

Abstract
Motivation: Cancer is an evolutionary process characterized by accumulating mutations. However, the precise timing and the order of genetic alterations that drive tumor progression remain enigmatic.
Results: We present a specific probabilistic graphical model for the accumulation of mutations and their interdependencies. The Bayesian network models cancer progression by an explicit unobservable accumulation process in time that is separated from the observable but error-prone detection of mutations. Model parameters are estimated by an Expectation-Maximization algorithm and the underlying interaction graph is obtained by a simulated annealing procedure. Applying this method to cytogenetic data for different cancer types, we find multiple complex oncogenetic pathways deviating substantially from simplified models, such as linear pathways or trees. We further demonstrate how the inferred progression dynamics can be used to improve genetics-based survival predictions which could support diagnostics and prognosis.
Availability: The software package ct-cbn is available under a GPL license on the web site cbg.ethz.ch/software/ct-cbn
Contact: moritz.gerstung@bsse.ethz.ch
1 INTRODUCTION
Cancer is a disease caused by alterations of the genome. Due to systematic analyses of tumor genomes in the last decade it became apparent that cancer is caused by the combined effect of multiple mutations rather than single mutations (Hanahan and Weinberg, 2000). These mutations accumulate slowly and tumors grow over a period of multiple years. Ever since the classic sequential diagrams of Fearon and Vogelstein (1990), researchers have thus been interested in linking the progression of cancer with the observed mutations. Because of the complexity of the mutation data, however, the process of accumulating mutations is likely to be more complex than what can be represented by a single path.
To account for this complexity, various mathematical and statistical models have been derived to describe the genetic progression of cancer. These models include oncogenetic trees (Desper et al., 2000; Jiang et al., 2000; von Heydebreck et al., 2004), tree mixtures (Beerenwinkel et al., 2005; Rahnenführer et al., 2005), hidden trees (Tofigh, 2009), probabilistic network models (Hjelm et al., 2006), principal components-based methods (Höglund et al., 2001, 2005) and clustering approaches (Liu et al., 2006). The latter two methods rely on general tools identifying the correlation of data and representing it in graphical terms. Oncogenetic trees and probabilistic network models, on the contrary, are generative probabilistic models based on structural assumptions about the carcinogenetic process in which mutations accumulate. Generalizing the analyses of Fearon and Vogelstein (1990), tree models allow for a branching of the accumulation process which gives rise to different mutational pathways. The tree structure is still substantially restricting the class of graphs, but enables efficient statistical inference. A generalization of tree models is the conjunctive Bayesian network (CBN; Beerenwinkel et al., 2006, 2007). The associated graphs allow for multiple parental nodes thereby modeling the synergistic effects of multiple events in promoting subsequent mutations. The continuous time CBN (Beerenwinkel and Sullivant, 2009) also includes an explicit timeline, making quantitative predictions about the speed of carcinogenesis.
In this work, we extend the CBN by including an error model accounting for observation errors arising from the limited resolution of available clinical data or technical noise. Errors that occur during the observation lead to a hidden accumulation process similar to a hidden Markov model. We apply this method to publicly available datasets from the Progenetix database (www.progenetix.net; Baudisand Cleary, 2001) and compare the results for different cancer types. We show that the resulting graphs deviate substantially from classic linear diagrams and from oncogenetic trees, therefore indicating a high degree of genetic complexity in the process of carcinogenesis.
2 METHODS
Our statistical model for the accumulation and detection of mutations is a specific Bayesian network, where the accumulation of mutations is modeled by a CBN. The CBN dynamics is hidden by an observation process; we therefore term the model hidden CBN (H-CBN). For the H-CBN, we derive an expectation-maximatization (EM) algorithm for the estimation of the continuous model parameters. Furthermore, we propose a simulated annealing algorithm to find the graph that maximizes the likelihood of the data.
2.1 Bayesian networks and the detection of cancer
The clinical detection of a tumor is a complex process, which, in the reductionist view, contains three elements: (i) the malignancy has developed; (ii) it is diagnosed in a clinical test; and (iii) the test is correct. Formalizing these notions, we define the following model: suppose the tumor develops in an initially healthy tissue after a time T. The waiting time is a random variable, because the exact occurrence of the tumor varies across patients. Diagnosis occurs at time Ts, which is also a random variable. Because the dependence of T and Ts is generally unknown, we assume that Ts is independent of T. Hence, the joint density factorizes, f(t, ts)=f(t)f(ts). The disease can only be detected if it is present at the time of observation. Let X∈{0, 1} denote the stochastic variable indicating whether the disease is present at diagnosis (X=1). The probability of X can be decomposed in a Bayesian fashion as
| (1) |
where the conditional probability Prob[X=1∣T=t, Ts=ts]=𝕀(t<ts) is simply given by the indicator function 𝕀.
So far we have assumed that the diagnosis is always correct. Suppose that, with a small probability ϵ, the disease might be overlooked (false negative) or misdiagnosed (false positive). Hence, the diagnosis is a probabilistic event Y that depends on X as Prob[Y]=∑X=0,1 Probϵ[Y∣X]Prob[X], with
| (2) |
and Prob[X] as defined in Equation (1).
The stochastic variables {T, Ts, X, Y} form a Bayesian network: the joint density factors into conditional densities according to the directed acyclic graph (DAG) shown in Figure 1A.
