

Abstract
The PKD1 and PKD2 genes are the genes that are mutated in patients suffering from autosomal dominant polycystic kidney disease. The human PKD2 gene codes for a 968-amino acid long membrane protein called polycystin-2 that represents a cation channel whose activity can be regulated by Ca2+ ions. By CD, fluorescence, and NMR spectroscopy, we have studied a 117-amino acid-long fragment of the cytoplasmic domain of polycystin-2, polycystin-2-(680–796) that was proposed to contain a Ca2+-binding site. NMR structure determination reveals the existence of two Ca2+-binding sites in polycystin-2-(680–796) arranged in a typical and an atypical EF-hand motif. In the absence of Ca2+ the protein forms a dimer that is dissociated by Ca2+ binding. This dissociation may be related to the Ca2+ inactivation observed earlier. The calcium affinity of the protein was determined by fluorescence and NMR spectroscopy. At 293 K, the KD values for the high and low affinity sites are 55 μm and 179 μm, respectively.
The PKD2 gene is one of the two genes mutated in patients with autosomal-dominant polycystic kidney disease (1). It encodes polycystin-2, a 968-amino acid-long protein with 6 putative membrane-spanning domains. According to structural predictions, both its N and C termini extend into the cytoplasm; furthermore, a pore-forming region has been postulated between the fifth and sixth membrane-spanning domains. By sequence comparison a coiled-coil domain and a Ca2+-binding EF-hand have been predicted in the C terminus of polycystin-2 (1) and a homology model has been published recently (2). However, so far only indirect evidence for the presence of these motifs has been presented. Electrophysiological measurements by several groups have demonstrated that polycystin-2 is a non-selective cation channel and that its channel activity is regulated by calcium. One report has shown the activation of polycystin-2 by the addition of 1 μm Ca2+ to the bath solution and its inhibition by millimolar concentrations of Ca2+ (3). An R742X mutant protein, however, did not respond to the addition of Ca2+. These initial observations were confirmed by other publications that reported that Ca2+ concentrations of up to 1.26 mm increase the probability for the open state of full-length polycystin-2 (4), whereas higher concentrations are inhibitory (4, 5). Again, the activity of a truncated protein, this time a 703-amino acid mutant, is not modulated by calcium (4).
From the above it seems obvious that the C terminus via binding of Ca2+ exerts a modulatory activity on the channel activity of polycystin-2. This part of the protein, however, also is of particular interest because it interacts with a wide variety of other proteins, among them polycystin-1, ion channels (inositol 1,4,5-trisphosphate receptor, polycystin-2 itself, and TRPC1), cytoskeletal proteins (α-actinin, CD2AP, mDia1, troponin I, and tropomyosin-1), intracellular trafficking proteins (PACS-1, PACS-2, and PIGEA-14) and even a transcription factor (Id2) (6). Whether all of these interactions are direct or require additional cofactors is not clear at present; furthermore, it is not known whether any of them are cooperative or exclusive. We have therefore decided to subject the C terminus of polycystin-2 to an extensive biochemical and structural analysis.
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant Polycystin-2
A fragment coding for amino acids 680–796 of human polycystin-2 (Fig. 1) was expressed in BL21(DE3) cells (Novagen) grown and adapted in a modified new minimal medium (NMM) (7) as described by Gronwald et al. (8). (1 liter contained 1 g of [15N]H4Cl (>98%, Cambridge Isotope Laboratories), 10 g of glucose, 7.5 g of Na2HPO4·2H2O, 3 g of KH2PO4, 0.5 g of NaCl, 0.25 g of MgSO4·7H2O, 14 mg of CaCl2·2H2O, 50 mg of ampicillin, 100 μg of ZnSO4·7H2O, 30 μg of MnCl2, 300 μg of H3BO3, 200 μg of CoCl2·6H2O, 10 μg of CuCl2·2H2O, 20 μg of NiCl2·6H2O, and 30 μg of Na2MoO4·2H2O, 5 mg of EDTA, and 2 mg of FeSO4·2H2O freshly dissolved in 1 ml of water.) The recombinant protein was purified as published earlier by us (9). Unlabeled, 15N-, 15N/13C-, and 2H/15N/13C-enriched samples were used in this study. The finally obtained polycystin-2-(680–796) fragment contains six additional amino acids (Gly-Ser-Thr-Ala-Ile-Gly) encoded by the vector.
FIGURE 1.

C-terminal domain of polycystin-2. Fragment of the C-terminal sequence of polycystin-2-(680–796) investigated in this report. Italics, amino acids from the thrombin cleavage site. An EF-hand is predicted by Prosite, with the central, metal binding loop in gray. Amino acids predicted to interact with Ca2+ ion are given in bold letters and labeled by (+). Helices predicted by Jpred3 (30) are labeled in yellow. Note that the three last amino acids EHE of the presumed EF-hand (gray) are also predicted as helical. A second EF-hand is obtained from the NMR structure analysis of this report. Residues of this hand show strong line broadening in the presence of manganese and thus are close to the metal binding site are labeled by (+). Blue, predicted coiled coil-region, red, predicted ER-retention signal (31).
Determination of the Protein Concentration
For quantitative experiments the initial protein concentration was determined photometrically by using a molar absorption coefficient ϵ280 of 4470 m−1 cm−1 calculated according to Pace et al. (10) for polycystin-2-(680–796). Alternatively, the protein concentration was checked by NMR in the samples by using the integral of well resolved protein resonances and the DSS3 methyl signal added to the solution. The concentrations obtained by the two methods were identical in the limits of error of ±5%.
Determination of the Ca2+ Content
Polycystin-2-(680–796) was dissolved at a concentration of 51.9 μm in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl. The amount of bound calcium ions was determined by inductively coupled plasma optical emission spectroscopy with a JY 70 Plus spectrometer (HORIBA Jobin Yvon, München, Germany) at a wavelength of 393.366 nm and under a gas pressure of 0.8 MPa. Alternatively, the inherent Ca2+ concentration was determined by titrating the sample with EDTA until the NMR signals of Ca2+-free EDTA were observed. Both methods gave identical results in the limits of error of ±10%.
NMR Spectroscopy
Data were recorded at a magnetic field strength of 14.1 and 18.8 tesla (T) using a Bruker AVANCE 600 and AVANCE 800 spectrometer equipped with TXI and TCI cryogenic probes operating at 1H resonance frequencies of 600.13 and 800.20 MHz. Usually, NMR spectra were acquired at 293 K. Unless stated otherwise, the NMR measurements were performed on a 0.5 mm protein sample in 10 mm potassium phosphate buffer, pH 6.8, 500 mm NaCl, 2 mm dithioerythritol, and 0.1 mm DSS containing 90% H2O and 10% D2O. 1H chemical shifts were referenced to DSS used as internal standard. 15N and 13C chemical shifts were indirectly referenced to DSS as described by Wishard et al. (11).
