

Abstract
Introduction
Dyggve–Melchior–Clausen (DMC) syndrome is a rare autosomal recessive type of skeletal dysplasia. It is characterized by the association of progressive spondyloepimetaphyseal dysplasia (SEMD), microcephaly, mental retardation (MR), and coarse facies. The radiographic appearance of generalized platyspondyly with double-humped end plates and the lace-like appearance of iliac crests are pathognomonic and distinctive of DMC syndrome. The disorder results from mutations in the DYM gene mapped in the 18q12-12.1 chromosomal region.
Materials and methods
In this report, we studied 15 Egyptian cases with DMC syndrome from nine unrelated families. We aimed to emphasize the characteristic clinical and radiological features in order to differentiate the condition from other SEMDs and mucopolysaccharidosis (MPS). Patients were subjected to detailed history taking, three-generation family pedigree analysis, complete physical examination, anthropometric measurements, quantitative estimation, and two-dimensional electrophoresis of glycosaminoglycans in the urine and measurement of α-l-iduronidase and galactose-6-sulfatase enzyme activities to exclude Hurler and Morquio diseases (MPS type I and MPS type IVA), respectively. Other investigations were carried out whenever indicated. All patients were the offspring of consanguineous apparently normal parents. Positive family history and similarly affected sibs were noted, confirming the autosomal recessive inheritance pattern of the syndrome. Short stature, microcephaly, variable degree of MR, and coarse facies were constant features. The frequency of characteristic orthopedic and radiological findings was reported. Orthopedic surgical intervention was carried out for two patients.
Conclusions
The study concluded that DMC syndrome may be more frequent in Egypt than previously thought, especially due to misdiagnosis. Characteristic facial dysmorphism, body habitus, and pathognomonic radiological signs suggest the diagnosis and differentiate it from other types of SEMDs and MPS for proper genetic counseling and management.
Keywords: Dyggve–Melchior–Clausen syndrome, Orthopedic manifestations, Pathognomonic radiological signs, Skeletal dysplasia, Glycosaminoglycans in urine
Introduction
Dyggve–Melchior–Clausen (DMC) syndrome is a rare autosomal recessive type of skeletal dysplasia. The first three cases with a condition resembling Morquio–Ullrich disease were described by Dyggve et al. [1]. Although some skeletal deformities in these cases were similar to Morquio disease, the absence of corneal clouding, the presence of mental retardation (MR), the absence of mucopolysacchariduria, and the characteristic radiological findings differentiate this syndrome from mucopolysaccharidosis (MPS) IV and other spondyloepimetaphyseal dysplasias (SEMDs). DMC syndrome was listed as a distinct Mendelian entity by the McKusick-Nathans Institute of Genetic Medicine in 1968 [2]. Since then, many reports have included patients with the syndrome.
DMC syndrome (OMIM: 223800) is characterized by the association of progressive SEMD, microcephaly, MR ranging from mild to severe, and coarse facies. The radiographic appearance of generalized platyspondyly with double-humped end plates and the lace-like appearance of iliac crests are pathognomonic and distinctive of DMC syndrome [3, 4].
The disorder results from mutations in the DYM gene mapped in the 18q12-12.1 chromosomal region [5]. Smith–McCort dysplasia (SMC) (OMIM: 607326), a rare variant of DMC syndrome without MR, was shown to be allelic to DMC syndrome [6]. Both have been mapped to the same region and mutations in the DYM gene were identified in both conditions [7]. Most mutations in DMC predict a loss of function, while those identified in SMC are mainly missense mutations, presumably associated with residual DYM activity that would be less deleterious to the brain with a less severe phenotype [8].
In this report, we studied 15 Egyptian cases with the DMC syndrome from nine unrelated families. Parental consanguinity was 100%. We aimed to emphasize the characteristic clinical, orthopedic, and radiological features in order to differentiate the condition from other SEMDs and MPS.
Subjects and methods
The study included 15 patients (seven males, eight females) from nine unrelated families originating from different governorates in Egypt. Patients were referred to us due to short stature, skeletal abnormalities, or associated MR. Their ages at presentation ranged from 3 to 17 years.
All cases were subjected to detailed history taking, three-generation family pedigree analysis, complete physical examination with emphasis on orthopedic findings, anthropometric measurements (height, weight, head circumference [HC], sitting height and upper/lower [U/L] segment ratio), and whole-body skeletal survey. Quantitative estimation and two-dimensional electrophoresis of glycosaminoglycans (GAGs) were studied in the urine of all patients to exclude the possibility of MPS [9, 10]. The measurement of α-l-iduronidase and galactose-6-sulfatase enzyme activities were done to exclude Hurler and Morquio diseases (MPS type I and MPS type IVA), respectively. Enzymatic assays were carried out according to the methods described by Kresse et al. [11]. Computed tomography of the brain, ophthalmologic examination including slit lamp, hearing assessment, IQ, echocardiography, abdomino-pelvic ultrasound, laboratory investigations including blood picture, blood glucose, and serum level of calcium, phosphorus, and alkaline phosphatase were carried out whenever indicated. Two cases were operated upon to relieve some of the associated orthopedic knee complications that interfered with proper walking.