Fig. 1.

(A) Simple Bayesian network for cancer detection. T denotes the waiting time for the tumor, Ts the time of diagnosis. The disease is present, X=1, if T<Ts. Yet the diagnosis Y may contain errors. (B) Graph of an H-CBN example. The waiting times Ti of the mutations i=1,…, 4 evolve according to the CBN depicted in the upper left: mutations 1 and 2 arise independently; mutation 3 can only occur after 1, T1<T3, mutation 4 occurs only if both 1 and 2 are present, T1, T2<T4. A mutation i is present in the genotype X=(X1,…, X4) if Ti<Ts, where Ts is the independent stopping time. The observations Y=(Y1,…, Y4) contain errors that occur independently for each mutation.
2.2 Conjunctive Bayesian networks
We now extend our model for the development of cancer. This process is driven by the accumulation of several genetic lesions. We therefore generalize the waiting time T=(T1,…, Tn) to incorporate the occurrence of n mutations. A model for the accumulation of multiple, possibly collinear mutations, is the CBN (Beerenwinkel and Sullivant, 2009).
Let n be the total number of possible mutations and define Ti as the waiting time for mutation i∈{1,…, n}=[n]. Furthermore, let pa(i) denote the set of mutations that need to be present before mutation i can fixate. We define Ti to be exponentially distributed with parameter λi conditioned on all mutations pa(i) being present,
| (3) |
The density of Ti, conditioned on the predecessors {Tj}j∈pa(i), is
| (4) |
where 𝕀 denotes the indicator function. The set of waiting times {Ti}i∈[n] forms a CBN with a partial temporal order Tj<Ti for all j∈pa(i) and all i∈[n]. The underlying algebraic structure of the mutations is a partially ordered set (poset) P, with the cover relations j→i for j∈pa(i). The cover relations of P correspond to the directed edges in the graph of the Bayesian network as illustrated in Figure 1B (top left). For the censoring, we assume that the waiting time Ts is independently exponentially distributed with parameter λs, Ts ∼ Exp(λs). We thus extend the poset P by the stopping event s, which does not have any order relation to the mutations i. This assumption resembles that the time of diagnosis is not bound to the presence of mutations.
In the previous section, we have introduced X as the binary event that the disease is present. Since we are now considering multiple mutations characterizing the transformation to malignancy, stopping generates a binary vector X=(X1,…, Xn)∈{0, 1}n, the genotype of the tumor. Using that the conditional density of X factorizes according to the Bayesian network structure, Prob[X∣ T, Ts]=∏i=1n Prob[Xi ∣ Ti, Ts] and Equation (1) one obtains:
| (5) |
Probλ,P[X] is invariant under rescalings of λ=(λs, λ1,…, λn); hence λi, i∈[n], can only be estimated up to the factor λs. Unless λs is known, we set λs=1.
2.2.1 H-CBN
Parameter estimation for the CBN requires that all mutations Xi are identified correctly. Because of experimental limitations, however, the observed genotype Y=(Y1,…, Yn) might contain errors. This could be because either a mutation is not functional (false positive) or below the limit of detection (false negative). We model the observation process by assuming that a mutation i is falsely observed with probability ϵ as in Equation (2). Because the conditioned variables Yi|Xi are independent for each i∈[n], the conditional probability of an observation Y given a genotype X is:
| (6) |
Here d(X, Y)=∑i=1n|Xi−Yi| denotes the Hamming distance between the genotype X and the observation Y. Hence, the dynamics of the accumulation process is a hidden process by two means: first, the dynamics is censored by a stopping process, and second, the observation contains errors. A schematic illustration of the H-CBN is shown in Figure 1B: the process of mutating is described by the waiting times Ti evolving according to partial order constraints. Genotypes X are generated by the censoring caused by Ts. Note that the mutations Xi are independent, conditioned on Ti and Ts. Finally, the observation process is erroneous, generating the observations Yi.
To estimate the model parameters, we must compute the posterior probability of observing the genotype X given an observation Y. The posterior can be computed by Bayes' theorem:
| (7) |
Here, Probλ,P[X] denotes the prior probability that the genotype X occurs according to Equation (5); J(P) is the lattice of order ideals, containing all genotypes compatible with the poset P (Beerenwinkel et al., 2007).
2.3 Parameter estimation
Although the dynamics of the H-CBN can only indirectly be observed, the model parameters ϵ and λ can be estimated by an EM algorithm. To estimate the set of relations P, we propose the method of simulated annealing.
2.3.1 EM algorithm
The joint probability of N independent observations Y=(Y(1),…, Y(N)) factorizes into the product Probϵ,λ,P[Y]=∏l=1NProbϵ,λ,P[Y(l)]=∏l=1N∑X∈J(P) Probϵ[Y(l)∣X]Probλ,P[X].
Hence, the log-likelihood of the data is:
| (8) |
We are interested in maximizing the log-likelihood ℓY(ϵ, λ, P) given observations Y. The likelihood depends on the observation error rate ϵ, the waiting time parameters λ and the relations in P. The parameters λ could be estimated by an EM algorithm if P and the true genotypes X=(X(1),…, X(N)) were known. In the case of hidden X and fixed P, this method can be embedded into a nested EM algorithm. The outer loop computes the parameter estimate 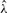 and the inner loop computes the error rate estimate
and the inner loop computes the error rate estimate  given the iterated value
given the iterated value 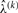 .