Sensitivity improved 1H-15N-HSQC experiments were recorded with 128 × 2048 complex data points using a sweep width of 8400 Hz in the 1H dimension and 2400 Hz in the 15N dimension. The sequential backbone assignment was performed on the 13C/15N-labeled protein sample of polycystin-2-(680–796) using HNCA, CBCA(CO)NH, HNCACB, HNCANNH, HNCO, 15N-TOCSY-HSQC, and 1H-15N-NOESY-HSQC three-dimensional spectra. Side-chain assignments are based on HCCH-TOCSY, 15N-TOCSY-HSQC, and two-dimensional NOESY spectra. NOE distance restraints were obtained from two-dimensional NOESY spectra and from 13C- or 15N-edited three-dimensional NOESY-HSQC spectra. For all NOESY spectra a mixing time of 120 ms was used.
Spectral Analysis, Structure Calculation, and Analysis
All NMR spectra were processed and analyzed using the TOPSPIN 2.1 software (Bruker Biospin) and AUREMOL (12). ϕ and ψ angle restraints were obtained from an analysis of the proton, nitrogen, and carbon chemical shifts with the program TALOS 1999.019.15.47 (13). Paramagnetic relaxation enhancement restraints were used only in a qualitative manner, because the protein contains two closely spaced metal binding sites. Hydrogen bonds were derived from the secondary structure prediction and confirmed by an analysis of the NOE patterns. For the ISIC refinement the x-ray structure of the calmodulin-like domain from soybean CDPK-α (pdb code 1S6I) (14) was selected. After sequence alignment residues 87–135 of 1S6I (corresponding to residues 415–463 of CDPK-α) show 30.6% sequence identity in the region 730–778 of polycystin-2.
Structure calculations were performed with the program CNS 1.1 (15) employing a simulated-annealing protocol for extended-strand-starting structures. High temperature torsional angle dynamics were run at 50,000 K for 1,000 steps with a time step of 0.015 ps. In the first cooling stage, torsional angle dynamics were used for 1,000 steps with a starting temperature of 50,000 K and a time step 0.015 ps. The second cooling stage was performed with 3,000 steps of Cartesian dynamics with a time step of 0.005 ps and a starting temperature of 3,000 K. Final energy minimization was performed for 2,000 steps.
The 10 lowest energy structures were refined using the ISIC procedure (16) on the basis of the NMR structural bundle of 1S6I. Out of 1000 calculated final structures, the 10 structures with the lowest pseudo-energies were further refined in explicit solvent (17, 18). The obtained structures are deposited in the Protein Data Bank (PDB) under the accession codes 2KLD and 2KLE.
Structure Analysis
Secondary structure elements, root mean square deviation values, and Ramachandran plots were calculated with the program MOLMOL 2K.2. The agreement of the NOESY data with the calculated structures was calculated from a two-dimensional-NOESY and a three-dimensional-NOESY-1H, 15N-HSQC with AUREMOL-RFAC-3D (19).
Diffusion Measurements
Longitudinal eddy current delay-stimulated echo (LED-STE) diffusion measurements (20) were performed with gradient sandwiches (21) (gradient lengths, 2 ms). In addition, during longitudinal evolution, spoiler gradients of 2- and 1-ms lengths were used. At least 32 scans were accumulated for each gradient strength. The data were evaluated as described by Munte et al. (22).
Ca2+ Binding Studied by Fluorescence Spectroscopy
The tyrosine fluorescence of polycystin-2 was measured by exciting the protein sample at 280 nm and measuring light emission from 290 nm through 310 nm using the fluorescence spectrophotometer Cary Eclipse (Varian). The slit-width used for excitation and emission was 5 nm. All fluorescence measurements were performed at 295 K. The polycystin-2-(680–796) fragment was dissolved at a concentration of 50 μm in buffer A (5 mm Tris-HCl, pH 6.8, 500 mm NaCl). A solution of 10 mm CaCl2 in buffer A was added in increments of 1 and 2 μl to 1 ml of the protein solution. Dilution effects were corrected by measuring a set of data using the same amounts of Ca2+-free buffer A. After each addition of CaCl2 the sample was mixed for 10 s and incubated for another 60 s before the fluorescence measurement was performed. Care was taken to use Ca2+-free solutions by using H2O additionally purified over a Chelex 100 column (Sigma).
Ca2+ Binding Studied by NMR Spectroscopy
41.5 μm unlabeled polycystin-2-(680–796) was dissolved in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl, 90% H2O, 10% D2O. A solution of CaCl2 (stock solution 10 mm) in the same buffer was titrated in 2-μl aliquots to the sample. At every titration step a proton experiment was carried out using a Watergate W5 water suppression (23). A recycle delay of 3.36 s was used with 1024 scans/free induction decay (FID) to obtain a reasonable signal-to-noise ratio.
Fitting of the Binding Data
KD values for the binding of Ca2+ ions were obtained from chemical shift changes Δδ or changes of signal volumes ΔI measured at different titration steps. The dissociation constant KD1 for a single independent binding site can be calculated from the chemical shift Δδ1 by Equation 1,
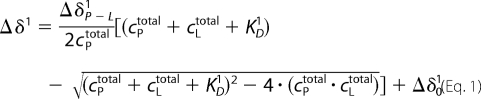 |
with cPtotal and cLtotal the total concentrations of the protein and the ligand (Ca2+ ion), respectively. Δδ1 is defined as δ1 − δ01, Δδ01 as δP1 − δ01, and ΔδP-L1 as δP-L1 − δP1. δ01, δP1, and δP-L1 are the chemical shifts at the lowest ligand concentration, in the ligand-free state, and the completely bound state, respectively. Equation 1 accounts for the case when spectra for the ligand-free state are not available, and Δδ01 is an additional fit parameter. An analogous equation holds for fluorescence intensity changes ΔI and cross-peak intensity changes when slow exchange conditions prevail. In Equation 2,
 |
ΔΙ is defined as I − I0, ΔΙ0 as IP − I01, and ΔΙP-L as IP-L − IP. I0, IP, and IP-L are the fluorescence intensities at the lowest ligand concentration, in the ligand-free state and the completely bound state, respectively, saturation. In case of slow exchange conditions in one-dimensional spectroscopy Equation 2 also holds but the intensity of the line under consideration in the absence of ligand must be zero. In case of two-dimensional HSQC spectra the situation is more complex, because relaxation effects during the mixing time may also influence the result (24).
The above functions were either entered in the fit program Origin, or, alternatively, the fluorescence data were evaluated with the specialized Origin software (MicroCal, LCC), which also assumes a single binding site model and gave the same results. The NMR data show that an additional weaker Ca2+-binding site exists. The corresponding intensity changes ΔΙ2 were fitted by Equation 3,
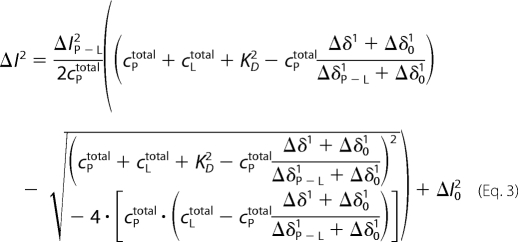 |
with KD2 the dissociation constant of the second binding site. The corresponding equation for the chemical shift changes is then Equation 4.
 |
In principle, these equations have to be solved simultaneously. However, the fitting procedure can also be used iteratively by improving KD1 with the obtained ΔI2P−L and so on.
