

Abstract
A solid lipid microparticle system containing budesonide was prepared by oil in water emulsification followed by spray drying. The solid lipid system was studied in terms of morphology, particle size distribution, crystallinity, thermal properties, aerosol performance, and dissolution/diffusion release. The microparticle system was also compared to conventional spray-dried crystalline and amorphous budesonide samples. The particle size distributions of the crystalline, amorphous, and solid lipid microparticles, measured by laser diffraction, were similar; however, the microparticle morphology was more irregular than the spray-dried drug samples. The thermal response of the solid lipid microparticles suggested polymorphic transition and melting of the lipid, glycerol behenate (at ~48°C and ~72°C). No budesonide melting or crystallisation peaks were observed, suggesting that the budesonide was integrated into the matrix. X-ray powder diffraction patterns of the crystalline and amorphous budesonide were consistent with previous studies while the solid lipid microparticles showed two peaks, at approximately 21.3 and 23.5 2θ suggesting the metastable sub-α and primarily β′ form. Analysis of the in vitro diffusion/dissolution of the formulations was studied using a flow through model and curves analysed using difference/similarity factors and fitted using the Higuchi model. Regression analysis of this data set indicated differences in the t0.5, where values of 49.7, 35.3, and 136.9 min were observed for crystalline, amorphous, and the solid lipid microparticles, respectively. The aerosol performance (<5 μm), measured by multistage liquid impinger, was 29.5%, 27.3%, and 21.1 ± 0.6% for the crystalline, amorphous, and the solid lipid microparticles, respectively. This study has shown that solid lipid microparticles may provide a useful approach to controlled release respiratory therapy.
Key words: controlled release, dry powder inhalation, solid lipid microparticles
INTRODUCTION
The delivery of respiratory medicines via inhalation has become the most popular route in the treatment of asthma and other respiratory illnesses. In addition, this mode of delivery shows significant promise as a method of treating local infection (1) and as a systemic portal (2). The use of dry powder inhalers (DPIs) has become a popular method of delivering inhalation medicines, since the active pharmaceutical ingredient (API) has greater chemical stability, the dosing options are broader, and the passive nature of the device ensures better patient compliance when compared to pressurised and nebulizer-based devices. However, in order to achieve efficient respiratory deposition, the API should have an aerodynamic diameter <6 μm (3), and the adhesion/cohesion between the drug and/or carrier particles should be less than the force imparted by the patient during inhalation.
In addition to these challenges, there is a potential for the development of a class of inhalation medicines, which possess controlled release properties. The advantages of such an approach include reduced dosing, effective therapy during sleep (asthma can be circadian in nature), increased effectiveness of rapidly cleared medicine, and enhanced residence time at the target site (for example in the treatment of infection). Many challenges exist in developing controlled release inhalation medicine, and to date, no commercial product exists; however, a series of advances have been made and approaches taken. These approaches are discussed in detail in two notable reviews (4,5).
One approach, which may provide a means of efficient controlled release inhalation therapy, is the use of solid lipid microparticles. Solid lipid microparticles are similar to conventional oil-in-water (o/w) emulsions; in which oil droplets, containing a hydrophobic API, are suspended in a polar solvent with a surfactant stabiliser. However, in comparison to conventional o/w emulsions, a higher molecular weight hydrophobic “oil” or saturated lipid component are used which is solid at room temperature. The solid lipid is melted, and the hydrophobic drug is dissolved into it. This melted solution is subsequently mixed into a polar phase and homogenised to form small o/w droplets that can then be cooled to form solid lipid microparticles. The polar phase can subsequently be removed via a drying, filtration, or sublimation leaving a solid lipid microparticle system with API incorporated into the matrix. Other methods of preparation include encapsulating the drug in a lipid “coat” via solvent emulsification–diffusion (6) or preparation of encapsulated materials via supercritical extraction methods (7). Recent reviews of process methodologies and choice (and application) of carrier vehicles can be found in the following reviews (8,9).