.
If both X and Y were known, the maximum likelihood (ML) estimator of the observation error rate would be the average distance per mutation, 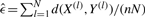 . Because X is hidden,
. Because X is hidden,  is computed iteratively by using the conditional expectation of the sufficient statistic d(X, Y(l)) (E-step) for computing the ML estimate (M-step):
is computed iteratively by using the conditional expectation of the sufficient statistic d(X, Y(l)) (E-step) for computing the ML estimate (M-step):
| (9) |
Doing this until the convergence yields an estimator  that locally maximizes
that locally maximizes 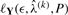 ; this value is in turn used to estimate λ.
; this value is in turn used to estimate λ.
For N realizations of the waiting times Ti, the ML estimator of the parameter λi is (Beerenwinkel and Sullivant, 2009):
| (10) |
As the waiting times Ti are censored, the denominator is replaced by the expected sufficient statistic 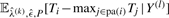 in the E-step of the outer EM algorithm. These values are computed from the Bayesian decomposition:
in the E-step of the outer EM algorithm. These values are computed from the Bayesian decomposition:
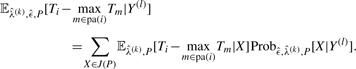 |
(11) |
The expectations 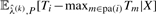 can be computed by dynamic programming. Yet, they need to be computed for all possible values of the hidden genotypes X∈J(P), imposing computational limitations in the case of many mutations. In the M-step of the outer EM-loop, the expected values in Equation (11) are then used for computing the next iteration step
can be computed by dynamic programming. Yet, they need to be computed for all possible values of the hidden genotypes X∈J(P), imposing computational limitations in the case of many mutations. In the M-step of the outer EM-loop, the expected values in Equation (11) are then used for computing the next iteration step 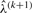 according to Equation (10). Iterating until the changes in
according to Equation (10). Iterating until the changes in 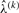 are sufficiently small gives the estimator
are sufficiently small gives the estimator 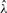 .
.
2.3.2 Simulated annealing
The EM algorithm locally maximizes the log-likelihood of the data, Equation (8), for a given poset P. In most of the situations, however, one is mainly interested in inferring P. Because the number of continuous parameters λi is fixed by the number of mutations and not by the number of relations in P, all models have the same degree of freedom. Therefore, we select the ML poset 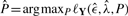 without an additional model selection criterion such as the Akaike or Bayesian information criterion (AIC and BIC, respectively). Yet due to the observation errors, there exists no direct analytical way to determine
without an additional model selection criterion such as the Akaike or Bayesian information criterion (AIC and BIC, respectively). Yet due to the observation errors, there exists no direct analytical way to determine 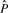 . Instead, we have to rely on heuristic ways to find the ML estimate. We do so by using a simulated annealing procedure (Kirkpatrick et al., 1983). In this algorithm, one computes
. Instead, we have to rely on heuristic ways to find the ML estimate. We do so by using a simulated annealing procedure (Kirkpatrick et al., 1983). In this algorithm, one computes 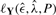 for a given poset P and the data Y; one then randomly generates a new poset P′ and accepts this if either
for a given poset P and the data Y; one then randomly generates a new poset P′ and accepts this if either 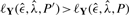 or, alternatively, with probability
or, alternatively, with probability 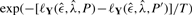 . The temperature T determines to which extend steps decreasing the log-likelihood are allowed, thus reducing the risk of remaining in local maxima. As T→0 only steps increasing
. The temperature T determines to which extend steps decreasing the log-likelihood are allowed, thus reducing the risk of remaining in local maxima. As T→0 only steps increasing 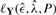 are accepted.
are accepted.
The efficiency of the algorithm relies on an adequate strategy for choosing the new poset P′. Our algorithm randomly removes or adds a cover relation to P. Because a poset defines a special DAG, we only consider the addition of relations yielding another poset. As the occurrence of a relation relies on the correlation of the observed data, we also allow for changing the direction of a relation. Moreover, a sequence i→k→j can be replaced by i→k and i→j, thereby changing two relations at once. To avoid inefficient moves, we use a preselection heuristic based on the fraction of data ρ without observation errors, which is a proxy for the likelihood. This computation is very fast as it does not require the nested EM algorithm. Moves are preselected with probability exp(−[ρ−ρ′]/0.05) if ρ′ < ρ and 1 otherwise. For moves having passed preselection, we then compute 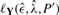 and proceed with the algorithm as stated above.
and proceed with the algorithm as stated above.
3 RESULTS
We first present results on simulated datasets illustrating the power of the algorithm. We then analyze cytogenetic data for different cancer types and demonstrate how the evolutionary model can be used for an improved survival analysis.
3.1 Simulations
To test our algorithms, we simulated data for different posets and parameter values of ϵ. We simulated 50 datasets each with N=800 observations and n=12 mutations. For ϵ, we chose parameter values of 0.01 and 0.1, respectively; for λ we used random values. As posets we selected the completely independent case, a linear chain 1→2→…, and two more complex posets shown in Figure 2A.
Fig. 2.