RESULTS
Expression and Spectroscopic Characterization of the C-terminal Fragments of Human Polycystin-2
The hydropathy analysis of polycystin-2 predicts 6 membrane-spanning domains of which the most C-terminal region ends with amino acid 679. Because amino acids 754–782 may represent an EF-hand motif (1), the channel activity of polycystin-2 is regulated by Ca2+ (3–5), and a coiled-coil motif has been identified in the region between amino acids 772–796 (25), we have started our structural characterization of polycystin-2 with a region extending from amino acids 680–796 (Fig. 1). This region was produced as a recombinant protein in Escherichia coli and purified to homogeneity (Fig. 2). The obtained 1H,15N-HSQC spectrum (Fig. 3, left panel) was poorly resolved and contains significantly less than the expected number of 126 amide back bone signals. After addition of 1 mm CaCl2 the spectral quality improved dramatically (Fig. 3, right panel). The spectrum showed satisfactory chemical shift dispersion, although still a rather large number of resonances were found at random-coil positions.
FIGURE 2.
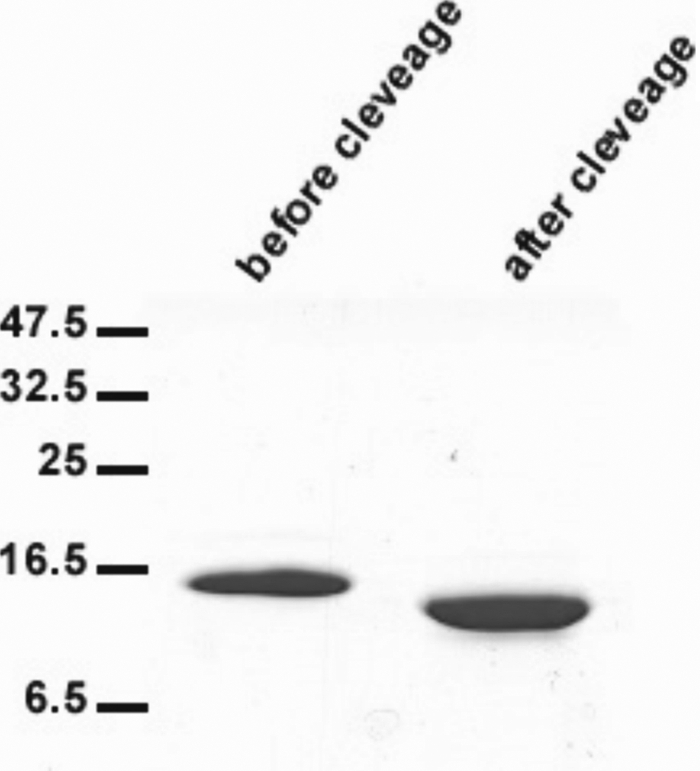
Gel electrophoresis of the C-terminal fragment of polycystin-2. PAGE of polycystin-2-(680–796) before and after removal of the His tag.
FIGURE 3.

1H-15N HSQC spectra of polycystin-2-(680–796). The sample conditions were: 0.25 mm polycystin-2-(680–796) in 10 mm potassium phosphate buffer, pH 6.8, 500 mm NaCl, 2 mm dithioerythritol, and 0.1 mm DSS containing 90% H2O and 10% D2O. Temperature 293 K. Before (left) and after (right) the addition of 1.00 mm CaCl2. In bold letters peaks of the predicted EF-hand, in thin and italic letters are those of the second EF-hand.
There are two mechanisms that would lead to a reduced spectral quality, the formation of large aggregates and the slow or intermediate exchange between different conformations. Because the spectral quality increased substantially after addition of CaCl2, a reduction of the effective molecular mass and/or the suppression of chemical exchange broadening must occur after calcium binding (Fig. 3).
Because the line broadening in the absence of divalent ions could be due to protein aggregation, diffusion measurements were performed (Fig. 4). Using DSS as reference apparent molecular masses of ∼24,300 g·mol−1 and 9,200 g·mol−1 are obtained for polycystin-2-(680–796) in the absence and presence of 2 mm CaCl2, respectively (Table 1). The value in the absence of CaCl2 is clearly larger than 13,515 g·mol−1 expected for a monomeric polycystin-2 fragment and corresponds closely to that expected for a dimer. The addition of CaCl2 reduces the relative hydrodynamic radius significantly, and the experimentally determined apparent mass is smaller than that calculated for a monomer. Addition of 2 mm MgCl2 does not have a significant effect on the diffusion constant determined and thus on the aggregation state of the protein.
FIGURE 4.

Aggregation state of polycystin-2-(680–796). Plot of I/Iref as a function of the relative gradient strength G with I denoting the signal intensity at gradient G and Iref the intensity at the lowest gradient strength. The samples contained 200 μm polycystin-2-(680–796) in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl, 0.1 mm DSS containing 90% H2O, and 10% D2O. As a reference DSS in buffer was used. Polyacrylamide (in 90% H2O, 10% D2O) was used to check the stability of the gradient system before and after the measurement. The signal of HDO in a sample of 99.9% D2O 0.1% H2O was used to calculate the diffusion constants D. *, polyacrylamide; +, HDO; ♢, DSS; ■, polycystin-2-(680–796) in the absence of divalent ions; ●, in the presence of 2 mm CaCl2; or ○, 2 mm MgCl2. Temperature was 298 K.
TABLE 1.
Apparent molecular masses and relative hydrodynamic radii of polycystin-2-(680–796) in the presence and absence of Ca2+ ions
The samples contained 200 μm polycystin-2-(680–796) in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl, 0.1 mm DSS in 90% H2O, and 10% D2O. Data were recorded in the absence and presence of 2 mm CaCl2. Data were evaluated as described by Munte et al. (22).
| Compound | Da | Rh/Rh,DSSb | Mexpc | Mcalcd |
|---|---|---|---|---|
| 10−12m2·s−1 | g·mol−1 | g·mol−1 | ||
| Polycystin-2-(680–796) | 1.51 ± 0.04 | 4.98 ± 0.14 | 24,249 ± 2,045 | 13,515 |
| Polycystin-2-(680–796)·2Ca2+ | 2.09 ± 0.09 | 3.60 ± 0.17 | 9,160 ± 1,297 | 13,595 |
| Polycystin-2-(680–796), MgCl2 | 1.47 ± 0.05 | 5.15 ± 0.17 | 26,742 ± 0,902 | 13,515 |
a The diffusion constant was quantified by measuring the signal decay in 99.9% D2O, 0.1% H2O as a function of the gradient strength and assuming a diffusion constant of HDO of 1.872·10−9·m2·s−1 at 298 K.
b Ratio of the hydrodynamic radii of polycystin-2 and DSS.
c Apparent molar mass of polycystin-2 experimentally obtained using DSS (196.34 g mol−1) as reference.
d Molar mass calculated from the sequence.
