514–662, and 374–662) were not recruited to Rab11 endosomes (Fig. 5, C and E, and supplemental Fig. S2, D and H), whereas all constructs containing both RGS domains (residues 120–662, 1–513, 120–513, 1–662, 1–659) were recruited (Fig. 5, B, D, and F, and supplemental Fig. S2, A and I). Co-expression of D-AKAP2 with Rab4 did not cause the same level of accumulation on endosomes, so the colocalization was less pronounced. Furthermore, the determinants of localization to Rab4 endosomes appeared to be distributed more widely throughout the protein. Compared with full-length D-AKAP2 (supplemental Fig. S2J), deletion of either the 119 residues amino-terminal to the RGS domains (Fig. 5G) or the 149 residues carboxyl-terminal to the RGS domains (Fig. 5I) decreased the extent of endosomal localization. Deleting both regions by expressing only the tandem RGS domains led to very faint colocalization (Fig. 5K) as did deleting the first 291 residues (Fig. 5H). These results suggest that localization of D-AKAP2 to endosomes may result from multiple protein interactions.
FIGURE 5.

The tandem RGS domains of D-AKAP2 mediate interactions with Rab4 and Rab11. A, domain structure of D-AKAP2 and its protein interactions. RGS-A is predicted to contain an insertion between residues 170 and 291 that splits it into two segments, RGS-A and RGS-A′. At the carboxyl terminus, the PDZ-binding motif can interact with PDZK1 and NHERF-1, and the A kinase-binding (AKB) motif of D-AKAP2 interacts with both the RI and RII regulatory (R) subunits of PKA, which bind to the catalytic (C) subunit. B–K, the mCherry-tagged truncations of D-AKAP2 containing residues 120–662 (B and G), 292–662 (C and H), 1–513 (D and I), 1–369 (E and J), and 120–513 (F and K) were co-expressed with either GFP-Rab11 (B–F) or FLAG-Rab4 stained with anti-FLAG antibodies (G–K). Scale bars, 10 μm. L, yeast two-hybrid analysis demonstrated that the tandem RGS domains (residues 120–513) are sufficient to interact with both Rab proteins. The RGS domains interacted with Rab4 WT, GTP-locked Rab4 Q67L, Rab11 WT, and GDP-locked Rab11 S25N. The individual RGS domains (residues 120–369 and 374–513) were not capable of interacting with either Rab.
To confirm these protein interactions and further investigate the binding determinants, we employed a split-ubiquitin yeast two-hybrid approach (Fig. 5L). The tandem RGS domains (residues 120–513) were sufficient to bind to both Rab4 and Rab11. In this assay, the RGS domains appeared to interact preferentially with GTP-bound Rab4 and GDP-bound Rab11, because they interacted with Rab4 Q67L and Rab11 S25N but not with Rab4 S22N or Rab11 S20V. It could be that we did not observe stable protein complexes between D-AKAP2 and Rab11 S25N in the co-immunoprecipitation experiments because of the inability of that Rab mutant to localize properly to the endosomes. Neither RGS-A (residues 120–369) nor RGS-B (residues 374–513) alone was able to interact with Rab4 or Rab11, suggesting that the two RGS domains form a single functional unit.
D-AKAP2 Regulates Trafficking through the Endocytic Recycling Compartment
Because co-expression with D-AKAP2 causes Rab11 to accumulate on enlarged endosomes, we asked what effect knocking down D-AKAP2 expression by RNA interference would have on the recycling compartment. Treating HeLa cells for 2 days with siRNA directed against D-AKAP2 effectively knocked down nearly all of the endogenous protein (Fig. 6A). Cells treated with siRNA were transfected with GFP-Rab4 and/or mCh-Rab11 and stained for endogenous EEA1, a marker of early endosomes (Fig. 6, B–G). Compared with the control siRNA, cells knocked down for D-AKAP2 showed an increased localization of mCh-Rab11 at the periphery, often at regions of membrane protrusion (Fig. 6, B–E). In some cells, Rab11 also appeared more condensed or more tubular than in control cells. GFP-Rab4 also became more peripheral in the D-AKAP2 knockdown cells but not to the same extent as Rab11 (Fig. 6, B, C, F, and G). Endogenous Rab11 and TfnR showed a movement to the periphery of the cell similar to that of mCh-Rab11 (Fig. 6, H and I). EEA1 staining of early endosomes appeared unchanged, for the most part, in all cells (Fig. 6, B–G). D-AKAP2 knockdown cells were also stained for other membrane markers. Although there was no obvious effect on the endoplasmic reticulum, peroxisomes, or lysosomes (data not shown), knocking down D-AKAP2 did appear to cause fragmentation of the Golgi complex, as seen by the staining of the cis-/medial-Golgi marker giantin and of γ-adaptin, a subunit of the AP-1 clathrin adaptor complex involved in vesicle formation both at the trans-Golgi network and on endosomes (Fig. 6, J and K). Rab11 is thought to play a role in traffic from endosomes to the Golgi (23).