Estimation on simulated data. (A) Simulated poset structures. (B) Boxplots of the estimates  for the true poset P. True parameter values are indicated by asterisks. (C) Distributions of the estimates
for the true poset P. True parameter values are indicated by asterisks. (C) Distributions of the estimates  after estimating the poset. (D) Boxplots of the waiting time parameter estimates
after estimating the poset. (D) Boxplots of the waiting time parameter estimates 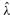 for the estimates of poset 1. (E) Boxplots of the numerically observed FPR and FNR for two values of ϵ. The sample size was N=800, and B=50 runs were performed.
for the estimates of poset 1. (E) Boxplots of the numerically observed FPR and FNR for two values of ϵ. The sample size was N=800, and B=50 runs were performed.
The simulations show that for a known poset P, the estimation of the error rate ϵ is highly accurate for both parameter values, with the highest variance arising in the independent case (Fig. 2B). The variance increases if the poset is also estimated by simulated annealing (Fig. 2C). Again the variance is largest in the independent cases with a bias toward larger values. For all other, more realistic, posets, however, the estimation of ϵ is very accurate. The same holds for the estimation of the waiting time parameters λ. The estimates after the annealing process have low variance, as long as the expected frequencies are larger than the noise level ϵ, as shown for poset 1 in Figure 2D (with similar results for poset 2). Outliers arise, most likely, if the estimated order relations of the corresponding mutations contain errors. If the noise level exceeds the expected frequency of a mutation, the variance of the associated waiting time estimator becomes large, because the true frequency cannot be accurately recovered. This is the case for the late-stage mutations 10, 11 and 12, as depicted in Figure 2D.
Slightly more complicated than estimating the parameters is finding the ML poset 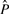 . The number of relations in a poset is given by the transitive closure of the cover relations, which are represented by edges in the corresponding DAG. The linear poset, for example, has exactly n−1 cover relations, but these sum up to a total number of r0=n(n−1)/2 relations. This number r0 is the maximal number of relations that can be found in any poset. We thus define the observed false positive rate
. The number of relations in a poset is given by the transitive closure of the cover relations, which are represented by edges in the corresponding DAG. The linear poset, for example, has exactly n−1 cover relations, but these sum up to a total number of r0=n(n−1)/2 relations. This number r0 is the maximal number of relations that can be found in any poset. We thus define the observed false positive rate 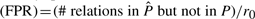 , and the false negative rate
, and the false negative rate 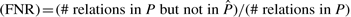 .
.
For all four structures, the estimation of P is very precise for ϵ=0.01, with median error rates < 0.05 (Fig. 2E). The distribution of false positive and false negative relations depends on the specific poset. For the independent case, the FNR is zero by definition, for the other posets both types of errors are possible. Both types of errors increase for the larger error rate, ϵ=0.1. In this case, we find median error rates of ∼0.1–0.2. Both the FPR and FNR increase monotonically as compared with their values at ϵ=0.01, showing that the structure imposes a distinct bias. The highest errors arise in the estimation of poset 1, which has the most complex structure. But still the median error rates are <17%. Importantly, the estimation of the error rate ϵ remains realistic despite inaccuracies in 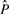 (Fig. 2C), making it possible to identify noisy data even without complete knowledge of the true poset.
(Fig. 2C), making it possible to identify noisy data even without complete knowledge of the true poset.
3.2 Renal cell carcinoma
Today, there exists a wealth of data on genetic alterations in cancer. The largest resource for whole-genome aberration data so far has been generated through cytogenetic (Mitelman et al., 2009) or molecular cytogenetic, e.g. chromosomal and array-based comparative genomic hybridization (CGH), techniques. Here, we apply our method to disease-specific CGH data from the Progenetix database (www.progenetix.net; Baudis and Cleary, 2001). A descriptive analysis of this data can be found in Baudis (2007).
We first apply our method to a dataset on renal cell carcinoma (RCC) from the Progenetix database. This dataset (N=251) has been published in parts before (Jiang et al., 2000), and contains clinical follow-up on patient survival for 82 cases. The most frequent losses for this cancer type are: –3p (59.4 %), –4q (29.9%), –6q (25.5%), –9p (24.4 %), –13q (23.1%), –14q (17.9 %), –8p (16.3%) and –18q (14.7%). Characteristic is the loss of the p arm on chromosome 3, which hosts the VHL gene, an important tumor suppressor. The most frequent gains are: +5q(31) (25.2%), +17q (21.2 %) and +7 (21.2%).
For our analysis, we restrict ourselves to the n=12 copy number alterations (CNAs) used by Jiang et al. (2000), which were selected by the method of Brodeur et al. (1982). These do not include the gain of chromosome 5p and the loss on 14q. Instead, the alterations of the X chromosome –X(p) (10.0%; often whole chromosome) and +X(p) (9.6%; often whole chromosome) get selected, as well as the gain on chromosome 17p (13.5%). Somewhat surprisingly, the estimated ML poset (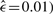 contains only two relations, –4q → –4p and +17q → +17p. That is, loss of 4q appears before the loss of the additional chromosome arm 4p, or the whole chromosome. The second relation exists between gain of chromosome 17q and the gain on the opposing chromosome arm. This could be the result of aneuploidy of chromosome 17, or of gains spanning both chromosome arms.
contains only two relations, –4q → –4p and +17q → +17p. That is, loss of 4q appears before the loss of the additional chromosome arm 4p, or the whole chromosome. The second relation exists between gain of chromosome 17q and the gain on the opposing chromosome arm. This could be the result of aneuploidy of chromosome 17, or of gains spanning both chromosome arms.