Because the diffusion constant is not only a function of the molecular mass but in addition also dependent on the macromolecular shape, the Ca2+-induced changes could also be caused at least partly by a change of the molecular shape. CD spectroscopy in the absence and presence of CaCl2 can be used to analyze the secondary structure of the protein (Fig. 5). The so-obtained CD spectra are indicative of a partially folded protein and reveal small changes of secondary structure on calcium binding. Without CaCl2 added the secondary structure analysis results in relative contents of α-helix, β-strand, turns, and unordered regions of 30, 16, 14 and 40%, respectively. In the presence of 2 mm CaCl2 the secondary structure is 47% α-helical, 12% β-strand, another 12% coiled, and 28% unordered conformation, that is, the α-helical content increases from 30% to 47% and the β-strand content decreases somewhat after addition of CaCl2.
FIGURE 5.

CD spectra of polycystin-2-(680–796). The sample contained 424 μm polycystin-2-(680–796) in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl, before (thin) and after (bold) the addition of 2 mm CaCl2. The data were fitted using the online server DICHROWEB (32) with the reference set SP 175 (33). With no CaCl2 added the secondary analysis gives a α-helix, β-strand, turns, and unordered content of 30%, 16%, 14, and 40%, respectively. In the presence of 2 mm CaCl2, 47% α-helix, 12% β-strand, 12% coiled, and 28% unordered are found.
Resonance Assignments and Secondary Structure of Polycystin-2-(680–796) in the Presence of CaCl2
The assignments of the resonance lines of polycystin-2-(680–796) were performed with heteronuclear methods (9) in the Ca2+-saturated state and are deposited in the BMRB (Biological Magnetic Resonance Bank) under accession number 16191.
From the 127 amino acids of the polycystin-2 sequence (not counting the 6 linker residues), it was possible to assign sequentially the backbone HN, Hα, N, C′, and Cα resonances of 92 amino acids. This corresponds to the backbone signals of ∼95% of all residues where an amide signals could be reliably detected in the HSQC spectrum. From these residues ∼80% of the corresponding side-chain resonances could be identified.
The combined backbone chemical shift changes (26) are plotted in Fig. 6a as function of the sequence. The N-terminal amino acids originating from the vector show very narrow lines and typical random coil chemical shifts. Amino acids 680–705 from polycystin-2 are characterized by somewhat broader lines and chemical shift values that are still close to the random-coil values. The amide signals of amino acids 707–724 are broadened beyond detection probably by slow or intermediate exchange processes. The following amino acids show large deviations from the random-coil chemical shifts and solely the last 6 amino acids show chemical shifts that are typically expected for an unstructured peptide. The heteronuclear NOE data fit into the picture (Fig. 6b): the first two N-terminal amino acids show a high mobility characterized by negative NOEs, the heteronuclear NOEs for the next amino acids slowly increase up to values of 0.6 as it is typical for mobile, weakly structured elements. In the central part NOEs typical for well folded structures are found. The 6 C-terminal amino acids show again decreasing NOEs that are typical for highly mobile peptides, whereas the terminal Leu-796 is again characterized by a negative NOE.
FIGURE 6.

Secondary structure from chemical shifts and back bone dynamics. a, the combined chemical shift changes Δδcomb of the HN, N, Hα, Cα, C′, and Cβ resonances were calculated according to Schumann et al. (26) using the Hamming distance from the random-coil values. The corrected standard deviation σ0corr was used as a measure for the deviation from random-coil conditions. b, 1H,15N NOEs plotted as a function of the sequence. Experiments were performed at 293 K in the presence of 5 mm CaCl2. c, the secondary structure elements were predicted from the Hα, Cα, Cβ, and C′ chemical shifts according to Wishart et al. (34). Residues not observable are indicated by (x and x-x). Negative values are indicative for α-helices (H2 and H3), positive values for β-pleated sheets (B1 and B2).
The chemical shift index (Fig. 6c) predicts two β-strands B1 and B2 from Lys-735 to Asn-737 and Glu-769 to Thr-771 and two α-helices H2 and H3 from Phe-738 to Gln-743 and from Ala-753 to Tyr-762, respectively. An analysis of the short and intermediate range NOE patterns reveals the existence of two additional helices H1 and H4, helix H1 from Leu-729 to Lys-735 and helix H4 from Thr-771 to His-775. According to this analysis, helix H2 is extending from Phe-738 to Leu-745 and helix H3 from Asp-752 to Tyr-762. This result fits into the picture of two paired helix-loop-helix (EF hand) motifs connected to each other by another small loop (Lys-746 to His-751). The structural elements are arranged in the topology αβααβα. The two β-strands B1 and B2 show the typical strong dαN(i,i+1) NOEs (Leu-736 Hα-Asn-737 HN, Asn-737 Hα-Phe-738 HN, Glu-769 Hα-Leu-770 HN, and Leu-770 Hα-Thr-771 HN). Additionally, the mutual arrangement of the β-strands is well defined by long range NOEs (Leu-736 HN-Glu-769 HN, Leu-736 HN-Leu-770 HN, Asn-737 HN-Leu-770 HN, and Asn-737 Hα-Leu-770 HN) between the two β-strands. This NOE pattern is typical for an antiparallel β-sheet.
Binding of Divalent Ions to Polycystin-2-(680–796)
Because we observed a strong effect of CaCl2 at submillimolar concentrations under high salt conditions (500 mm NaCl), polycystin-2-(680–796) must have at least one binding site for Ca2+ ions. Titrations with EGTA show that the protein samples always contain some calcium even if they are prepared in calcium free buffers. This was also confirmed by inductively coupled plasma optical emission spectroscopy measurements. In the freshly prepared samples the protein has initially 0.1–0.2 calcium ions bound per polycystin-2-(680–796) even if calcium was not added to the sample.
Human polycystin-2-(680–796) contains three tyrosine residues at positions 684, 708, and 762; one of them (Tyr-762) is located close to the predicted calcium binding site, the two others in the unfolded regions of the protein. It can be expected that the tyrosine fluorescence intensity changes when Ca2+ ions are bound. Indeed, the fluorescence intensity increased when CaCl2 was added (Fig. 7). Taking into account that initially 8.4 μm calcium is contained in the sample as determined by optical emission spectroscopy, the data can be fitted well by Equation 2 (“Experimental Procedures”) that assumes a single calcium binding site. This is a good approximation even if a second binding site exists as long as the affinity of the second site is significantly lower. From the fit of the data a dissociation constant KD of 68 ± 2 μm−1 was obtained (Table 2).
FIGURE 7.

Calcium binding of polycystin-2-(680–796) detected by fluorescence spectroscopy. The sample contained 51.9 μm polycystin-2-(680–796) in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl. The initial calcium concentration of the sample was 8.4 μm. Data were measured at 295 K and fitted with Equation 2. The line represents a fit of the data with a KD of 68 ± 2 μm−1.
TABLE 2.
Ca2+ affinity of polycystin-2-(680–796) measured by NMR and fluorescence spectroscopy
The samples contained 51.9 μm polycystin-2-(680–796) in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl, 2 mm dithioerythritol. For NMR spectroscopy DSS and D2O to end concentrations of 0.1 mm and 10% were added. The temperature was 295 K.
a Data were fitted assuming two binding sites by Equations 3 and 4.
b Data were fitted assuming a single calcium binding site by Equation 2.