Results
Genetic data
Table 1 shows the genetic data of the studied cases. All patients were the offspring of consanguineous apparently normal parents. Pregnancy and delivery histories were irrelevant in all cases. Delayed motor and mental development were noted by parents during the first few years of age in all cases.
Table 1.
Genetic data of the studied cases
| Case numbers | Sex | Age at presentation (years) | Consanguinity | Other similarly affected family members |
|---|---|---|---|---|
| Family 1 | ||||
| Case 1 | F | 17 | + | Affected sibs |
| Case 2 | M | 16 | + | Affected sibs |
| Case 3 | F | 14 | + | Affected sibs |
| Case 4 | F | 13 | + | Affected sibs |
| Family 2 | ||||
| Case 5 | F | 16 | + | Affected sib and history of an affected cousin, offspring of consanguineous parents |
| Case 6 | M | 14 | + | Affected sib and history of an affected cousin |
| Family 3 | ||||
| Case 7 | F | 10 | + | History of two affected cousins, offspring of consanguineous parents |
| Family 4 | ||||
| Case 8 | F | 4 | + | – |
| Family 5 | ||||
| Case 9 | M | 10 | + | – |
| Family 6 | ||||
| Case 10 | M | 3 | + | History of an affected cousin, offspring of consanguineous parents |
| Family 7 | ||||
| Case 11 | F | 8 | + | Affected sib |
| Case 12 | M | 7 | + | Affected sib |
| Family 8 | ||||
| Case 13 | M | 11 | + | History of two affected sibs |
| Family 9 | ||||
| Case 14 | M | 17 | + | Affected sib |
| Case 15 | F | 5 | + | Affected sib |
F female; M male; + positive consanguinity
Clinical data
Short stature was a common finding, with a mean standard deviation score (SDS) of height of −6.8 SD (ranging between −3.7 and −12.6 SD). The U/L segment ratio was decreased in 11 cases, denoting more affection of the trunk. Microcephaly was present in all cases with a mean SDS of head circumferences of −4.1 SD (ranging between −3.0 and −5.3 SD). The mean SDS of the patient weight was −3.53 SD.
MR was variable, ranging from moderate to severe. All cases had low hair line, narrow forehead, simple prominent ears, and coarse features in the form of long philtrum, macrostomia, thick lips, prominent premaxilla, and macrognathia. Protruding abdomen and generalized hirsutism were common findings in most patients. No corneal clouding, hearing deficit, cardiac anomalies, or organomegaly were detected in any case. Tone and reflexes were normal. External genitalia were normal in all cases.
Orthopedic manifestations and their frequencies are shown in Table 2. Figures 1 and 2 illustrate the clinical manifestations, including coarse facies and skeletal abnormalities, in DMC syndrome.
Table 2.
Frequency of the main orthopedic manifestations in the studied Dyggve–Melchior–Clausen (DMC) syndrome cases
| Finding | No. of cases with positive finding | Frequency % |
|---|---|---|
| Pigeon-shaped chest | 15/15 | 100 |
| Flaring of lower ribs | 15/15 | 100 |
| Exaggerated lumbar lordosis | 15/15 | 100 |
| Broad metaphyses of long bones | 15/15 | 100 |
| Broad interphalangeal joints | 15/15 | 100 |
| Brachydactyly | 15/15 | 100 |
| Short neck | 14/15 | 93 |
| Short chest | 14/15 | 93 |
| Rhizomelic shortening of the upper limbs | 14/15 | 93 |
| Elevated shoulder joints | 13/15 | 86 |
| Pectus carinatum deformity | 13/15 | 86 |
| Genu valgum | 14/15 5 cases had asymmetric affection |
93 |
| Scoliosis | 11/15 | 73 |
| Kyphosis | 8/15 | 53 |
| Limited extension of joints | 7/15 | 46 |
| Knee joints | 4/15 | |
| Elbow joint | 1/15 | |
| Interphalangeal joints | 2/15 | |
| Genu varus | 1/15 | 6 |
| Others | ||
| Bilateral clinodactyly of the fifth fingers | 5/15 | 33 |
| Flat feet | 3/15 | 20 |
| Prominent heels | 3/15 | 20 |
| Camptodactyly of the fingers | 1/15 | 6 |
| Genu varus | 1/15 | 6 |
| Talipes equinovarus | 1/15 | 6 |
| Bowing of the femur and tibia | 1115 | 6 |
Fig. 1.