FIGURE 6.

Knockdown of D-AKAP2 by RNA interference causes the Rab11 compartment to become more peripheral. A, siRNA of D-AKAP2 effectively knocked down the levels of endogenous protein in HeLa cells after 2 days as judged by immunoblotting of whole-cell extracts. B–K, HeLa cells were treated with either control (B, D, F, H, and J) or D-AKAP2 siRNA (C, E, G, I, and K). B–G, cells treated with siRNA were transfected with GFP-Rab4 WT and mCh-Rab11 WT (B and C), mCh-Rab11 WT (D and E), or GFP-Rab4 WT (F and G). Fixed coverslips were stained for endogenous EEA1 to mark early endosomes. Rab11 and, to a lesser extent, Rab4 became more peripheral in D-AKAP2 knockdown cells, whereas EEA1 staining remained relatively unchanged. H–K, HeLa cells treated with siRNA were fixed and stained for endogenous Rab11 and transferrin receptor (H and I) or γ-adaptin and giantin (J and K). Endogenous Rab11 and TfnR displayed similar shifts to the periphery of cells treated with D-AKAP2 siRNA. Knocking down D-AKAP2 also caused fragmentation of the Golgi as visualized by staining the cis/medial-Golgi marker giantin, as well as γ-adaptin, which localizes to the trans-Golgi network and to endosomes. Scale bars, 10 μm.
We next tested whether the changes in the morphology of the recycling compartment were linked to altered kinetics of receptor recycling by performing a pulse-chase experiment with fluorescently labeled transferrin. HeLa cells were pulsed for 20 min with fluorescent transferrin and then chased in the presence of unlabeled transferrin and the iron chelator deferroxamine for 0, 15, or 30 min. In control cells, the pulse-chased transferrin tended to accumulate in the perinuclear region, whereas in D-AKAP2 knockdown cells, it showed a greater accumulation near the plasma membrane (Fig. 7, A–F). To quantify the rate of transferrin recycling, using flow cytometry we measured the total amount of fluorescent transferrin remaining in each cell after different chase times (Fig. 7G). After the 20-min pulse (0 min chase), cells treated with D-AKAP2 siRNA contained 33.3 ± 6.2% more transferrin than cells treated with control siRNA (control = 100, D-AKAP2 siRNA = 133.3; p = 0.0022, two-tailed t test), whereas after a 30-min chase they contained 31.0 ± 8.5% less transferrin (control = 30.9, D-AKAP2 siRNA = 21.3; p = 0.023). Fitting all of the data to an exponential decay function yielded a rate of transferrin recycling that was 73 ± 21% faster in the D-AKAP2 knockdown cells than in control cells (2.11 ± 0.24/h for control cells and 3.64 ± 0.12/h for D-AKAP2 knockdown cells). The increased internalization of transferrin during the pulse was not due to a change in the rate of endocytosis but rather to increased surface expression of the receptor, presumably caused by the faster recycling (data not shown). These results suggest that endogenous D-AKAP2 may promote the accumulation of transferrin in the slow recycling pathway of the Rab4/Rab11-containing ERC. In the D-AKAP2 knockdown cells, the increased rate of traffic to the plasma membrane may exceed the rate of exocytosis, resulting in increased localization of internalized transferrin near the cell surface (Fig. 7, E and F).
FIGURE 7.