Comparing this result with the oncogenetic tree models of Jiang et al. (2000), one finds that the tree contains more relations, but it also has a much smaller likelihood (likelihood ratio  ). Interestingly, the tree occurs close to a local maximum of the likelihood. Performing a local search for the ML H-CBN starting from the tree revealed a poset with Λ=0.004(ϵ=0.08; Fig. 3). This value is on the order of changes of a single relation, hence the statistical difference is small. Moreover, the relations appear to be in better agreement with the pathways reported previously in the literature. For example, it is known that the VHL gene on 3p plays an important initializing role in RCC (Gnarra et al., 1994). In the poset shown in Figure 3, the initializing events are –3p and –4q. The mutation –3p induces a pathway including –9p and –18q, which has been previously reported by Höglund et al. (2004). A second pathway involves both –3p and –4q, which induce –4p and –6q, as well as +17q and –13q, ultimately leading to –Xp. This progression is similar to the one proposed by the tree models of Jiang et al. (2000); yet the poset includes nodes with multiple incoming edges such as –Xp or –6q, which cannot be represented by a tree. In the analysis of Höglund et al. (2004), the losses on chromosome 4 are, in general, a late-stage event. Our approach recovers the same grouping, but assigns an initializing role to –4q, in agreement with the work of (Jiang et al., 2000). Höglund et al. (2004) also report an independent pathway involving the gains on chromosome 17, eventually leading to –4. Our analysis suggests that those alterations occur independently from –3p, but only after being initialized by –4q. Note, however, that the likelihood ratios of some edges are relative large; hence the statistical evidence for those relations is weak.
). Interestingly, the tree occurs close to a local maximum of the likelihood. Performing a local search for the ML H-CBN starting from the tree revealed a poset with Λ=0.004(ϵ=0.08; Fig. 3). This value is on the order of changes of a single relation, hence the statistical difference is small. Moreover, the relations appear to be in better agreement with the pathways reported previously in the literature. For example, it is known that the VHL gene on 3p plays an important initializing role in RCC (Gnarra et al., 1994). In the poset shown in Figure 3, the initializing events are –3p and –4q. The mutation –3p induces a pathway including –9p and –18q, which has been previously reported by Höglund et al. (2004). A second pathway involves both –3p and –4q, which induce –4p and –6q, as well as +17q and –13q, ultimately leading to –Xp. This progression is similar to the one proposed by the tree models of Jiang et al. (2000); yet the poset includes nodes with multiple incoming edges such as –Xp or –6q, which cannot be represented by a tree. In the analysis of Höglund et al. (2004), the losses on chromosome 4 are, in general, a late-stage event. Our approach recovers the same grouping, but assigns an initializing role to –4q, in agreement with the work of (Jiang et al., 2000). Höglund et al. (2004) also report an independent pathway involving the gains on chromosome 17, eventually leading to –4. Our analysis suggests that those alterations occur independently from –3p, but only after being initialized by –4q. Note, however, that the likelihood ratios of some edges are relative large; hence the statistical evidence for those relations is weak.
Fig. 3.

Renal cell carcinoma. Locally optimal poset close to the tree of Jiang et al. (2000). Nodes correspond to specific recurrent mutations (≳20%). Small numbers at each edge denote the fold change Λ of the likelihood if the corresponding relation is left out.
3.2.1 Survival analysis
For 82 cases of the RCC dataset, clinical follow-up data with survival information was available. The standard method for survival analysis is the Cox proportional hazards model (Cox, 1972). Here, the risk associated with a genotype X is given by the hazard function
| (12) |
where λ0(t) denotes the baseline hazard. The contribution of each mutation to the risk is given by the coefficients β, which are estimated from the survival data. A multivariate survival analysis on all 12 CNAs does not reveal a significant association of any of the selected CNAs with survival (P=0.185, likelihood ratio test). This might be due to erroneous observations. We therefore calculated the maximum a posteriori (MAP) estimator 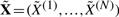 of the hidden data X. For each observation Y(l), it is defined as:
of the hidden data X. For each observation Y(l), it is defined as:
| (13) |
where 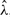 , and
, and  are the model parameters estimated on the complete dataset (N=251). Based on the dynamics of the CBN, this strategy selects the most probable hidden genotype. For the sparse poset, the MAP estimates are almost identical to the observations (mean Hamming distance
are the model parameters estimated on the complete dataset (N=251). Based on the dynamics of the CBN, this strategy selects the most probable hidden genotype. For the sparse poset, the MAP estimates are almost identical to the observations (mean Hamming distance 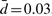 , maximal distance dmax=1). The poset shown in Figure 3 introduces a stronger deviation (
, maximal distance dmax=1). The poset shown in Figure 3 introduces a stronger deviation (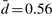 , dmax=4). The average distances per mutation are denoted in Table 1. Interestingly, most CNAs have a higher frequency in the observed data, except for –3p and –4q. This effect could be a result of the coarse-graining to chromosome arms, which erroneously includes alterations in non-functional bands.
, dmax=4). The average distances per mutation are denoted in Table 1. Interestingly, most CNAs have a higher frequency in the observed data, except for –3p and –4q. This effect could be a result of the coarse-graining to chromosome arms, which erroneously includes alterations in non-functional bands.