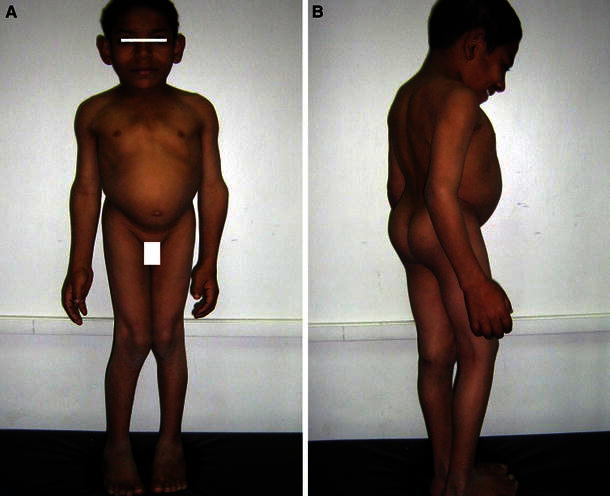
Case 13 at the age of 11 years. a Frontal view. b Lateral view
Fig. 2.
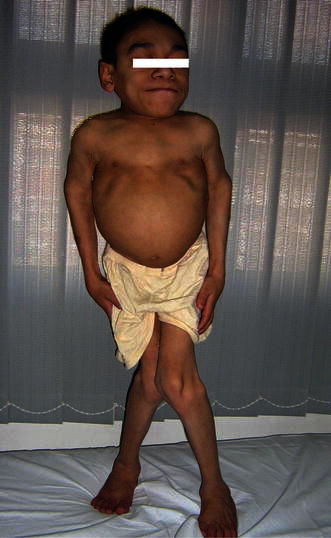
Case 14 at the age of 16 years. Note coarse facies and skeletal anomalies
Radiological data
The frequencies of the main radiological findings in the studied cases are shown in Table 3. The characteristic microcephaly, platyspondyly with central depression of end plates exhibiting a double-hump shape, broad ribs, narrow small pelvis with fine sclerotic bony irregularity giving a lace-like appearance of iliac crests, irregular acetabular cavities, wide symphysis pubis, and medially rotated femoral heads are shown in Fig. 3.
Table 3.
Frequency of the main radiological features in the studied DMC syndrome cases
| Finding | No. of cases with positive finding | Frequency % |
|---|---|---|
| Microcephaly | 15/15 | 100 |
| Misshaped long bones | 15/15 | 100 |
| Short humeri with flattened heads | 15/15 | 100 |
| Small irregular delayed ossification of epiphyses | 15/15 | 100 |
| Broad metaphyses with irregular surface | 15/15 | 100 |
| Short metacarpals and phalanges with broad metaphyses | 15/15 | 100 |
| Medially deviated femoral necks | 15/15 | 100 |
| Abnormally shaped pointed femoral head | 15/15 | 100 |
| Small iliac bone | 15/15 | 100 |
| Irregular surface of iliac bones (lace-like appearance) | 15/15 | 100 |
| Irregular shallow acetabular roof | 15/15 | 100 |
| Wide symphysis pubis | 15/15 | 100 |
| Platyspondyly (rectangular shape) | 15/15 | 100 |
| Upper and lower surface constriction of the vertebral body | 15/15 | 100 |
| Exaggerated lumbar lordosis | 15/15 | 100 |
| Disfigured chest | 14/15 | 93 |
| Broad ribs | 14/15 | 93 |
| Hanging clavicles | 13/15 | 86 |
| Scoliosis | 11/15 | 73 |
| J-shaped sella | 9/15 | 60 |
| Subluxation of the hip or dislocated femoral heads | 8/15 | 53 |
| Kyphosis | 8/15 | 53 |
| Small deep sacrosciatic notch | 6/15 | 40 |
| Thick calvaria | 4/15 | 26 |
| Osteoporosis | 4/15 | 26 |
| Others: | ||
| Increased intracranial digital markings | 2/15 | 13 |
| Odontoid hypoplasia | 2/15 | 13 |
| Healed fracture of long bones | 2/15 | 13 |
| Subluxation of the left fibular head and bowing of the fibula | 2/15 | 13 |
| Lateral tibial subluxation | 1/15 | 6 |
| Abnormal talocalcaneal articulation | 1/15 | 6 |
Fig. 3.