Knockdown of D-AKAP2 increases the rate of transferrin recycling. A–F, after 2 days of treatment with control (A–C) or D-AKAP2 siRNA (D–F), HeLa cells were incubated for 20 min with fluorescently labeled transferrin and then chased with unlabeled transferrin for 0 (A and D), 15 (B and E), or 30 min (C and F) before being washed and fixed. Pulse-chased transferrin showed a more perinuclear localization in control cells and a more peripheral localization in D-AKAP2 knockdown cells, similar to the change seen with the Rab11 compartment. G, to quantify transferrin recycling, HeLa cells were pulse-chased in the same manner as described in A–F but were analyzed by flow cytometry. Fluorescence values ± S.E. are normalized to the value for the control cells at 0 min. p values were determined by a two-tailed t test. H, proposed model for the localization and function of D-AKAP2 in protein recycling with respect to Rab proteins as adapted from Sönnichsen et al. (13). Endocytosed cargo recycles to the plasma membrane either directly from the Rab5/Rab4 sorting endosome (SE) by the fast recycling pathway or via the Rab4/Rab11 ERC in the slow recycling pathway. D-AKAP2 is proposed to localize to Rab4/Rab11-positive membranes and to negatively regulate trafficking of Rab11 recycling vesicles to the surface.
One mechanism for how knockdown of D-AKAP2 might increase the rate of recycling would be by decreasing the amount of cargo entering the slower, Rab11-dependent, recycling pathway through the ERC and shunting more into the faster direct pathway. Another mechanism would be by increasing the rate of trafficking from the ERC to the plasma membrane. The former model would predict a decrease in the amount of colocalization of internalized transferrin with Rab11. The fact that the knockdown of D-AKAP2 had similar effects on the membrane compartments of endogenous Rab11 and TfnR (Fig. 6I), which continued to colocalize, suggests that D-AKAP2 is not required for trafficking from the sorting endosomes to the ERC. Likewise, pulse-chased transferrin continued to colocalize with mCh-Rab11 in knockdown cells (data not shown). We therefore prefer a model in which the rate of traffic from the ERC to the plasma membrane is increased in the D-AKAP2 knockdown, leading to a shift in the localization of TfnR and Rab11 toward the periphery of the cell (Fig. 7H).
DISCUSSION
Although the mechanisms that regulate recycling of endocytosed proteins back to the plasma membrane are poorly understood, the small GTPases Rab4 and Rab11 are known to play key roles. Here we report that D-AKAP2 can bind to both Rab4 and Rab11 through its two RGS domains, recruiting D-AKAP2 from the cytosol onto endosomes of the recycling pathway. This represents a novel interaction for RGS domains, which typically interact with heterotrimeric G proteins rather than with Ras-like G proteins.
D-AKAP2 appears to act as a traditional effector for Rab4, binding most stably to GTPγS-bound Rab4 or to its GTP-locked mutant form. Rab11, on the other hand, interacts in a more nontraditional manner. Three observations support the presence of this protein complex: 1) Rab11 co-immunoprecipitated with D-AKAP2 from human cells, 2) the RGS domains interacted with Rab11 in a yeast two-hybrid assay, and 3) overexpression of Rab11 recruited D-AKAP2 to Rab11-positive endosomes. In the yeast two-hybrid assay, D-AKAP2 interacted with both dominant negative and wild-type Rab11, but only wild-type Rab11, and not the GDP- and GTP-locked forms, formed stable complexes in mammalian cells. Rab effectors are often recruited to membranes by the coordinated interactions of multiple proteins (11). Therefore, the preference of D-AKAP2 for wild-type Rab11 in mammalian cells could be because the mutant forms do not localize to the proper membrane microdomains to make additional stabilizing interactions. The lack of binding to the GTP-locked form could also indicate that D-AKAP2 may have guanine nucleotide exchange factor activity toward Rab11. If this were the case, D-AKAP2 could serve to activate and recruit Rab11 to membranes containing active Rab4 or to maintain Rab11 in an active state while in the Rab4/Rab11-containing ERC.
Because the interactions of a number of effectors with Rab proteins are mediated by coiled coils or helical hairpins (24–26), the helical RGS domains could present a similar binding interface. The RGS domains of D-AKAP2 may constitute a single functional unit, as neither domain alone showed Rab binding by yeast two-hybrid analysis (Fig. 5L) or showed strong recruitment to endosomes in HeLa cells (Fig. 5, E and J, and supplemental Fig. S2, H and R). It is possible that simultaneous interactions with both Rab proteins may be important for the localization of D-AKAP2 to endosomes. No protein is currently known to bind simultaneously to both Rab4 and Rab11. The Rab-coupling protein was found to interact with both Rab4 and Rab11 (27), but subsequent results indicated that in vitro they share the same binding site and that in vivo Rab11 is probably the dominant interacting partner (28, 29).