Although solid lipid microparticles have been extensively studied as oral, parenteral, and topical formulations (8,10), they have received little attention with respect to pulmonary delivery. In one previous study, Jaspart et al. studied the release of a model drug, salbutamol acetonide, from a glycerol behenate-based solid lipid system (11). Although, in this previous study, the microparticles were of a suitable size for inhalation therapy, the aerosol performance was not studied. Also, the API had to be modified to ensure solubility in the melted lipidic phase, and thus, the clinical relevance was not directly evident. In another study, Sebti and Amighi studied the performance of budesonide-loaded lipid microparticles, containing cholesterol and phospholipids, and reported improved aerosolisation efficiency, when compared to commercially available DPIs (12). In this study, however, the effect on drug release was not performed, and the method of preparation was via direct spray drying from organic solvent. Cook et al. modified the solid lipid microparticle approach by incorporating solid hydrophilic nanoparticles into the lipidic phase (13). While this study was not a bona fide solid lipid microparticle system, the incorporation of terbutaline sulphate nanoparticles into a tripalmitin/glycerol behenate lipid phase, coated with dipalmitoylphosphatidylcholine, produced a controlled release system with satisfactory aerosol performance (13).
One issue relating to the use of hydrophobic matrices in pulmonary drug delivery is the potential toxicological, accumulative, and inflammatory affects. While significant research is required in the area of pulmonary toxicology of controlled release excipients, a recent study by Sanna et al. suggested that glycerol behenate solid lipid microparticles (stabilised using poloxamer emulsifiers) showed no significant change in alveolar macrophage, lymphocyte, and polymorphonuclear neutrophil counts, in murine lungs, 72 h postinfusion, indicating no significant inflammatory response (14).
In this study, we investigate the potential of solid lipid microparticles as a means of delivering a controlled release steroidal formulation to the respiratory tract. Particles containing budesonide were prepared via solvent-free emulsification and studied in terms of their morphology, thermal properties, aerosol performance, and controlled release profile. In addition, the solid lipid microparticles were compared to crystalline and amorphous budesonide powders of similar size.
MATERIALS AND METHODS
Materials
Budesonide was obtained from Yicheng Chemical Corp. (Jiangsu, China). Glycerol behenate (15) (Compritol 888) was supplied by Gattefossé SAS (Saint-Priest Cedex, France). Pluronic F-68 (Pluronic® BASF Corp), magnesium chloride, potassium chloride, and sodium phosphate monobasic monohydrate were supplied by Sigma-Aldrich (St. Louis, USA). Methanol (high-performance liquid chromatography (HPLC) grade) was obtained from Lab-Scan Analytical Sciences (Bangkok, Thailand). Calcium chloride dihydrate, sodium chloride, and sodium sulphate were purchased from AnalaR®-BDH (Kilsyth, Victoria, Australia); sodium acetate anhydrous, disodium hydrogen orthophosphate, sodium hydrogen carbonate, and ethyl alcohol were supplied by Ajax Chemicals (Sydney, NSW, Australia); sodium citrate was supplied by Standard Laboratories Pty (Melbourne, Victoria, Australia). Water (>2 MΩ cm resistivity at 25°C) was purified using a Modulab Type II Deionization System (Continental Water Systems, Sydney, Australia). All compounds were of analytical grade and were used as received.
Preparation of the Solid Lipid Microparticles
Solid lipid microparticles, containing budesonide, were prepared by o/w emulsification via a phase inversion technique. Briefly, hot (90°C) water (50 ml), containing 0.3% (w/v) of surfactant (Pluronic F-68) was added to a melted lipid phase (2 g of Compritol 888) in which budesonide (0.08 g) had been dissolved. The ratio of budesonide to lipid was chosen to produce a ~400 μg drug loading when ~10 mg of powder was aerosolised (equivalent to commercially available therapeutic doses for this steroid). The mixture was maintained at 90°C and subjected to high-shear mixing (10,000 rpm for 2 min, L4RT, Silveston Machines Limited, UK). In addition, the o/w emulsion was sonicated at a constant duty cycle (20 kHz) using a probe (Model 450, Branson Ultrasonics Corporation, USA; power input of 450 W).