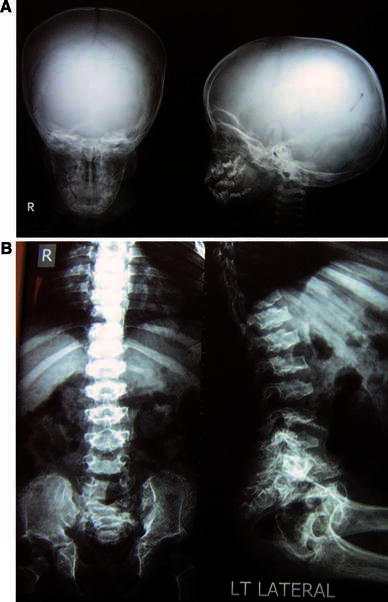
Radiological manifestations of case 13 (family 8) at the age of 12 years. a Skull X-ray: anteroposterior (AP) and lateral view. b Spine X-ray: AP and lateral view
Computed tomography of the brain was normal in all cases, except for one case with central and cortical atrophic changes, who had dysarthric speech and stereotyped movements with no apparent environmental insult or associated condition.
Biochemical investigations
GAGs in urine were elevated in six patients. Two-dimensional electrophoretic separations of GAGs revealed normal pattern. The enzyme activity of α-l-iduronidase and galactose-6-phosphatase was normal in all cases. Other laboratory tests, including blood count, blood glucose, urine analysis, and serum calcium, phosphorus, and alkaline phosphatase were normal in all cases.
Discussion
Our study included nine unrelated Egyptian families with 15 affected patients consistent with the clinical and radiological findings of DMC syndrome. The presence of consanguinity in all families, affected males and females, and the occurrence of affected sibs confirm the autosomal recessive pattern of inheritance in the syndrome. Segregation analysis and study of consanguineous data confirm that DMC syndrome is an autosomal recessive disorder [12].
Growth parameters in our cases indicated disproportionate short stature with more affection of the upper segment in most cases. Rhizomelic shortening of limbs, mainly upper limbs, was noted in all cases and microcephaly was a universal finding. Stature was less affected in patients below 5 years of age, which might be attributed to genetic potential or the progression of short stature with age. Follow up every 6 months was scheduled to study the growth and development over time, which will provide a better idea of growth parameters in the syndrome with the progression of age. The degree of MR was variable in our studied cases. Most cases were severely retarded, in agreement with previous reports. Mild MR was reported by Galasso et al. [13].
All of the studied patients had variable degrees of coarse facies, most notably with increased age and more severe in males than in females. Prominent mandible was described in DMC syndrome [3, 4, 14]. Although characteristic skeletal manifestations were described in the syndrome, this study is the first to analyze of the frequency and degree of orthopedic deformities in 15 cases with DMC syndrome. Pigeon-shaped chest with flaring of the lower ribs, exaggerated lumbar lordosis, broad interphalangeal joints, and brachydactyly were present in 100% of patients. Short neck with elevated shoulder joints, rhizomelic shortening of the upper limbs, short deformed chest with pectus carinatum deformity, and genu valgum (asymmetric in one-third of cases) were present in almost all cases. Scoliosis and kyphosis were present in more than 50% of cases. The severity of skeletal deformities was not the same on both sides in the same patient and there were different degrees of affection in different patients. Limited extension of variable joints was present in 46% of our cases; this limited movement was reported in the syndrome, unlike hyperextensibility, which is noted with Morquio disease [3].
One case in this series had additional unreported manifestations, including dysarthric speech, stereotyped movements, and cortical brain atrophy, with no apparent associated disorder or history of environmental insult, which may expand the phenotypic spectrum of the syndrome.
Most characteristic radiological features in DMC syndrome were documented in our patients. These included microcephaly with facial bones disproportionately large with respect to the cranium, epiphyseal dysplasia, irregular metaphyses, proximal involvement of the limb bones with variable shortening of their shafts, and metaphyseal widening [4]. There is generalized platyspondyly with central constriction of vertebral bodies [15]. The changes are most prominent between the ages of 8 and 12 years. In adults, the vertebral bodies become more rectangular in shape [3]. A pathognomonic radiological sign is small iliac wings with lacy appearance of iliac crests, which was found to be caused by bone tissue deposited in a wavy pattern at the osteochondral junction [16]. Other reported signs in the pelvis and hip joint are small ilium with diminished vertical height, wide irregular sacroiliac joint, small sacroiliac notch, hypoplastic acetabulum, delayed hypoplastic deformed femoral head and neck, femoral neck beaked medially, bilateral dislocation of the hip joint, and wide symphysis pubis [3]. Carbonell et al. [17] used magnetic resonance imaging (MRI) to evaluate DMC cases and concluded that MRI provides a much clearer definition of the aspects of bone dysplasia that are related to the syndrome, including morphologic changes in the soft tissue and cartilage of spine, hips, and knees.