The spectral changes induced by Ca2+ can also be used to calculate binding constants from the NMR spectra. The one-dimensional proton spectrum (Fig. 8a) as well as the two-dimensional HSQC spectrum of polycystin-2-(680–796) without Ca2+ ions (Fig. 3a) are typical for a partially structured protein. Addition of CaCl2 leads to shifts of resonance lines as well as to the appearance of new resonance lines. At the highest calcium concentration the total intensity of the signals has substantially increased in the HSQC spectra of 15N-enriched protein (Fig. 3, right panel) as well as in the one-dimensional spectra of unlabeled protein (Fig. 8a).
FIGURE 8.

Ca2+ affinity measurements by NMR spectroscopy. a, Ca2+-induced spectral changes observed in a sample of 40 μm polycystin-2-(680–796) in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl, 2 mm dithioerythritol, and 0.1 mm DSS containing 90% H2O and 10% D2O. The temperature was 293 K. The initial calcium concentration was 9.2 μm, the concentrations of CaCl2 added are indicated. The position of the amide proton resonance frequency of Asp-752 at 9.2 μm CaCl2 is depicted by a vertical line crossing all one-dimensional spectra. b, chemical shift change Δδ1 of the amide proton of Asp-752 was plotted as a function of the Ca2+ concentration. c, the change of the peak volume ΔδI2 of the amide resonance of Phe-738 is plotted as a function of the calcium concentration. The curves shown represent a fit of the data assuming a two site model (Equations 3 and 4).The resulting parameters are given in Table 2.
As an example for calcium-dependent chemical shift changes the amide proton resonance of Asp-752 has been selected. It is located in helix H3. It can easily be identified in the one-dimensional spectra and thus can serve as a reporter group for calculating the KD value for calcium. However, the 1H and 15N chemical shifts of a number of residues that can easily be followed in the two-dimensional HSQC spectra show an analogous dependence on the Ca2+ concentration (Gln-743, Asp-744, Glu-754, and Glu-772). The chemical shift changes of Asp-752 are plotted as a function of the calcium concentration (Fig. 8b). A fit of the data with the simple one binding site model (Equation 1) gives a KD of 72 ± 7 μm for binding site 1 sensed by Asp-752. The resonance of Phe-738 (located in β-strand B1) is barely visible at low calcium concentrations, its intensity increases strongly with the calcium concentration. A similar behavior in the two-dimensional-HSQC-spectra is observed for the vast majority of residues between positions 732 and 777, which include the proposed EF-hands 1 and 2 as well as the linker sequence joining the two Ca2+-binding motifs. Although the Ca2+ concentration dependences of the HSQC peak volumes vary between individual residues, we did not attempt to analyze the data quantitatively, because peak volumes are potentially modulated by additional factors (e.g. polarization transfer efficiency) apart from conformational exchange broadening. A plot of the intensity of the resonance line of Phe-738 as a function of the calcium concentration is shown in Fig. 8c. The obtained curve is less steep and reaches a plateau much later. This indicates that polycystin-2 has at least two different binding sites for calcium. Fitting the data with Equation 2, which assumes a single binding site, results in a KD of 172 ± 11 μm for the second binding site. In this calculation the fact was taken into account that the sample contained already some calcium as determined by ORS and NMR.
A more rigorous evaluation is provided by fitting the data with a two-binding site model. Here, a KD for site 1 of 55 ± 12 μm and KD of 179 ± 29 μm is obtained (Table 2). The binding sites are specific for calcium, because the addition of MgCl2 to the solution does not lead to significant spectral changes in the concentration range up to 10 mm.
Divalent manganese ions usually can replace divalent ions such as Mg2+ or Ca2+ at their binding sites. Because Mn(II) is paramagnetic it can be used to map the binding sites by the induced paramagnetic relaxation enhancement. We have mapped the binding sites by titration of the 15N-enriched polycystin-2 fragment with MnCl2 (Fig. 9). Fig. 9 displays the results merely for the C-terminal part of our polycystin-2 fragment (amino acids 730–776), because significant relaxation enhancement was absent in the N-terminal part. The strongest effects are observed for Gln-731, Asn-737, Gln-743, Asp-744, Leu-745, Glu-754, Ile-755, Asp-765, and Asp-767.
FIGURE 9.

Manganese binding to polycystin-2-(680–796). To a sample of 500 μm unlabeled polycystin-2-(680–796) in 5 mm Tris-HCl, pH 6.8, 100 mm NaCl, 2 mm dithioerythritol, 0.1 mm DSS, 5 mm CaCl2 in 90% H2O, 10% D2O a concentrated solution of MnCl2 was added, and 1H-1H NOESY spectra were recorded. The ratio of the cross-peak volumes in the absence (I0) and the presence (IMn2+) of MnCl2 was determined. For every residue the intra-residual cross-peak with the lowest ratio of IMn2+/I0 at a MnCl2 concentration of 0.4 mm was plotted as a function of the sequence. Residues where intra-residual cross-peaks are too weak to be observed in the absence of MnCl2 are labeled by x.
Tertiary Structure of Polycystin-2-(680–796)
Based on the hetNOE data and the NOESY patterns it is evident that polycystin-2-(680–796) is only partly folded, even in the presence of calcium. This was also in line with the results of a first structure calculation that lacked well defined conformations in the N-terminal fragment from residues 680 to 724 (data not shown). Hence, our efforts to determine three-dimensional structures were limited to the C-terminal fragment of the protein sequence between residues 724 and 796. A set of three-dimensional structures was calculated on the basis of 534 NOE restraints, 73 angle restraints, and 4 hydrogen bonds (Table 3). After water refinement it shows an αβααβα structure typical for two consecutive EF-hands. The first helix H1 is not well defined, the second helix H2 extends from Phe-738 to Leu-745, the third helix H3 from Asp-752 to Tyr-762, and the fourth helix H4 from Glu-772 to Gln-777. The β-pleated sheet consisting of strand B1 (residue Leu-736/Asn-737) and strand B2 (residues Glu-769/Leu-770) is well formed (Fig. 10). In the well folded part of the protein (amino acids 728–778) the density of restraints is 13 NOEs per residue and thus high enough (Table 3) to obtain a sufficiently well defined structure. The experimental restraints are sufficiently well fulfilled in the computed structural bundle. After water refinement the structural parameters are in the expected range, e.g. the Ramachandran plot is indicative of a rather good structural quality (only 3.2% of the ϕ,ψ-angles are in the energetically disfavored range) (Table 4). The root mean square deviation values are 0.19 nm for the backbone atoms and 0.26 nm for all heteroatoms. Outside the region from amino acid 728 to 778 only 52 additional experimental restraints are available; therefore, the density of experimental restraints is much too small to obtain a well defined structure in these parts of the structure.
TABLE 3.
Experimental restraints for the NMR structure of polycystin-2-(680–796)
Values in parentheses, restraints in the well folded part of polycystin-2 (amino acids 728–778).
| Type of restraint | Number of restraints |
|---|---|
| NOE restraints | 534 (496) |
| Intraresidual (i, i) | 228 (207) |
| Sequential (i, i+1) | 185 (168) |
| Short distance (i, i + j; 2 ≤ j ≤ 4) | 108 (103) |
| Long distance (i, i + j; j ≥ 5) | 136 (136) |
| φ- and ψ-angles from TALOS-analysis | 73 (64) |
| Hydrogen bonds | 4 (4) |
| Total | 734 (682) |
FIGURE 10.