Table 1.
Average distances (in percent) of the estimated hidden data to the observed data, 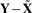 , for the RCC poset shown in Figure 3
, for the RCC poset shown in Figure 3
| –3p | –4p | –4q | –6q | –8p | –9p | –13q | –18q | –Xp | +17p | +17q | +Xp |
|---|---|---|---|---|---|---|---|---|---|---|---|
| –2 | 1 | –2 | 8 | 3 | 4 | 6 | 7 | 7 | 5 | 6 | 6 |
Re-estimating the risk coefficients for the estimated hidden data 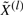 , we find a somewhat stronger overall effect (P=0.10; likelihood ratio test). To pinpoint the relevant CNAs, we selected the best covariate subset by applying a LASSO version of the Cox proportional hazards model (Park and Hastie, 2007; Tibshirani, 1997). Here, the sparseness of the solution can be controlled, by imposing an L1 penalty on the likelihood. The optimal penalization parameter is chosen by maximization of the cross-validated partial likelihood. Applying this method to the estimated hidden data
, we find a somewhat stronger overall effect (P=0.10; likelihood ratio test). To pinpoint the relevant CNAs, we selected the best covariate subset by applying a LASSO version of the Cox proportional hazards model (Park and Hastie, 2007; Tibshirani, 1997). Here, the sparseness of the solution can be controlled, by imposing an L1 penalty on the likelihood. The optimal penalization parameter is chosen by maximization of the cross-validated partial likelihood. Applying this method to the estimated hidden data 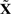 reveals a combination of –3p, –4q and –Xp as the best predictor subset. This result is confirmed by the subsets selected by the BIC (excluding –Xp) and AIC (including –6q) model selection criteria. For the LASSO selection, the risk is balanced between the relieving effect of –3p (
reveals a combination of –3p, –4q and –Xp as the best predictor subset. This result is confirmed by the subsets selected by the BIC (excluding –Xp) and AIC (including –6q) model selection criteria. For the LASSO selection, the risk is balanced between the relieving effect of –3p (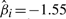 ) and the malignant effects of –4q and –Xp (
) and the malignant effects of –4q and –Xp (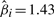 and
and 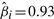 , respectively). A positive effect of VHL mutations on 3p has been reported previously for clear-cell RCC (Yao et al., 2002). The Kaplan–Meyer plot of the data is shown in Figure 4. Depicted are three groups according to the overall risk given by Equation (12) with the LASSO estimates
, respectively). A positive effect of VHL mutations on 3p has been reported previously for clear-cell RCC (Yao et al., 2002). The Kaplan–Meyer plot of the data is shown in Figure 4. Depicted are three groups according to the overall risk given by Equation (12) with the LASSO estimates 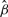 and the MAP covariates
and the MAP covariates 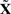 . The groups are clearly separated with 5 year survival rate of <20% for patients in the highest risk group. On the contrary, those in the lowest risk group have a 10 year survival of 70%. Similar results are obtained using the posterior expectations 𝔼[X∣Y(l)] instead of the MAP estimates (data not shown).
. The groups are clearly separated with 5 year survival rate of <20% for patients in the highest risk group. On the contrary, those in the lowest risk group have a 10 year survival of 70%. Similar results are obtained using the posterior expectations 𝔼[X∣Y(l)] instead of the MAP estimates (data not shown).
Fig. 4.

Kaplan–Meier plot of the RCC dataset. Shown are three risk groups according to the coefficients 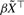 from the LASSO selection in the Cox model.
from the LASSO selection in the Cox model. 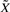 denotes the MAP estimate of the hidden genotypes.
denotes the MAP estimate of the hidden genotypes.
3.3 Breast and colorectal cancer
We continue by exploring the poset structure of other cancer data available in the Progenetix database. For this purpose, we chose breast and colorectal cancer as two prominent examples.
3.3.1 Breast cancer
The data for breast cancer found in the Progenetix database consists of 817 cases. The most frequent (>20%) gains are: +1(q31) (59.7%), +8(q23) (48.0%), +17q (36.2%), +20(q) (31.7%), +16(p) (25.1%), +11q13 (24.5%) and +3q (22.4%). Highly recurrent losses (> 20%) are: –16(q) (29.0%), –8p (27.8%) and –13q (24.7%). The graph of the ML poset (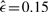 ) estimated by our method is shown in Figure 5A. The gain+1q acts as a central initializing event, inducing –8p, +3q and –16q in conjunction with +8q. Independently of this pathway, there exists a pathway involving +17q, +16p, +20q and –13q.
) estimated by our method is shown in Figure 5A. The gain+1q acts as a central initializing event, inducing –8p, +3q and –16q in conjunction with +8q. Independently of this pathway, there exists a pathway involving +17q, +16p, +20q and –13q.
Fig. 5.

Estimated posets for breast cancer (A) and colorectal cancer (B). Nodes correspond to specific recurrent mutations (≳ 20%). Numbers at each edge denote the likelihood change Λ if the corresponding relation is left out.