The peculiar radiographic signs in the pelvis, as well as in the spine, represent the most consistent skeletal findings in DMC syndrome. In SEMDs, the double hump with central constriction is absent. The vertebrae show anterior pointing and narrowed disk spaces with premature degeneration changes, sclerotic irregular end plates, and no odontoid hypoplasia, while in Morquio disease, there is anterior pointing with thoracolumbar hypoplasia of the vertebrae, gibbus formation, distal radial, and ulnar obliquity at the wrist joint and proximal metacarpal pointing.
Biochemical analysis of our studied cases showed an increased amount of GAGs in the urine in six cases with normal pattern of electrophoresis and enzyme assays, excluding the presence of mucopolysaccharidosis. Histochemical studies were carried out by Horton and Scott [18], showing fibrous resting cartilage matrix that consisted in many areas of randomly oriented bundles of loosely woven fibers stained as collagen. The chondrocytes were excessively vacuolated and many contained cytoplasmic inclusions that stained as protein. Engfeldt et al. [19] found increased amounts of GAGs in the cartilage in cases with DMC syndrome and indicated that the ability of proteoglycan monomers to reaggregate to hyaluronic acid chains was decreased, suggesting that DMC dysplasia is due to disturbance in proteoglycan metabolism. On the other hand, Beck et al. [20] investigated DMC fibroblasts for abnormal proteoglycan degradation and confirmed normal function. Elevated pipecolic acid levels in plasma and urine with normal results of other studies of peroxisomal function were detected in a child with DMC syndrome by Roesel et al. [21]. To date, the lysosomal pathway appears normal in DMC patients and biochemical analysis failed to reveal any enzymatic deficiency or accumulated substrate [22, 23].
Linkage studies using homozygosity mapping have led to the localization of the disease-causing gene (DYM) on chromosome 18q12-q21.1 [5, 24]. Most mutations identified in DMC syndrome predict a loss of function of a gene proposed by El Ghouzzi et al. [25] as Dymeclin, which may have a role in the process of intracellular digestion of proteins. The DMC gene transcript is widely distributed, but appears abundant in chondrocytes and the fetal brain [25]. DYM mutations associated with DMC result in mis-localization and the subsequent degradation of Dymeclin. Osipovich et al. [26] established Dymeclin as a novel protein involved in Golgi organization and intracellular traffic. Dimitrov et al. [27] stated that permeabilization assays revealed that Dymeclin is not a transmembrane but a novel peripheral protein of the Golgi apparatus which shuttles rapidly between the cytosol and mature Golgi membranes.
The abnormal gene has a wide geographical distribution and has been encountered in families from different countries. The fact that many reported cases came from an Arab origin, cases from Morocco, Gaza [3], Lebanon [8, 14, 28], and Egypt [29, 30], suggests a relatively high frequency of the DYM gene in Arabs, which is a finding that needs further confirmation. This was also pointed out by Teebi [31]. It would be of interest to characterize the mutations of our cases.
Characteristic facial dysmorphism and body habitus suggest the diagnosis of DMC syndrome, which should be confirmed by radiological findings. Differentiation from MPS (types I and IV) and other types of SEMDs is important in order to provide accurate genetic counseling and proper management. Rodríguez Rodríguez et al. [32] pointed to the diagnostic difficulty of DMC syndrome due to its similarity to Morquio disease (MPS type IV). The initial clinical presentation may be similar, but the intellectual prognosis is different [33]. The absence of corneal clouding, deafness, and cardiac anomalies in addition to the characteristic radiological findings of DMC helps in differentiation. Enzyme assays are essential for the definite diagnosis of MPS. A distinct form of SEMD with microcephaly and MR but no lacy appearance of iliac wings was described only once by Bieganski et al. [34] in three boys in a pattern consistent with X-linked recessive inheritance (OMIM: 300232). Another type of SEMD with MR, microcephaly, ataxia, and dysmorphic features without lacy iliac pelvic crest and absence of mutation in the DYM gene was described by Geneviève et al. [35]. Khosravi et al. [36] reported three sibs with some radiographic and chondro-osseous morphologic findings resembling DMC syndrome, yet, the clinical course did not fit due to progressive central nervous system (CNS) degeneration, resulting in early death during infancy. The fact that no enzyme deficiency has yet been discovered should be explained to the patients, indicating the unavailability at present of enzyme replacement therapy, which is now the treatment of choice for certain types of MPS.