Well folded domain (residues 728–778) of the three-dimensional structure of polycystin-2-(680–796). a, sausage representation of structural ensemble obtained without AUREMOL-ISIC refinement (dark blue) and with AUREMOL-ISIC refinement (red). The spline radius is proportional to the average displacement of the Cα atoms from the mean structure of the ensemble. b, structural ensembles before (dark blue) and after (red) AUREMOL-ISIC refinement. Only the backbone atoms are shown. c, structural bundle after AUREMOL-ISIC refinement. All atoms are shown. d, comparison of the lowest energy structure after AUREMOL-ISIC refinement and the ROBETTA model PC2-EF by Celic et al. (2). The alignment was made by superposition of the backbone atoms of EF-hand 2.
TABLE 4.
Structural statistics
| Structural statistics before ISIC refinementa | |
|---|---|
| Energies (average in the ten lowest-energy structures) | kJ mol−1 |
| Etotal | −9548 ± 312 |
| Ebond | 224 ± 15 |
| Eangle | 681 ± 52 |
| Edihe | 196 ± 20 |
| ENOE | 4 ± 1 |
| NOE violations > 0.05 nm | 0.1 ± 0.3 |
| φ,ψ-angles in Ramachandran plot | |
| Residues in most favored regions | 74.5% |
| Residues in additional allowed regions | 17.2% |
| Residues in generously allowed regions | 5.1% |
| Residues in disallowed regions | 3.2% |
| Number of non-glycine and non-proline residues | 100.0% |
| Root mean square deviation values for the ten lowest energy structures | nm |
| Backbone atoms Cα, C', N in the well folded region (residues 728–778) | 0.192 ± 0.062 |
| Heavy atoms in the well folded region (residues 728–778) | 0.262 ± 0.069 |
| Combined NMR R-factorb | 0.798 |
| Structural statistics after ISIC refinementc | |
|---|---|
| Root mean square deviation values for the ten lowest energy structures | nm |
| Backbone atoms Cα, C′, and N in the well folded region (residues 728–778) | 0.034 ± 0.008 |
| Heavy atoms in the well folded region (residues 728–778) | 0.094 ± 0.009 |
| φ,ψ-angles in Ramachandran plot | |
| Residues in most favored regions | 73.1% |
| Residues in additional allowed regions | 20.9% |
| Residues in generously allowed regions | 4.5% |
| Residues in disallowed regions | 1.5% |
| Number of non-glycine and non-proline residues | 100.0% |
| Combined NMR R-factorb | 0.756 |
a The structure was determined as described under “Experimental Procedures,” including a refinement in explicit water using the protocol proposed by Linge et al. (17) and by Nabuurs et al. (18).
b The combined NMR R-factor (19) was calculated from a two-dimensional 1H,1H NOESY and a three-dimensional 15N-NOESY-HSQC spectrum.
The unbiased structure refinement method ISIC (16) allows to improve NMR structures by other structures of related proteins. By sequence alignment the metal binding site of the calmodulin-like domain of the calcium-dependent soybean protein kinase-α (14) (pdb entry 1S6I) was identified that shows 31% identity with amino acids 702–778 of polycystin-2. The bundle of NMR structures of the calmodulin-like domain of protein kinase-α was used to find an improved solution consistent with our experimental data (shown in red in Fig. 10a, b). Indeed, the ISIC-refined structural bundle (Fig. 10c) led to a reduction of the NMR R-factor and improved structural features (Table 4).
For a comparison with the structural prediction by Celic et al. (2), the lowest energy structure after ISIC refinement is depicted in Fig. 10d (in dark blue) and directly compared with the ROBETTA model PC2-EF by Celic et al. (2) shown in light blue. While the overall topologies of the experimental structure and the model are comparable, the exact positioning of the individual helices within the protein sequence, their lengths, and relative orientations are notably different. One obvious reason for the observed differences is the fact that the ROBETTA protocol does not take into account the presence of Ca2+ ions, whereas the NOE-derived distances used to calculate the experimental structure were obtained in the presence of saturating concentrations of Ca2+. Moreover, addition of paramagnetic Mn2+ to Ca2+ saturated polycystin-2 enabled us to locate the positions of the two Ca2+ binding sites by observing paramagnetic enhancement of nuclear relaxation on 1H nuclei in sufficient proximity to protein-bound Mn2+ (Fig. 11). The data are in reasonable agreement with the predictions for Ca2+ binding residues shown in Fig. 1 and provide structural evidence for the existence of two paired EF-hands able to bind divalent ions.
FIGURE 11.

Calcium-binding sites of polycystin-2-(680–796). a, ribbon presentation of the lowest energy conformation of polycystin-2-(680–796) obtained after AUREMOL-ISIC refinement. The side-chain atoms of residues predicted to interact with Ca2+ are displayed in blue and green for the atypical EF-hand 1 and the canonical EF-hand 2, respectively. The positions of residues affected by enhanced relaxation of some of their nuclei due to nearby paramagnetic Mn2+ are indicated using spheres. b, Van der Waals surface of polycystin-2-(680–796). Predicted Ca2+-interaction sites (shaded in blue) are compared with the experimentally determined binding sites (in yellow). Experimental setup and molecular view are as in a.
DISCUSSION
Three-dimensional Structure of Polycystin-2-(680–796)
The C-terminal fragment of polycystin-2, polycystin-2-(680–796), shows an NMR spectrum that is typical for a partially folded protein. Addition of CaCl2 to the sample leads to a strong increase of the diffusion constant that would be in line with a monomerization of the protein. Using DSS as standard, the obtained molecular mass is even somewhat smaller than that calculated for a monomer. This could be due to the more compact structure of the protein and associated reduction of the shape factor. Strong conformational changes can also influence the hydrodynamic radius and thus the calculated diffusion constants. However, neither CD spectroscopy nor NMR spectroscopy support such an interpretation. Because information theory requires the diffusion constants were calculated from the directly measured data, not from the derived logarithmic plot of the data. A further benefit from this presentation is the fact that deviations from the expected functional behavior can be seen more easily (Fig. 4). Indeed, at high gradient strengths the diffusion curve for polycystin-2 in the presence of calcium displays such a systematic deviation from the expected functional description: the signal intensity is higher than expected. This would be typical for the presence of some dimers in solution.
The most likely interpretation is a Ca2+-induced monomerization of the protein. The observed reduced line widths would be in good agreement with this view. The addition of MgCl2 does not have significant effects on diffusion constants or line widths. This indicates that the observed structural transitions are specific for calcium ions.
In the presence of calcium, the N-terminal and a few of the C-terminal residues are rather mobile, and the central part from amino acid 728 to 782 has an internal mobility that would be expected for a well folded protein. The region from amino acid 706 to 727 cannot be observed in the NMR spectrum, the corresponding amino acids are most probably broadened by chemical exchange processes. This can be due to conformational exchange in the monomeric structure, possibly by an exchange between two conformations where this part is either extended or back folded to the central part of the protein. Another (or additional) reason could be a still existing exchange between monomeric and dimeric (polymeric) structures with this part of the protein being involved in the protein-protein interaction process.