The obtained emulsion was rapidly cooled to room temperature, using an ice bath, under magnetic stirring until solidification of the microparticles occurred. The formed particles were recovered by spray drying using a Bϋchi Mini Spray Dryer B-290 (Bϋchi Laboratory-Techniques, Switzerland) with the following conditions: inlet temperature, 65°C; outlet temperature, 39–41°C; spraying air flow, 800 l/h; drying air flow rate, 40 m3 h−1; solution feed rate, 2.9 ml min−1; nozzle size, 0.5 mm. The concentration of budesonide, in a known sample mass, was measured using a validated high-performance liquid chromatography method, and the encapsulation efficiency was calculated, based upon the theoretical percentage loading. Analysis of the solid lipid microparticles using methods described previously (16) indicated a 2.9 ± 0.3% w/w budesonide component (n = 3), corresponding to an encapsulation efficiency of 77.9 ± 7.5%. In addition to the budesonide solid lipid microparticles, a formulation containing only lipid and surfactant was prepared using identical methodology. All powders were stored in airtight containers over silica gel for a minimum of 48 h prior to use. It is important to note that the method of preparation for the solid lipid microparticles is temperature-controlled emulsification, and the spray-drying step was only used to remove the continuous phase. Subsequently, the final particle morphology will not be dependent on parameters such as temperature, concentration, and molecular mobility in the drying droplet; as observed in the conventional solution-based spray drying used to produce the crystalline and amorphous budesonide samples.
Preparation of Spray-Dried Crystalline and Amorphous Powder Budesonide
Primarily amorphous or crystalline budesonide powders were produced via spray drying. Optimised conditions were used to produce particles of similar size to the solid lipid microparticles. Crystalline budesonide particles were prepared by spray drying a 1.75% w/w ethanolic (95%) solution of budesonide using the following conditions: inlet temperature, 100°C; outlet temperature, ~66°C; spraying N2 flow, 380 l/h; drying N2 flow rate, 40 m3/h; solution feed rate, 8.9 ml/min; nozzle size, 0.5 mm. Amorphous budesonide particles were prepared by spray drying the same solution under the same conditions, but with a reduced inlet temperature of 77°C and outlet temperature of ~48°C. The spray-drying conditions for both particulate systems were optimised using an internally developed database. Again, it is important to note that the spray dryer was utilised to produce dried powders from an ethanolic solution in this case (in comparison to the solid lipid microparticles where it was used as a method of powder “filtration”).
High-Performance Liquid Chromatography
Chemical analysis of budesonide was performed using a Waters 600 model HPLC Controller, 2487 model dual-wavelength absorbance detector, 3.9 × 150 mm Waters Nova-Pak® C18 column, and 515 model pump equipped with a 717plus model Autosampler. The HPLC settings were as follows: detection wavelength 280 nm, flow rate 1.0 ml min−1, injection volume 100 μl, and retention time 6 min. A 65:35 (v/v) methanol:water solution was used as mobile phase and ethanol as diluent. A calibration curve between 1 and 100 μg ml−1 was established. Budesonide encapsulated in the microparticles was obtained via sonication of samples in ethanol for 5 min.
Particle Size Analysis
The particle size distribution of each formulation was analysed using laser diffraction (Malvern Mastersizer 2000, Malvern Instruments Ltd., UK). Samples of powder were dispersed using the Scirocco dry dispersion unit (Malvern, UK) at a feed pressure of four bars and feed rate of 50%. All samples were analysed in triplicate with an obscuration value between 0.3% and 10%. Particle size distributions were calculated using a reference refractive index of 1.6 for both lipid and budesonide.
Scanning Electron Microscopy
The morphology of each of powder was studied using a field emission scanning electron microscope at 5 keV (Zeiss Ultra plus, Carl Zeiss Pty Ltd, Sydney, Australia). Images were taken at random locations, at three magnifications (×1,000, ×5000, and ×10,000). Prior to imaging, samples were dispersed onto carbon sticky tabs and coated with gold to a thickness of approximately 15–20 nm.
Differential Scanning Calorimetry
The thermal response of each powder was analysed using a differential scanning calorimeter (DSC; DSC823e, Mettler-Toledo GmbH, Schwerzenbach, Switzerland). Approximately 5 mg of powder was weighed into DSC sample pans, crimp-sealed and lid-pierced. Each sample was analysed over a 20°C to 300°C temperature range using a ramp rate of 10°C min−1.