Although the prognosis for survival is better in DMC syndrome than in Morquio disease and the physical disability is less, the associated MR makes it a more handicapping disease. Patients with DMC syndrome may require orthopedic femoral osteotomy, total hip arthroplasty, early meniscectomy, realignment osteotomy, or posterior cervical spine fusion. Treatment with temporary epiphysiodesis using a two-hole plate was performed in a male patient at the age of 8 years of this series to treat genu valgum deformity. Another case, a 16-year-old boy, was suffering from severe valgus knee deformity and was treated by femoral and tibial osteotomies and gradual correction. Atlantoaxial instability-induced spinal cord compression is a serious and preventable complication that should be considered. Kandziora et al. [37] presented anterior transarticular atlantoaxial screw fixation as an alternative if posterior and transoral fixation are impossible. Hosny and Fabry [38] performed Chiari pelvic osteotomy to halt hip subluxation in a Belgian patient with DMC syndrome, but follow up showed that progressive lateral migration of the femoral head seemed to have been unaffected by the procedure.
Despite the expected difficult airway due to short neck, macroglossia, and the disturbance of neck flexion, Eguchi et al. [39] experienced no complications, including malignant hyperthermia, with anesthetic management of a patient with DMC syndrome. Two cases in this series had been operated upon and did not suffer from these complications.
DMC syndrome may be more frequent in Egypt than previously thought, especially due to misdiagnosis. The high consanguinity rate in our society favors autosomal recessive disorders. Accurate diagnosis is a pre-requisite for proper genetic counseling and management. Awareness of the characteristic features of the syndrome will prevent misdiagnosis. Molecular studies and the identification of DYM gene mutations are important steps to provide an insight into the genetic heterogeneity of the families and improve the management of patients with DMC syndrome.
References
- 1.Dyggve HV, Melchior JC, Clausen J. Morquio-Ullrich’s disease: an inborn error of metabolism? Arch Dis Child. 1962;37:525–534. doi: 10.1136/adc.37.195.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Online Mendelian Inheritance in Man (OMIM). McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, and the National Center for Biotechnology Information, National Library of Medicine, Bethesda, MD. Home page at: http://www.ncbi.nlm.nih.gov/omim/
- 3.Schorr S, Legum C, Ochshorn M, Hirsch M, Moses S, Lasch EE, El-Masri M. The Dyggve–Melchior–Clausen syndrome. Am J Roentgenol. 1977;128:107–113. doi: 10.2214/ajr.128.1.107. [DOI] [PubMed] [Google Scholar]
- 4.Beighton P. Dyggve–Melchior–Clausen syndrome. J Med Genet. 1990;27:512–515. doi: 10.1136/jmg.27.8.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thauvin-Robinet C, El Ghouzzi V, Chemaitilly W, Dagoneau N, Boute O, Viot G, Mégarbané A, Sefiani A, Munnich A, Le Merrer M, Cormier-Daire V. Homozygosity mapping of a Dyggve–Melchior–Clausen syndrome gene to chromosome 18q21.1. J Med Genet. 2002;39:714–717. doi: 10.1136/jmg.39.10.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spranger J, Bierbaum B, Herrmann J. Heterogeneity of Dyggve–Melchior–Clausen dwarfism. Hum Genet. 1976;33:279–287. doi: 10.1007/BF00286853. [DOI] [PubMed] [Google Scholar]
- 7.Ehtesham N, Cantor RM, King LM, Reinker K, Powell BR, Shanske A, Unger S, Rimoin DL, Cohn DH. Evidence that Smith–McCort dysplasia and Dyggve–Melchior–Clausen dysplasia are allelic disorders that result from mutations in a gene on chromosome 18q12. Am J Hum Genet. 2002;71:947–951. doi: 10.1086/342669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neumann LM, El Ghouzzi V, Paupe V, Weber HP, Fastnacht E, Leenen A, Lyding S, Klusmann A, Mayatepek E, Pelz J, Cormier-Daire V. Dyggve–Melchior–Clausen syndrome and Smith–McCort dysplasia: clinical and molecular findings in three families supporting genetic heterogeneity in Smith–McCort dysplasia. Am J Med Genet A. 2006;140:421–426. doi: 10.1002/ajmg.a.31090. [DOI] [PubMed] [Google Scholar]