CD spectroscopy reveals an α-helical content of 47% (corresponding to 58 residues out of 123 residues) that is somewhat smaller than the predicted 64% (79 residues, Fig. 1) by Jpred3. The secondary structure analysis of amino acids 728 to 778 included in the structure calculation (Fig. 10b) recognizes 28 residues as α-helical. For the region 706–727 not observable in the spectra a predominantly helical structure has been predicted by Jpred3, a fact that at least partly could explain the discrepancy between the helical content found by NMR spectroscopy and CD analysis. Indeed, the non-visible region is flanked by residues with Cα-chemical shifts typical for α-helices (these residues were not assigned by CSI to be in α-helical conformation, probably because the number of residues is below the minimal threshold needed for this algorithm).
Celic et al. (2) have analyzed three different fragments of polycystin-2, PC2-C, PC2-EF, and PC2-CC that, according to our nomenclature, correspond to polycystin-2-(704–968), polycystin-2-(720–797), and polycystin-2-(828–927). One of the constructs, polycystin-2-(720–797) is closely related to the polycystin-2 fragment 680–796 used in the present study. The analysis of CD spectra by Celic et al. gave an α-helical content of 27 and 45% in the absence and presence of calcium, respectively. This is close to the values found for our fragment that also shows an increase of α-helicity from 30% to 47% and indicates that overall the two fragments have similar structural features (even though the secondary structure distributions in the two constructs may be different). However, the experimentally determined three-dimensional structure has only weak similarities with the three-dimensional structure calculated by ROBETTA (Fig. 10).
Calcium Binding Sites
Our data show that polycystin-2-(680–796) contains two binding sites with different calcium affinities that form a pair of EF-hand structures. One of the EF-hands was predicted earlier (1), and a homology model of the fragment polycystin-2-(704–797) has been published recently (2). According to the latter study polycystin-2-(719–800) has a single calcium site with a KD of 214 μm by isothermal titration calorimetry (ITC) (25 mm Tris-HCl, pH 7.5, 250 mm NaCl). A shorter construct, polycystin-2-(772–796), has a much higher affinity with a KD of 12 μm, although it binds calcium with a stoichiometry of only 0.4. The calcium affinity can be destroyed by mutations in the predicted EF-hand (EF-hand 2). In contrast, we can identify two calcium binding sites with KD values of 55 μm (EF-hand 2) and 179 μm (EF-hand 1) in 5 mm Tris-HCl, pH 6.8, 500 mm NaCl, 295 K, respectively. The detection of the low affinity site by ITC or fluorescence spectroscopy is often difficult when the ΔH changes or fluorescence changes induced by binding to the second site are small. In fact, the second site could also not be detected directly by fluorescence spectroscopy in this study. The differences in the KD of the canonical high affinity site may be partly due to the different experimental conditions, because higher ionic strength should reduce the Ca2+-affinity. An alternative explanation might be that the study of Celic et al. (2) was performed at a different temperature and pH. pH may be an important regulatory factor by itself, because the second EF-hand contains a histidine residue that may decrease dramatically the calcium affinity when positively charged at lower pH values. A third reason for altered Ca2+ binding behavior could be conformational changes associated to the protein fragment lengths, which were chosen differently in the two studies.
NMR structural analysis shows two paired EF-hands that are able to bind divalent ions. One of the predicted EF-hands has a typical metal recognition site. A sign of an intact EF-hand motif in NMR is a strong downfield shift (>2 ppm) of an amide proton signal located in the middle of the EF-hand (consensus position 6) that forms a hydrogen bond to the side-chain carbonyl of the acidic residue at consensus position 1 (27, 28). Such a highfield shift can indeed be observed in polycystin-2-(680–796) for Gln-768 in consensus position 6 (δ(HN) 10.1 ppm). For the first, non-canonical calcium binding site such a downfield shifted residue was not observed.
Calcium Coordination
The manganese paramagnetic relaxation enhancement-measurements should delineate the residues directly involved in the coordination of the physiologically occurring Ca2+ ions. However, because the majority of residues are visible only after Ca2+ saturation, the experiments had to be performed as competition experiments. Under these conditions unspecific manganese binding may also occur. A further difficulty is that the two Ca2+-binding sites are close in the tree-dimensional structure, the manganese binding on one site can also influence the line widths of the second binding site. Therefore, these data can only be evaluated qualitatively.
The high affinity calcium binding site is expected to have a higher specificity for calcium binding. In line with this expectation, the manganese effects are smaller for this site than for the EF-hand 1 (Fig. 9). Strong line broadening can be observed Gln-731, Asn-737, Gln-743, Asp-744, and Leu-745 that most probably form binding site 1. For the canonical binding site strong effects can be observed especially for Asp-764 and Asp-766. The 6 residues probably involved in the calcium coordination in each EF-hand are labeled in Fig. 1. The last residue in the position usually denoted as -Z must be an aspartate or a glutamate residue and usually forms a bidentate complex with the calcium ion via its side chain carboxylate group. In EF-hand 1 it would be Asp-744 and in EF-hand 2 Glu-774. For the second, canonical EF-hand motif the residues involved in the interaction with the metal ion can safely be predicted from the amino acid sequence itself and agree well with the manganese data. For the first EF-hand an unequivocal prediction is not possible, but the scheme given in Fig. 1 is consistent with all experimental data.
CONCLUSION
The C terminus of polycystin-2 contains a pair of EF-hands that bind Ca2+ ions with different affinities. The KD value of the first, atypical binding site is 179 μm, that of the second site with 55 μm is clearly smaller. In polycystin-2-(680–796) Ca2+ binding to the low affinity site leads to a monomerization of the protein. A coiled-coil structure (a typical dimerization motif) has been predicted in the region of the second EF-hand. In the absence of calcium ions the coiled-coil may exist, but a possible coiled-coil structure is necessarily destroyed by the formation and stabilization of the EF-hand structure on calcium binding. This mechanism would explain the observed monomerization.
In vivo, the functional state of polycystin-2 in the endoplasmic reticulum or the plasma membrane is not known, although a homodimerization has been observed (29). Saturation of the weak calcium-binding site in polycystin-2-(680–796) leads to monomerization of the domain that may be responsible for the observed calcium-dependent inactivation of the channel (4, 5). Occupation of the second EF-hand by calcium may also have a functional role by inducing local or global structural changes in the C-terminal domain that change the opening rate of the channel and/or influence the binding of other proteins that are identified as possible interacting partners (see the introduction). However, the molecular details of these interactions are not known, and it is even not known if they preferentially interact with the monomeric or dimeric state of polycystin-2.
The experimentally determined calcium affinities are much higher than the physiological levels in the cytoplasm where the C terminus is located. However, millimolar calcium concentrations exist in the extracellular space and can occur in the endoplasmic reticulum. Because polycystin-2 is also a calcium channel, it is possible that also at the cytoplasmic site of the membrane transiently rather high local calcium concentrations occur. In addition, protein-protein interaction may also modulate the calcium sensitivity and may explain the observed activation of the channel by 1 μm calcium (3).