X-ray Powder Diffraction
The X-ray powder diffraction pattern for each powder was analysed using a D5000 XRD (Siemens, Munich, Germany). Samples were dispersed. Measurements were conducted at room temperature using Cu Kα radiation at 30 mA and 40 kV, with an angular increment of 0.04°/s and count time of 2 s.
In Vitro Drug Diffusion/Dissolution Studies
A modified British Pharmacopoeia (BP) apparatus 4 was utilised to assess budesonide release/dissolution rates from the different powder formulations (17). The method followed that described by Salama et al. (18). Approximately 25 mg of solid lipid microparticles or 1.0 mg of budesonide powder was accurately weighed and spread onto a membrane filter (Pall HT Tuffryn 0.2 μm membrane disc filters: Pall Corporation, New York, USA). A second membrane filter was used to sandwich the powder, and the assembly was secured between the metal mesh screens inside the filter adaptor. Fifty millilitre of dissolution medium (heated to 37.0 ± 1°C in a temperature-controlled water bath) was passed through the flow-through cell and recirculated in a closed-loop configuration using Tygon® tubing (I.D. 1.59 mm, Saint Gobain Performance Plastics, USA) and a peristaltic pump (pump speed stability of ±1%; Gilson MiniPuls3, USA) at 1.5 ml min−1. The flow exposure area was 2.5 cm in diameter, and the flow direction was maintained vertically through the assembly to avoid dead volumes. At fixed time intervals, 1 ml aliquots of the medium were withdrawn and replaced with an equal volume of fresh release medium. At the end of the experimental procedure, the flow cell was disassembled, and the remaining particulates were dissolved and tested for total mass recovery (data was corrected for total volume replacement). Two dissolution media were studied: (1) phosphate buffer (0.05 M phosphate-buffered saline pH 7.4) and (2) simulated lung fluid (without DPPC l-alpha-phosphatidylcholine (DPPC)) at pH 7.4 (19,20). Each formulation was tested five times.
In Vitro Aerosol Performance of Powder Formulations
The aerosolisation efficiency of the budesonide solid lipid microparticles and spray-dried budesonide powders were evaluated using the multistage liquid impinger (MSLI; Copley Scientific Ltd, Nottingham, UK) using the procedures set out in the British Pharmacopoeia (21). The design of the MSLI is such that at a flow rate of 60 l min−1, the aerodynamic cutoff diameters of stages 1, 2, 3, and 4 are 13, 6.8, 3.1, and 1.7 µm, respectively. Stage 5 contains a filter housing for capturing particles less than 1.7 µm. Prior to testing, 20 ml of ethanol was added to stages 1 through 4 and the flow rate through the MSLI set to 60 l min−1 using a GAST Rotary vein pump (Erweka GmbH, Germany) and calibrated flowmeter (TSI 3063, TSI instruments Ltd., Buckinghamshire, UK). Approximately 11 mg of sample was accurately weighed into a size 3 hydroxy-proyl-methyl-cellulose capsule (Capsugel, Sydney, Australia), which was placed into the sample compartment of an Aerolizer™ DPI (Novartis, Basel, Switzerland). The device was activated, connected to a mouthpiece adapter, inserted into a United States Pharmacopeia (USP) throat (connected to the MSLI), and tested for 4 s at 60 l min−1. The procedure was repeated using a further two capsules. After actuation of all three capsules, the device and capsules, throat, and all sample stages were washed into separate volumetrics using ethanol and sonicated for 5 min to ensure disruption of the microparticles and solubilisation of the budesonide (22). Recovered ethanol samples from the MSLI after filtered were quantified using HPLC. Both the spray-dried budesonide and solid lipid microparticles were tested in triplicate.
Statistical Analysis
Data were subjected to analysis of variance. Significant differences between formulations were analysed using post hoc multiple comparisons, and p values of <0.05 (Fisher pairwise) were considered to be significant.