- 9.Whitley CB, Draper KA, Dutton CM, Brown PA, Severson SL, France LA. Diagnostic test for mucopolysaccharidosis. II. Rapid quantification of glycosaminoglycan in urine samples collected on a paper matrix. Clin Chem. 1989;35:2074–2081. [PubMed] [Google Scholar]
- 10.Meikle PJ, Ranieri E, Ravenscroft EM, Hua CT, Brooks DA, Hopwood JJ. Newborn screening for lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 11.Kresse H, von Figura K, Klein U, Glössl J, Paschke E, Pohlmann R. Enzymic diagnosis of the genetic mucopolysaccharide storage disorders. Methods Enzymol. 1982;83:559–572. doi: 10.1016/0076-6879(82)83052-8. [DOI] [PubMed] [Google Scholar]
- 12.Toledo SP, Saldanha PH, Lamego C, Mourão PA, Dietrich CP, Mattar E. Dyggve–Melchior–Clausen syndrome: genetic studies and report of affected sibs. Am J Med Genet. 1979;4:255–261. doi: 10.1002/ajmg.1320040308. [DOI] [PubMed] [Google Scholar]
- 13.Galasso C, Fabbri F, Pagnotta G, Palusci A, Sanna ML, Serrao Arnone D, Scirè G. Dyggve–Melchior–Clausen syndrome: description of 2 further cases. Pediatr Med Chir. 1995;17:573–576. [PubMed] [Google Scholar]
- 14.Naffah J. The Dyggve–Melchior–Clausen syndrome. Am J Hum Genet. 1976;28:607–614. [PMC free article] [PubMed] [Google Scholar]
- 15.Spranger JW, Maroteaux P, Der Kaloustian VM. The Dyggve–Melchior–Clausen syndrome. Radiology. 1975;114:415–421. doi: 10.1148/114.2.415. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura K, Kurokawa T, Nagano A, Nakamura S, Taniguchi K, Hamazaki M. Dyggve–Melchior–Clausen syndrome without mental retardation (Smith–McCort dysplasia): morphological findings in the growth plate of the iliac crest. Am J Med Genet. 1997;72:11–17. doi: 10.1002/(SICI)1096-8628(19971003)72:1<11::AID-AJMG3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 17.Carbonell PG, Fernández PD, Vicente-Franqueira JR. MRI findings in Dyggve–Melchior–Clausen syndrome, a rare spondyloepiphyseal dysplasia. J Magn Reson Imaging. 2005;22:572–576. doi: 10.1002/jmri.20414. [DOI] [PubMed] [Google Scholar]
- 18.Horton WA, Scott CI. Dyggve–Melchior–Clausen syndrome a histochemical study of the growth plate. J Bone Joint Surg Am. 1982;64:408–415. [PubMed] [Google Scholar]
- 19.Engfeldt B, Bui TH, Eklöf O, Hjerpe A, Reinholt FP, Ritzen EM, Wikström B. Dyggve–Melchior–Clausen dysplasia. Morphological and biochemical findings in cartilage growth zones. Acta Paediatr Scand. 1983;72:269–274. doi: 10.1111/j.1651-2227.1983.tb09710.x. [DOI] [PubMed] [Google Scholar]
- 20.Beck M, Lücke R, Kresse H. Dyggve–Melchior–Clausen syndrome: normal degradation of proteodermatan sulfate, proteokeratan sulfate and heparan sulfate. Clin Chim Acta. 1984;141:7–15. doi: 10.1016/0009-8981(84)90161-X. [DOI] [PubMed] [Google Scholar]
- 21.Roesel RA, Carroll JE, Rizzo WB, van der Zalm T, Hahn DA. Dyggve–Melchior–Clausen syndrome with increased pipecolic acid in plasma and urine. J Inherit Metab Dis. 1991;14:876–880. doi: 10.1007/BF01800466. [DOI] [PubMed] [Google Scholar]
- 22.Burns C, Powell BR, Hsia YE, Reinker K. Dyggve–Melchior–Clausen syndrome: report of seven patients with the Smith–McCort variant and review of the literature. J Pediatr Orthop. 2003;23:88–93. [PubMed] [Google Scholar]
- 23.Paupe V, Gilbert T, Le Merrer M, Munnich A, Cormier-Daire V, El Ghouzzi V. Recent advances in Dyggve–Melchior–Clausen syndrome. Mol Genet Metab. 2004;83:51–59. doi: 10.1016/j.ymgme.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 24.Cohn DH, Ehtesham N, Krakow D, Unger S, Shanske A, Reinker K, Powell BR, Rimoin DL. Mental retardation and abnormal skeletal development (Dyggve–Melchior–Clausen dysplasia) due to mutations in a novel, evolutionarily conserved gene. Am J Hum Genet. 2003;72:419–428. doi: 10.1086/346176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Ghouzzi V, Dagoneau N, Kinning E, Thauvin-Robinet C, Chemaitilly W, Prost-Squarcioni C, Al-Gazali LI, Verloes A, Le Merrer M, Munnich A, Trembath RC, Cormier-Daire V. Mutations in a novel gene dymeclin (FLJ20071) are responsible for Dyggve–Melchior–Clausen syndrome. Hum Mol Genet. 2003;12:357–364. doi: 10.1093/hmg/ddg029. [DOI] [PubMed] [Google Scholar]