The +1q/+8q pathway corresponds roughly to a previously reported path of breast cancer (Höglund et al., 2002b). A putative oncogene on chromosome arm 8q is MYC. Despite its high recurrence, there is no obvious candidate oncogene on chromosome 1q. Furthermore, the progression into the –16q state has been associated with high differentiation and good prognosis (Roylance et al., 1999). The initializing event of the latter path, 17q, is the locus of ERBB2, a well-known oncogene; typically gains of this chromosome correspond to a poor prognosis (Buerger et al., 1999). Targets on 20q and 13q are AURKA and BRCA2, respectively, which are both involved in the maintenance of genome stability.
3.3.2 Colon cancer
For colorectal cancer, 570 cases were filed in the Progenetix database. The gains recurring most frequently (≳20) are: +20(q13) (46.7%), +13q (37.9%), +8(q24), +7(q) (32.8%) and +X(q24) (30.4%). The most frequent losses are: –18(q22) (44.4%), –8p(22) (34.2%), –17p12 (25.3%), –4(q) (23.3%), –15q (19.2%) and –1p (18.8%). The estimated poset (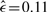 ) is shown in Figure 5B. For this type of cancer, +20q appears to be the central initializing event. This chromosome arm harbors the putative oncogene AURKA, which is known to cause genetic instability (Bischoff et al., 1998). This instability-causing role agrees well with an initializing role found by our approach. Loss of 18q then appears to play a central role in the upcoming stages of tumor progression by triggering a variety of subsequent losses. The q arm of chromosome 18 is locus of the tumor suppressor SMAD4, which indicates an important role in tumor development.
) is shown in Figure 5B. For this type of cancer, +20q appears to be the central initializing event. This chromosome arm harbors the putative oncogene AURKA, which is known to cause genetic instability (Bischoff et al., 1998). This instability-causing role agrees well with an initializing role found by our approach. Loss of 18q then appears to play a central role in the upcoming stages of tumor progression by triggering a variety of subsequent losses. The q arm of chromosome 18 is locus of the tumor suppressor SMAD4, which indicates an important role in tumor development.
This result agrees with previous findings based on PCA (Höglund et al., 2002a). Those authors report two overlapping pathways in colorectal tumors, one dominated by losses, the other mostly involving chromosomal gains, whereas for adenomas, the patterns are less clear. In the gain pathway, an intermediate role was assigned to +20q, whereas in our analysis it is a main trigger in agreement with its putative biological role. The other pathway reported by Höglund et al. (2002a) is triggered by –1p and involves –17p, –8p, –18q and –15q as downstream events. Our analysis recovers this grouping, however, in the opposite order: –18q induces the other alterations. Our model also elucidates a possible overlap of the two pathways through the events –18q and +8q.
4 DISCUSSION
We have developed a statistical method for the inference of partial temporal orders of cancer mutations. Our method is based on a waiting time model of cancer progression allowing for temporal constraints in terms of a continuous time CBN. We have extended this model to account for observation errors and presented algorithms to infer the ML model parameters.
Similar to the CBN, oncogenetic trees were developed to model the dependencies among accumulating mutations (Desper et al., 2000; Jiang et al., 2000; von Heydebreck et al., 2004). H-CBN extends the concept of oncogenetic trees in two ways: first, the CBN substantially extends the class of possible graphs by allowing for more than one parent per node. Biologically this allows to include direct dependencies on multiple mutations. Second, H-CBN includes an observation process. Therefore, a fraction of data deviating from the CBN can be explained by observation errors. It thus provides a direct interpretation for the fraction of data not matching the graph. This is in contrast with mixture models, where the mixture process is less intuitive. Another interpretation of our error model is that it enables approximating more general accumulation processes by the closest CBN.
A further improvement on our model could be to use different parameters ϵ+ and ϵ− for false positives and false negatives in the error model, as used in the context of longitudinal data (Beerenwinkel and Drton, 2007). This would refine the error process and give more detailed information about the nature of mismatches. Another modification of the model would be to use disjunctive instead of a conjunctive action of multiple incoming edges (Beerenwinkel et al., 2006). This model would drastically enlarge the class of possible graphs; however, we would expect only a limited statistical power given the size of available data. The same limitation would also apply to a full Bayesian network approach on the complete set of DAGs.
Our analysis of cancer CGH data reveals complex structures of cancer progression. Our results indicate that there typically exist multiple independent events triggering complex downstream pathways. This generalizes the classic sequential model of cancer progression by Fearon and Vogelstein (1990). For the RCC dataset, we have also shown that the prognostic value of CNAs can be increased by correcting for observation errors using the MAP estimates of the genotypes. This approach revealed the combination of –4q, –3p and –Xp as the best genetic predictor subset for RCC.
In this work, we have applied our data to available CGH mutation data. This data is binary and simply denotes the presence of a certain chromosomal alteration. Due to the limited resolution, however, important information about small-scale mutation such as point mutations may be missing. Also epigenetic information is not covered. We emphasize that our method is in principle suitable for the analysis of such data, including data on differentially expressed genes. Also clinical variables like treatment, tumor subtypes and patient information can be easily integrated into our Bayesian network approach.
Conflict of Interest: none declared.