RESULTS AND DISCUSSION
Particle Size Distribution
Particle size distributions of the two spray-dried budesonide and the solid lipid microparticles are shown in Fig. 1. The median particle diameters for the spray-dried crystalline and amorphous budesonide particles were 3.22±0.09 μm and 3.13±0.12 μm, respectively. The median diameter for the budesonide-containing solid lipid microparticles was 3.45±0.27 μm.
Fig. 1.

Particle size distribution of the spray-dried crystalline, amorphous budesonide, and budesonide solid lipid microparticles
Scanning Electron Microscopy
Scanning electron micrographs of the crystalline, amorphous, and solid lipid microparticles are shown in Fig. 2a–c, respectively. Qualitative analysis of the two spray-dried budesonide samples suggested a spherical particle shape, characteristic of a spray-drying process, with diameters within the same range as that observed using laser diffraction. Analysis of the solid lipid microparticles indicated a similar size range; however, the particle morphology was irregular. Interestingly, the morphology appeared similar to that reported in previous studies by Sanna et al. (14), however, in this previous case, the size was polydispersed over three orders of magnitude (0.1–100 μm).
Fig. 2.

Scanning electron microscopy of amorphous and crystalline budesonide particles and the solid lipid microparticles
Differential Scanning Calorimetry
The thermal response of raw budesonide, spray-dried budesonide samples, and the solid lipid microparticles (either with or without budesonide) are shown in Fig. 3.
Fig. 3.

DSC thermograms of “as supplied” (raw) budesonide, spray-dried crystalline, and amorphous budesonide (upper) and solid lipid microparticles (lower; with and without encapsulated budesonide). Inset is magnified region between 25°C and 85°C
Analysis of the thermograms for the “as supplied” and spray-dried crystalline budesonide showed both samples to have only one single endothermic peak at 259°C indicative of melting. Such observations are in good agreement with previous studies (23,24) of crystalline budesonide, indicating that spray-dried crystalline budesonide could be prepared with a similar size to the solid lipid microparticles. In comparison, the thermogram for the spray-dried amorphous budesonide indicated a single exothermic peak at around 110°C followed by an endothermic peak at 258°C. Such observations are indicative of crystallisation and melting suggesting that amorphous spray-dried budesonide, with a similar size to the solid lipid microparticles, had been produced successfully. Analysis of the solid lipid microparticles containing lipid alone showed two endothermic peaks (a small peak at ~48°C and a larger peak at ~72°C), which are likely to be due to polymorphic transition and melting endotherms of the glycerol behenate. Glycerol behenate is a mixture of mono-, bi-, and triglycerides which can form a mixture of polymorphs: sub-α, α β, and β′ (25). The lower endothermic peak can be related to the monotropic polymorphic transition of the sub-α form to α form, present due to rapid cooling during production (25). The melting endotherm at ~72°C corresponds with data reported previously for Compritol 888 (25–28). The budesonide-containing solid lipid microparticles had a similar thermal response to that of the lipid only sample with two endothermic peaks relating to polymorphic transition and melting. No exothermic peak of amorphous budesonide crystallisation or endothermic peak of a budesonide melt was observed in the budesonide solid lipid microparticle samples. Subsequently, it can be concluded that budesonide became miscible in the melted lipid component. This observation was further confirmed by analysis of a physical mixture (4% w/w budesonide/lipid), which showed a similar endothermic curve to that of the solid lipid microparticles.
X-ray Powder Diffraction
X-ray diffraction patterns for the “as supplied”, crystalline and amorphous, spray-dried, and solid lipid microparticles are shown in Fig. 4. Analysis of the diffraction for the “as supplied” (raw) and spray-dried crystalline budesonide suggested a diffraction pattern consistent with that observed in previous studies (23,29). In comparison, analysis of the spray-dried amorphous budesonide suggested a single diffuse peak characteristic of a primarily amorphous material. Analysis of the solid lipid microparticles (with or without budesonide) showed two peaks, at approximately 21.3 and 23.5 2θ (corresponding to spacing of 4.2 and 3.8 Å, respectively). Such observations suggest a metastable sub-α and primarily β′ form as reported by Brubach et al. (25). In addition, the solid lipid microparticles, containing budesonide, contained two small peaks at 15.2 and 15.8 (Fig. 4; inset) suggesting partial budesonide crystallinity within/on the matrix.