- 26.Osipovich AB, Jennings JL, Lin Q, Link AJ, Ruley HE. Dyggve–Melchior–Clausen syndrome: chondrodysplasia resulting from defects in intracellular vesicle traffic. Proc Natl Acad Sci USA. 2008;105:16171–16176. doi: 10.1073/pnas.0804259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimitrov A, Paupe V, Gueudry C, Sibarita JB, Raposo G, Vielemeyer O, Gilbert T, Csaba Z, Attie-Bitach T, Cormier-Daire V, Gressens P, Rustin P, Perez F, El Ghouzzi V. The gene responsible for Dyggve–Melchior–Clausen syndrome encodes a novel peripheral membrane protein dynamically associated with the Golgi apparatus. Hum Mol Genet. 2009;18:440–453. doi: 10.1093/hmg/ddn371. [DOI] [PubMed] [Google Scholar]
- 28.Bonafede RP, Beighton P. The Dyggve–Melchior–Clausen syndrome in adult siblings. Clin Genet. 1978;14:24–30. doi: 10.1111/j.1399-0004.1978.tb02056.x. [DOI] [PubMed] [Google Scholar]
- 29.Elsayed SM. Dyggve–Melchior–Clausen syndrome: case report. Egypt J Med Hum Genet. 2005;6:67–72. [Google Scholar]
- 30.Temtamy SA, Aglan MS, El-Gammal MA, Hosny LA, Ashour AM, El-Badry TH, Awad SA, Fateen E. Genetic heterogeneity in spondylo-epi-metaphyseal dysplasias: a clinical and radiological study. Egypt J Med Hum Genet. 2007;8:147–171. [Google Scholar]
- 31.Teebi AS. Introduction. In: Teebi AS, Farag TI, editors. Genetic disorders among Arab populations. New York: Oxford University Press; 1997. p. 11. [Google Scholar]
- 32.Rodríguez Rodríguez CM, Pineda Marfa M, Duque R, Cormier-Daire V. Dyggve–Melchior–Clausen syndrome, diagnostic difficulty due to it similarity to Morquio disease. Neurologia. 2007;22:126–129. [PubMed] [Google Scholar]
- 33.Coëslier A, Boute-Bénéjean O, Moerman A, Fron D, Manouvrier-Hanu S. Dyggve–Melchior–Clausen syndrome: differential diagnosis of mucopolysaccharidosis type IV or Morquio disease. Arch Pediatr. 2001;8:838–842. doi: 10.1016/S0929-693X(01)00544-9. [DOI] [PubMed] [Google Scholar]
- 34.Bieganski T, Dawydzik B, Kozlowski K. Spondylo-epimetaphyseal dysplasia: a new X-linked variant with mental retardation. Eur J Pediat. 1999;158:809–814. doi: 10.1007/s004310051211. [DOI] [PubMed] [Google Scholar]
- 35.Geneviève D, Héron D, El Ghouzzi V, Prost-Squarcioni C, Le Merrer M, Jacquette A, Sanlaville D, Pinton F, Villeneuve N, Kalifa G, Munnich A, Cormier-Daire V. Exclusion of the dymeclin and PAPSS2 genes in a novel form of spondyloepimetaphyseal dysplasia and mental retardation. Eur J Hum Genet. 2005;13:541–546. doi: 10.1038/sj.ejhg.5201339. [DOI] [PubMed] [Google Scholar]
- 36.Khosravi M, Weaver DD, Bull MJ, Lachman R, Rimoin DL. Lethal syndrome of skeletal dysplasia and progressive central nervous system degeneration. Am J Med Genet. 1998;77:63–71. doi: 10.1002/(SICI)1096-8628(19980428)77:1<63::AID-AJMG14>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 37.Kandziora F, Neumann L, Schnake KJ, Khodadadyan-Klostermann C, Rehart S, Haas NP, Mittlmeier T. Atlantoaxial instability in Dyggve–Melchior–Clausen syndrome. Case report and review of the literature. J Neurosurg. 2002;96:112–117. doi: 10.3171/spi.2002.96.1.0112. [DOI] [PubMed] [Google Scholar]
- 38.Hosny GA, Fabry G. Treatment of hip subluxation in Dyggve–Melchior–Clausen syndrome. J Pediatr Orthop B. 1998;7:32–34. doi: 10.1097/01202412-199801000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Eguchi M, Kadota Y, Yoshida Y, Masuda M, Masuyama T, Kammura Y. Anesthetic management of a patient with Dyggve–Melchior–Clausen syndrome. Masui. 2001;50:1116–1117. [PubMed] [Google Scholar]