Acknowledgments
We thank E. Besl for her valuable contributions to protein purification, K.-J. Tiefenbach and P. Babinger for help with CD and fluorescence spectroscopy, R. Sterner for giving access to the CD spectrometer, M. Thomm for giving access to the OES spectrometer, and H. Huber for the inductively coupled plasma optical emission spectroscopy analysis.
This work was supported by the Deutsche Forschungsgemeinschaft (Grant SFB 699) and the Fonds der Chemischen Industrie.
- DSS
- 2,2-dimethyl-2-silapentane-5-sulfonic acid
- CDPK-α
- calmodulin-like domain of the calcium dependent soybean protein kinase α
- HSQC
- heteronuclear single quantum coherence
- ISIC
- intelligent structural information combination
- TOCSY
- total correlation spectroscopy
- NOE
- nuclear Overhauser effect
- NOESY
- nuclear Overhauser effect spectroscopy.
REFERENCES
- 1.Mochizuki T., Wu G., Hayashi T., Xenophontos S. L., Veldhuisen B., Saris J. J., Reynolds D. M., Cai Y., Gabow P. A., Pierides A., Kimberling W. J., Breuning M. H., Deltas C. C., Peters D. J., Somlo S. (1996) Science 272, 1339–1342 [DOI] [PubMed] [Google Scholar]
- 2.Celić A., Petri E. T., Demeler B., Ehrlich B. E., Boggon T. J. (2008) J. Biol. Chem. 283, 28305–28312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vassilev P. M., Guo L., Chen X. Z., Segal Y., Peng J. B., Basora N., Babakhanlou H., Cruger G., Kanazirska M., Ye C. p., Brown E. M., Hediger M. A., Zhou J. (2001) Biochem. Biophys. Res. Commun. 282, 341–350 [DOI] [PubMed] [Google Scholar]
- 4.Koulen P., Cai Y., Geng L., Maeda Y., Nishimura S., Witzgall R., Ehrlich B. E., Somlo S. (2002) Nat. Cell Biol. 4, 191–197 [DOI] [PubMed] [Google Scholar]
- 5.González-Perrett S., Kim K., Ibarra C., Damiano A. E., Zotta E., Batelli M., Harris P. C., Reisin I. L., Arnaout M. A., Cantiello H. F. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 1182–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witzgall R. (2007) Handb. Exp. Pharmacol. 179, 363–375 [DOI] [PubMed] [Google Scholar]
- 7.Budisa N., Steipe B., Demange P., Eckerskorn C., Kellermann J., Huber R. (1995) Eur. J. Biochem. 230, 788–796 [DOI] [PubMed] [Google Scholar]
- 8.Gronwald W., Huber F., Grünewald P., Spörner M., Wohlgemuth S., Herrmann C., Kalbitzer H. R. (2001) Structure 9, 1029–1041 [DOI] [PubMed] [Google Scholar]
- 9.Schumann F. H., Hoffmeister H., Schmidt M., Bader R., Besl E., Witzgall R., Kalbitzer H. R. (2009) Biomol. NMR Assign. 3, 141–144 [DOI] [PubMed] [Google Scholar]
- 10.Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wishart D. S., Bigam C. G., Yao J., Abildgaard F., Dyson H. J., Oldfield E., Markley J. L., Sykes B. D. (1995) J. Biomol. NMR 6, 135–140 [DOI] [PubMed] [Google Scholar]
- 12.Gronwald W., Kalbitzer H. R. (2004) Prog. NMR Spectr. 44, 33–96 [Google Scholar]
- 13.Cornilescu G., Delaglio F., Bax A. (1999) J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 14.Weljie A. M., Vogel H. J. (2004) J. Biol. Chem. 279, 35494–35502 [DOI] [PubMed] [Google Scholar]
- 15.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 16.Brunner K., Gronwald W., Trenner J. M., Neidig K. P., Kalbitzer H. R. (2006) BMC Struct. Biol. 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linge J. P., Williams M. A., Spronk C. A., Bonvin A. M., Nilges M. (2003) Proteins 50, 496–506 [DOI] [PubMed] [Google Scholar]
- 18.Nabuurs S. B., Nederveen A. J., Vranken W., Doreleijers J. F., Bonvin A. M., Vuister G. W., Vriend G., Spronk C. A. (2004) Proteins 55, 483–486 [DOI] [PubMed] [Google Scholar]
- 19.Gronwald W., Brunner K., Kirchhöfer R., Trenner J., Neidig K. P., Kalbitzer H. R. (2007) J. Biomol. NMR 37, 15–30 [DOI] [PubMed] [Google Scholar]
- 20.Gibbs S. J., Johnson C. (1991) J. Magn. Reson. 93, 395–402 [Google Scholar]
- 21.Wider G., Dötsch V., Wüthrich K. (1994) J. Magn. Reson. 108, 255–258 [Google Scholar]
- 22.Munte C. E., Gäde G., Domogalla B., Kremer W., Kellner R., Kalbitzer H. R. (2008) FEBS J. 275, 1163–1173 [DOI] [PubMed] [Google Scholar]
- 23.Lui M., Mao X., Ye C., Huang H., Nicholson J. K., Lindon J. C. (1998) J. Magn. Reson. 132, 125–129 [DOI] [PubMed] [Google Scholar]
- 24.Maurer T., Meier S., Kachel N., Munte C. E., Hasenbein S., Koch B., Hengstenberg W., Kalbitzer H. R. (2004) J. Bacteriol. 186, 5906–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian F., Germino F. J., Cai Y., Zhang X., Somlo S., Germino G. G. (1997) Nat. Genet. 16, 179–183 [DOI] [PubMed] [Google Scholar]
- 26.Schumann F. H., Riepl H., Maurer T., Gronwald W., Neidig K. P., Kalbitzer H. R. (2007) J. Biomol. NMR 39, 275–289 [DOI] [PubMed] [Google Scholar]
- 27.Strynadka N. C., James M. N. (1989) Annu. Rev. Biochem. 58, 951–998 [DOI] [PubMed] [Google Scholar]
- 28.Wright N. T., Varney K. M., Ellis K. C., Markowitz J., Gitti R. K., Zimmer D. B., Weber D. J. (2005) J. Mol. Biol. 353, 410–426 [DOI] [PubMed] [Google Scholar]
- 29.Gallagher A. R., Hoffmann S., Brown N., Cedzich A., Meruvu S., Podlich D., Feng Y., Könecke V., de Vries U., Hammes H. P., Gretz N., Witzgall R. (2006) J. Am. Soc. Nephrol. 17, 2719–2730 [DOI] [PubMed] [Google Scholar]
- 30.Cole C., Barber J. D., Barton G. J. (2008) Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai Y., Maeda Y., Cedzich A., Torres V. E., Wu G., Hayashi T., Mochizuki T., Park J. H., Witzgall R., Somlo S. (1999) J. Biol. Chem. 274, 28557–28565 [DOI] [PubMed] [Google Scholar]
- 32.Whitmore L., Wallace B. A. (2004) Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lees J. G., Miles A. J., Wien F., Wallace B. A. (2006) Bioinformatics 22, 1955–1962 [DOI] [PubMed] [Google Scholar]
- 34.Wishart D. S., Sykes B. D. (1994) J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]