Fig. 4.

X-ray powder diffraction patterns of “as supplied” (raw) budesonide, spray-dried crystalline, and amorphous budesonide (upper) and solid lipid microparticles (lower; with and without budesonide)
In Vitro Drug Diffusion/Dissolution
The in vitro diffusion/dissolution of the amorphous budesonide, crystalline budesonide, and budesonide-containing solid lipid microparticles is shown in Fig. 5. It is important to note that standardised in vitro methods for measuring the dissolution and/or diffusion of drug-particulate systems in the lung is yet to be established, and many experimental protocols and tests have been reported in the literature (5; with limited or no in vitro–in vivo correlation). Subsequently, a standardised pharmacopoeia methodology, recommended for the testing of controlled release microparticle systems (the flow through apparatus), was chosen as an appropriate model (17). Experiments were conducted in both phosphate buffer and simulated lung fluid. The influence of both formulation and media on the release rate of budesonide was statistically tested and analysed using a model-independent fit factor method (18,30,31) and is presented in Table I. In general, the fit factor directly compares the difference between percentage drug released per unit time between a reference and test formulation. The difference factor (f1) and similarity factor (f2) are calculated using Eqs. 1 and 2, respectively.
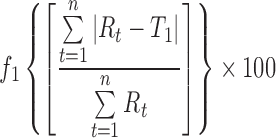 |
1 |
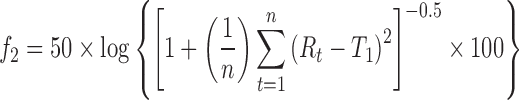 |
2 |
where n is the number of dissolution sample times and Rt and Tt the mean percent drug released at each time point t. It is generally considered, for curves to be statistically similar, f1 should be close to zero (<15) and f2 close to 100 (>50).
Fig. 5.

In vitro release of diffusion/dissolution of budesonide from amorphous and crystalline spray-dried particles and solid lipid microparticles (n = 5). PBS phosphate buffer media, SLF simulated lung fluid
Table I.
Statistical Analysis of Diffusion/Dissolution Release Profiles of Budesonide from Different Formulations in Different Media (Each Formulation n = 5)
| Constant | Reference | Test | Difference (f 1) (>15 indicates difference) | Similarity (f 2) (>50 indicates similarity) |
|---|---|---|---|---|
| Amorphous | Phosphate buffer | Simulated lung fluid | 3.76 | 72.74 |
| Crystalline | Phosphate buffer | Simulated lung fluid | 8.69 | 55.85 |
| Solid lipid microparticles | Phosphate buffer | Simulated lung fluid | 10.77 | 58.85 |
| Phosphate buffer | Amorphous | Crystalline | 5.169 | 65.77 |
| Phosphate buffer | Amorphous | Solid lipid microparticles | 50.41a | 28.13a |
| Phosphate buffer | Crystalline | Solid lipid microparticles | 30.30a | 31.44a |
| Simulated lung fluid | Amorphous | Crystalline | 1.55 | 85.91 |
| Simulated lung fluid | Amorphous | Solid lipid microparticles | 44.24a | 29.36a |
| Simulated lung fluid | Crystalline | Solid lipid microparticles | 30.72a | 29.37a |
a f 1 > 15 and f 2 < 50 suggest statistical differences
Analysis of the difference and similarity factors when comparing simulated lung fluid or phosphate buffer with each formulation suggested no significant difference between the media used. Such observations are in good agreement with previous studies by Davies and Feddah (20) where crystalline budesonide of similar size to those studied here showed no difference in dissolution rates, when studied using a flow-through cell with either phosphate buffer or simulated lung fluid.
Comparison of the dissolution rates of crystalline and amorphous budesonide, using either phosphate buffer or simulated lung fluid, suggested no difference in the drug release curves. Such observations are interesting, since it would be expected that the amorphous budesonide to have a different dissolution profile compared to the crystalline budesonide. In comparison, the release profile of budesonide from the solid lipid microparticles was significantly less than both the crystalline and amorphous in both dissolution media. Such observations are likely due to the encapsulation of budesonide inside the acylglycerol matrix.