While this manuscript was in preparation, it was reported that a large-scale yeast two-hybrid screen for targets of Rab proteins had identified D-AKAP2 as an interacting partner for Rab proteins 4A/B, 14, 19, 40A/B/C, and 41 (30). The fragments of D-AKAP2 pulled from the mouse cDNA library appear to be similar to the 292–662 truncation, which showed faint localization to GFP-Rab4 endosomes but did not accumulate with Rab11 on enlarged endosomes (Fig. 5, C and H). Taken together, these results suggest that the second half of RGS-A may combine with RGS-B (residues 292–513) to form the Rab4-binding domain, whereas the first half of RGS-A (residues 120–291) may be necessary but not sufficient for Rab11 binding. RGS-A is atypical among RGS proteins for its large insertion predicted to be between helices α4 and α5 (8).
D-AKAP2 appears to regulate the morphology of the Rab11-containing compartment, with co-expression causing enlarged Rab11 endosomes (Fig. 2A) and knockdown causing movement of Rab11 to the periphery (Fig. 6, C, E, and I) and an accompanying increase in the rate of transferrin recycling (Fig. 7G). D-AKAP2 may act in a fashion analogous to Rabaptin-5, a divalent Rab effector that is also primarily cytosolic but can be recruited onto specific endosomal domains by simultaneous interactions with Rab4 and Rab5 (31). Rabaptin-5 inhibits the formation of recycling vesicles in vitro, whereas its depletion accelerates their formation (32). Overexpression of Rabaptin-5 leads to enlarged endosomes and delayed recycling of transferrin (31, 33). Although Rabaptin-5 would be expected to regulate recycling from Rab5/Rab4-positive sorting endosomes, D-AKAP2 may similarly inhibit recycling from Rab4/Rab11 domains.
Given its carboxyl-terminal interactions, it seems likely that D-AKAP2 helps mediate PKA- or PDZ-dependent regulation of trafficking. D-AKAP2 has been shown to interact with the PDZ-containing proteins NHERF-1 and PDZK1/NHERF-3 through a carboxyl-terminal type I PDZ ligand, TKL (3). Such PDZ ligands can act as transplantable “recycling sequences” that are sufficient to cause endocytosed receptors such as the β2-adrenergic receptor to recycle rapidly to the plasma membrane (34, 35). We found that D-AKAP2 could recruit PDZK1 to endosomes but not if its PDZ ligand was removed (Fig. 3, F and G). At endogenous protein levels, the PDZ domain proteins may help recruit D-AKAP2 to membrane domains rich in proteins containing PDZ-binding motifs.
PKA tethered to D-AKAP2 may have several effects. Binding either type I or type II PKA could affect the binding of other proteins to D-AKAP2 and vice versa. PKA could modulate D-AKAP2 function by phosphorylating its conserved consensus phosphorylation site (residue 554). Proteins binding to D-AKAP2 could also be targets, because phosphorylation of PDZK1 by PKA has been shown to cause increased protein expression of a PDZK1 binding partner, scavenger receptor class B type I (36). Finally, PKA anchored by D-AKAP2 could phosphorylate recycling membrane proteins directly. Phosphorylation of aquaporin-2 by PKA, for instance, is required for its vasopressin-induced translocation from intracellular vesicles to the plasma membrane (37).
Although the biological function of D-AKAP2 has remained unknown for many years, it has been linked genetically to disease in humans. A single nucleotide polymorphism (SNP) causing an I646V transition in the PKA-binding motif of D-AKAP2 shows the strongest correlation with morbidity or mortality of 6,500 SNPs studied (38). The I646V SNP, which increases the affinity for the RI regulatory subunit of PKA (38), is associated with myocardial infarction (39, 40), heart rate dysregulation (41), and familial breast cancer (42). In mice, a gene disruption deleting the last 51 residues of D-AKAP2 acts in a dominant negative fashion to increase cardiac arrhythmia, cholinergic sensitivity, and premature adult deaths (41). Our results show that the interactions of D-AKAP2 with Rab4 and Rab11 allow it to regulate protein recycling and suggest that these disease phenotypes could be the result of altered surface expression of various receptors or channels. D-AKAP2 may help fill several gaps in our understanding of membrane trafficking, including how Rab11 links to Rab4, how PDZ ligands link to the recycling machinery, and how PKA regulates trafficking.