REFERENCES
- Baudis M. Genomic imbalances in 5918 malignant epithelial tumors: an explorative meta-analysis of chromosomal CGH data. BMC Cancer. 2007;7:226. doi: 10.1186/1471-2407-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudis M, Cleary ML. Progenetix.net: an online repository for molecular cytogenetic aberration data. Bioinformatics. 2001;17:1228–1229. doi: 10.1093/bioinformatics/17.12.1228. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N, Drton M. A mutagenetic tree hidden Markov model for longitudinal clonal HIV sequence data. Biostatistics. 2007;8:53–71. doi: 10.1093/biostatistics/kxj033. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N, Sullivant S. Markov models for accumulating mutations. Biometrika. 2009;96:645–661. [Google Scholar]
- Beerenwinkel N, et al. Mtreemix: a software package for learning and using mixture models of mutagenetic trees. Bioinformatics. 2005;21:2106–2107. doi: 10.1093/bioinformatics/bti274. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N, et al. Evolution on distributive lattices. J. Theor. Biol. 2006;242:409–420. doi: 10.1016/j.jtbi.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N, et al. Conjunctive Bayesian networks. Bernoulli. 2007;13:893–909. [Google Scholar]
- Bischoff JR, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM, et al. Statistical analysis of cytogenetic abnormalities in human cancer cells. Cancer Genet. Cytogenet. 1982;7:137–152. doi: 10.1016/0165-4608(82)90010-3. [DOI] [PubMed] [Google Scholar]
- Buerger H, et al. Different genetic pathways in the evolution of invasive breast cancer are associated with distinct morphological subtypes. J. Pathol. 1999;189:521–526. doi: 10.1002/(SICI)1096-9896(199912)189:4<521::AID-PATH472>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Cox DR. Regression models and life-tables. J. R. Stat. Soc. Ser. B Methodol. 1972;34:187–220. [Google Scholar]
- Desper R, et al. Distance-based reconstruction of tree models for oncogenesis. J. Comput. Biol. 2000;7:789–803. doi: 10.1089/10665270050514936. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Gnarra JR, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hjelm M, et al. New probabilistic network models and algorithms for oncogenesis. J. Comput. Biol. 2006;13:853–865. doi: 10.1089/cmb.2006.13.853. [DOI] [PubMed] [Google Scholar]
- Höglund M, et al. Multivariate analyses of genomic imbalances in solid tumors reveal distinct and converging pathways of karyotypic evolution. Genes Chromosomes Cancer. 2001;31:156–171. doi: 10.1002/gcc.1129. [DOI] [PubMed] [Google Scholar]
- Höglund M, et al. Dissecting karyotypic patterns in colorectal tumors: two distinct but overlapping pathways in the adenoma-carcinoma transition. Cancer Res. 2002a;62:5939–5946. [PubMed] [Google Scholar]
- Höglund M, et al. Multivariate analysis of chromosomal imbalances in breast cancer delineates cytogenetic pathways and reveals complex relationships among imbalances. Cancer Res. 2002b;62:2675–2680. [PubMed] [Google Scholar]
- Höglund M, et al. Dissecting karyotypic patterns in renal cell carcinoma: an analysis of the accumulated cytogenetic data. Cancer Genet. Cytogenet. 2004;153:1–9. doi: 10.1016/j.cancergencyto.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Höglund M, et al. Statistical behavior of complex cancer karyotypes. Genes Chromosomes Cancer. 2005;42:327–341. doi: 10.1002/gcc.20143. [DOI] [PubMed] [Google Scholar]
- Jiang F, et al. Construction of evolutionary tree models for renal cell carcinoma from comparative genomic hybridization data. Cancer Res. 2000;60:6503–6509. [PubMed] [Google Scholar]
- Kirkpatrick S, et al. Optimization by simulated annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. Distance-based clustering of CGH data. Bioinformatics. 2006;22:1971–1978. doi: 10.1093/bioinformatics/btl185. [DOI] [PubMed] [Google Scholar]
- Mitelman F, et al. Mitelman Database of Chromosome Aberrations in Cancer. 2009 Available at http://cgap.nci.nih.gov/Chromosomes/Mitelman (last accessed date August 31, 2009) [Google Scholar]
- Park MY, Hastie T. L1-regularization path algorithm for generalized linear models. J. R. Stat. Soc. Ser. B Stat. Methodol. 2007;69:659–677. [Google Scholar]
- Rahnenführer J, et al. Estimating cancer survival and clinical outcome based on genetic tumor progression scores. Bioinformatics. 2005;21:2438–2446. doi: 10.1093/bioinformatics/bti312. [DOI] [PubMed] [Google Scholar]
- Roylance R, et al. Comparative genomic hybridization of breast tumors stratified by histological grade reveals new insights into the biological progression of breast cancer. Cancer Res. 1999;59:1433–1436. [PubMed] [Google Scholar]
- Tibshirani R. The lasso method for variable selection in the Cox model. Stat. Med. 1997;16:385–395. doi: 10.1002/(sici)1097-0258(19970228)16:4<385::aid-sim380>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Tofigh A. PhD Thesis, KTH School of Computer Science and Communication. Stockholm, Sweden: 2009. Using trees to capture reticulate evolution. [Google Scholar]
- von Heydebreck A, et al. Maximum likelihood estimation of oncogenetic tree models. Biostatistics. 2004;5:545–556. doi: 10.1093/biostatistics/kxh007. [DOI] [PubMed] [Google Scholar]
- Yao M, et al. VHL tumor suppressor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J. Natl Cancer Inst. 2002;94:1569–1575. doi: 10.1093/jnci/94.20.1569. [DOI] [PubMed] [Google Scholar]