The percentage of budesonide in solution as a function of time was fitted to a series of popular release kinetic models (Table II). Based on a correlation coefficient criterion, R2, the best overall mathematical function to describe the release rate, in phosphate buffer, was the Higuchi model, where R2 values of 0.93, 0.91, and 0.99 were observed for the crystalline, amorphous, and solid lipid microparticles, respectively. Furthermore, regression analysis of this data set indicated differences in the time for 50% release (t0.5); where t0.5 values of 49.7, 35.3, and 136.9 min were observed for crystalline, amorphous, and solid lipid microparticles, respectively. Such observations are expected, since the amorphous budesonide would have a moderately increased rate of dissolution when compared to the crystalline budesonide. In comparison, the release of budesonide from the solid lipid microspheres was significantly longer due to the more complex diffusion process.
Table II.
Mathematical Functions Describing Release Rate of Budesonide
| Function | Equation |
|---|---|
| Zero order |
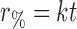
|
| First order |
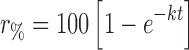
|
| Hixon–Crowell |
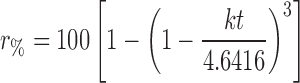
|
| Higuchi |
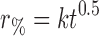
|
r % percentage drug released at time t, k rate constant
In Vitro Aerosol Performance of Powder Formulations
The percentage drug deposition of budesonide from each formulation is shown in Fig. 6. The total drug recovery from all formulations was within 5% of the theoretical loaded dose. In general, the crystalline budesonide had the highest aerosol efficiency, mainly due to the increased deposition of drug on stage 3 (corresponding to particles with an aerodynamic diameter between 3.1 and 6.8 μm). When comparing the crystalline formulation to the amorphous formulation, such observations are expected; both formulations are spherical, with similar size distributions, however, amorphous material is generally reported as having a higher surface energy (and thus, cohesion/adhesion) than its crystalline counterpart, due to its metastable state (32,33). This is also evident from the higher mean device/capsule retention and associated error.
Fig. 6.

In vitro aerosol efficiency of amorphous and crystalline budesonide particles and the solid lipid microparticles, measured by MSLI (n = 5)
When comparing the spray-dried budesonide samples to the solid lipid microparticles, it becomes more difficult to hypothesise reasons for the difference in performance. Firstly, the microparticles are made up of a combination of mono-, di-, and triglycerides as well as budesonide and a poloxamer component. Secondly, the morphology of the particles is more irregular which may either reduce contact area or promote mechanical interlocking between contiguous surfaces. In general, the device retention of the solid lipid microparticles was significantly less than the spray-dried budesonide samples. This may be due to reduced adhesion due to particle roughness and/or reduced surface energy. The greatest difference, in terms of MSLI stage deposition, is on stage 1, where significantly more solid lipid microparticles are deposited when compared to both spray-dried budesonide samples. This stage represents particles with an aerodynamic diameter greater than 13 μm and thus, suggests that the aerosolisation process is not great enough to fully break up agglomerated particles. However, there was still a relatively high deposition of the solid lipid microparticles on the remaining stages.
To quantify the performance of each formulation, linear regression analysis of cumulative percentage deposition vs. logarithmic stage cutoff diameter was used to calculate the mass of particles with an aerodynamic diameter <5 μm. This mass was represented as the percentage of the total dose (fine particle fraction; FPF) and was considered representative of particles that would have a therapeutic respiratory affect (3). The FPF of the crystalline and amorphous budesonide particles was 29.5 ± 0.3% and 27.3 ± 2.1%, respectively. The difference between the FPF for amorphous and crystalline samples, again, may be due to the difference metastability. The FPF of the solid lipid microparticles was 21.1 ± 0.6%, which is a respectable percentage for devices of this generation (where FPF values of <20% are routinely observed; 34). As with the overall aerosol performance, the lower FPF of the solid lipid microparticles when compared to the spray-dried budesonide samples may be due to multiple factors. Firstly, the solid lipid microparticles undergo a series of phase transformations at relatively low temperatures. The underlying drive for this phase change may result in greater instability and thus, higher interparticulate adhesion when compared to the budesonide alone. Secondly, the solid lipid microparticles were irregular in morphology (compared to the spherical nature of the spray-dried budesonide solutions). Subsequently, the drug packing and contact geometry of the lipid-based system may promote greater particle interlocking and thus, a lower aerosol performance as observed in this study.