Supplementary Material
Acknowledgment
We thank Nicolas Prévost for valuable assistance with the flow cytometry experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant DK054441.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Movies 1 and 2.
C. T. Eggers, R. K. Sunahara, and A. Krumins, unpublished data.
- D-AKAP2
- dual-specific A-kinase-anchoring protein 2
- ERC
- endocytic recycling compartment
- EEA1
- early endosome antigen 1
- TfnR
- transferrin receptor
- RGS
- regulator of G protein signaling
- PKA
- protein kinase A (cAMP-dependent protein kinase)
- siRNA
- small interfering RNA
- mAb
- monoclonal antibody
- pAb
- polyclonal antibody
- HEK
- human embryonic kidney
- GFP
- green fluorescent protein
- GST
- glutathione S-transferase
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- DMEM
- Dulbecco's modified Eagle's medium
- PBS
- phosphate-buffered saline
- BSA
- bovine serum albumin
- SNP
- single nucleotide polymorphism
- WT
- wild type
- mCh
- mCherry.
REFERENCES
- 1.Huang L. J., Durick K., Weiner J. A., Chun J., Taylor S. S. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 11184–11189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L., Sunahara R. K., Krumins A., Perkins G., Crochiere M. L., Mackey M., Bell S., Ellisman M. H., Taylor S. S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3220–3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gisler S. M., Madjdpour C., Bacic D., Pribanic S., Taylor S. S., Biber J., Murer H. (2003) Kidney Int. 64, 1746–1754 [DOI] [PubMed] [Google Scholar]
- 4.Neubig R. R., Siderovski D. P. (2002) Nat. Rev. Drug Discov. 1, 187–197 [DOI] [PubMed] [Google Scholar]
- 5.Zheng B., De Vries L., Gist Farquhar M. (1999) Trends Biochem. Sci. 24, 411–414 [DOI] [PubMed] [Google Scholar]
- 6.Berman D. M., Wilkie T. M., Gilman A. G. (1996) Cell 86, 445–452 [DOI] [PubMed] [Google Scholar]
- 7.Watson N., Linder M. E., Druey K. M., Kehrl J. H., Blumer K. J. (1996) Nature 383, 172–175 [DOI] [PubMed] [Google Scholar]
- 8.Hamuro Y., Burns L., Canaves J., Hoffman R., Taylor S., Woods V. (2002) J. Mol. Biol. 321, 703–714 [DOI] [PubMed] [Google Scholar]
- 9.Segev N. (2001) Curr. Opin. Cell Biol. 13, 500–511 [DOI] [PubMed] [Google Scholar]
- 10.Zerial M., McBride H. (2001) Nat. Rev. Mol. Cell Biol. 2, 107–117 [DOI] [PubMed] [Google Scholar]
- 11.Pfeffer S. (2003) Cell 112, 507–517 [DOI] [PubMed] [Google Scholar]
- 12.Maxfield F. R., McGraw T. E. (2004) Nat. Rev. Mol. Cell Biol. 5, 121–132 [DOI] [PubMed] [Google Scholar]
- 13.Sönnichsen B., De Renzis S., Nielsen E., Rietdorf J., Zerial M. (2000) J. Cell Biol. 149, 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheff D. R., Daro E. A., Hull M., Mellman I. (1999) J. Cell Biol. 145, 123–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Sluijs P., Hull M., Webster P., Mâle P., Goud B., Mellman I. (1992) Cell 70, 729–740 [DOI] [PubMed] [Google Scholar]
- 16.Ullrich O., Reinsch S., Urbé S., Zerial M., Parton R. G. (1996) J. Cell Biol. 135, 913–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urbé S., Huber L. A., Zerial M., Tooze S. A., Parton R. G. (1993) FEBS Lett. 334, 175–182 [DOI] [PubMed] [Google Scholar]
- 18.Lapierre L. A., Avant K. M., Caldwell C. M., Ham A. J., Hill S., Williams J. A., Smolka A. J., Goldenring J. R. (2007) Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1249–G1262 [DOI] [PubMed] [Google Scholar]
- 19.Bolte S., Cordelières F. P. (2006) J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 20.Wang X., Kumar R., Navarre J., Casanova J. E., Goldenring J. R. (2000) J. Biol. Chem. 275, 29138–29146 [DOI] [PubMed] [Google Scholar]
- 21.Ren M., Xu G., Zeng J., De Lemos-Chiarandini C., Adesnik M., Sabatini D. D. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 6187–6192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casanova J. E., Wang X., Kumar R., Bhartur S. G., Navarre J., Woodrum J. E., Altschuler Y., Ray G. S., Goldenring J. R. (1999) Mol. Biol. Cell 10, 47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilcke M., Johannes L., Galli T., Mayau V., Goud B., Salamero J. (2000) J. Cell Biol. 151, 1207–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eathiraj S., Pan X., Ritacco C., Lambright D. G. (2005) Nature 436, 415–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostermeier C., Brunger A. T. (1999) Cell 96, 363–374 [DOI] [PubMed] [Google Scholar]
- 26.Zhu G., Zhai P., Liu J., Terzyan S., Li G., Zhang X. C. (2004) Nat. Struct. Mol. Biol. 11, 975–983 [DOI] [PubMed] [Google Scholar]
- 27.Lindsay A. J., Hendrick A. G., Cantalupo G., Senic-Matuglia F., Goud B., Bucci C., McCaffrey M. W. (2002) J. Biol. Chem. 277, 12190–12199 [DOI] [PubMed] [Google Scholar]
- 28.Lindsay A. J., McCaffrey M. W. (2004) FEBS Lett. 571, 86–92 [DOI] [PubMed] [Google Scholar]
- 29.Peden A. A., Schonteich E., Chun J., Junutula J. R., Scheller R. H., Prekeris R. (2004) Mol. Biol. Cell 15, 3530–3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuda M., Kanno E., Ishibashi K., Itoh T. (2008) Mol. Cell. Proteomics 7, 1031–1042 [DOI] [PubMed] [Google Scholar]
- 31.Nagelkerken B., Van Anken E., Van Raak M., Gerez L., Mohrmann K., Van Uden N., Holthuizen J., Pelkmans L., Van Der Sluijs P. (2000) Biochem. J. 346, 593–601 [PMC free article] [PubMed] [Google Scholar]
- 32.Pagano A., Crottet P., Prescianotto-Baschong C., Spiess M. (2004) Mol. Biol. Cell 15, 4990–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deneka M., Neeft M., Popa I., van Oort M., Sprong H., Oorschot V., Klumperman J., Schu P., van der Sluijs P. (2003) EMBO J. 22, 2645–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao T. T., Deacon H. W., Reczek D., Bretscher A., von Zastrow M. (1999) Nature 401, 286–290 [DOI] [PubMed] [Google Scholar]
- 35.Gage R. M., Kim K. A., Cao T. T., von Zastrow M. (2001) J. Biol. Chem. 276, 44712–44720 [DOI] [PubMed] [Google Scholar]
- 36.Nakamura T., Shibata N., Nishimoto-Shibata T., Feng D., Ikemoto M., Motojima K., Iso-o N., Tsukamoto K., Tsujimoto M., Arai H. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13404–13409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fushimi K., Sasaki S., Marumo F. (1997) J. Biol. Chem. 272, 14800–14804 [DOI] [PubMed] [Google Scholar]
- 38.Kammerer S., Burns-Hamuro L. L., Ma Y., Hamon S. C., Canaves J. M., Shi M. M., Nelson M. R., Sing C. F., Cantor C. R., Taylor S. S., Braun A. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 4066–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishihama K., Yamada Y., Matsuo H., Segawa T., Watanabe S., Kato K., Yajima K., Hibino T., Yokoi K., Ichihara S., Metoki N., Yoshida H., Satoh K., Nozawa Y. (2007) Int. J. Mol. Med. 19, 129–141 [PubMed] [Google Scholar]
- 40.Yoshida T., Yajima K., Hibino T., Kato K., Matsuo H., Segawa T., Watanabe S., Oguri M., Yokoi K., Nozawa Y., Kimura G., Yamada Y. (2007) Int. J. Mol. Med. 20, 581–590 [PubMed] [Google Scholar]
- 41.Tingley W. G., Pawlikowska L., Zaroff J. G., Kim T., Nguyen T., Young S. G., Vranizan K., Kwok P. Y., Whooley M. A., Conklin B. R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 8461–8466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wirtenberger M., Schmutzhard J., Hemminki K., Meindl A., Sutter C., Schmutzler R. K., Wappenschmidt B., Kiechle M., Arnold N., Weber B. H., Niederacher D., Bartram C. R., Burwinkel B. (2007) Carcinogenesis 28, 423–426 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}