CONCLUSIONS
This study has demonstrated the potential use of solid lipid microparticles for the controlled release of steroids used in the treatment of asthma. Particles of a suitable size range, aerosol performance, and controlled release profile have been prepared and tested using in vitro methodologies. Future studies should be conducted to evaluate the effectiveness of these systems in vivo, in terms of both release profile and toxicity.
References
- 1.Traini D, Young PM. Delivery of antibiotics to the respiratory tract for pulmonary infection: an update. Expert Opinion on Drug Delivery, Sep 2009, Vol. 6, No. 9 doi:10.1517/17425240903110710 [DOI] [PubMed]
- 2.Scheuch G, Kohlhaeufl MJ, Brand P, Siekmeier R. Clinical perspectives on pulmonary systemic and macromolecular delivery. Adv Drug Deliv Rev. 2006;58(9–10):996–1008. doi: 10.1016/j.addr.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Pritchard JN. The influence of lung deposition on clinical response 1. J Aerosol Med. 2001;14:S19–S26. doi: 10.1089/08942680150506303. [DOI] [PubMed] [Google Scholar]
- 4.Zeng XM, Martin GP, Marriott C. The controlled delivery of drugs to the lung. Int J Pharm. 1995;124(2):149–164. doi: 10.1016/0378-5173(95)00104-Q. [DOI] [Google Scholar]
- 5.Salama R, Traini D, Chan HK, Young PM. Recent advances in controlled release pulmonary therapy. Curr Drug Discov. 2009;6:404–414. doi: 10.2174/156720109789000546. [DOI] [PubMed] [Google Scholar]
- 6.Trotta M, Debernardi F, Caputo O. Preparation of solid lipid nanoparticles by a solvent emulsitication-diffusion technique. Int J Pharm. 2003;257(1–2):153–160. doi: 10.1016/S0378-5173(03)00135-2. [DOI] [PubMed] [Google Scholar]
- 7.Dos Santos IR, Richard J, Pech B, Thies C, Benoit JP. Microencapsulation of protein particles within lipids using a novel supercritical fluid process. Int J Pharm. 2002;242(1–2):69–78. doi: 10.1016/S0378-5173(02)00149-7. [DOI] [PubMed] [Google Scholar]
- 8.Jaspart S, Piel G, Delattre L, Evrard B. Solid lipid microparticles: formulation, preparation, characterisation, drug release and applications. Expert Opin Drug Deliv. 2005;2(1):75–87. doi: 10.1517/17425247.2.1.75. [DOI] [PubMed] [Google Scholar]
- 9.Zhang LJ, Qian Y, Long CX, Chen Y. Systematic procedures for formulation design of drug-loaded solid lipid microparticles: selection of carrier material and stabilizer. Ind Eng Chem Res. 2008;47(16):6091–6100. doi: 10.1021/ie7017806. [DOI] [Google Scholar]
- 10.Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery—a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. doi: 10.1016/S0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 11.Jaspart S, Bertholet P, Piel G, Dogne JM, Delattre L, Evrard B. Solid lipid microparticles as a sustained release system for pulmonary drug delivery. Eur J Pharm Biopharm. 2007;65(1):47–56. doi: 10.1016/j.ejpb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Sebti T, Amighi K. Preparation and in vitro evaluation of lipidic carriers and fillers for inhalation. Eur J Pharm Biopharm. 2006;63(1):51–58. doi: 10.1016/j.ejpb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Cook RO, Pannu RK, Kellaway IW. Novel sustained release microspheres for pulmonary drug delivery. J Control Release. 2005;104(1):79–90. doi: 10.1016/j.jconrel.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Sanna V, Kirschvink N, Gustin P, Gavini E, Roland I, Delattre L, et al. Preparation and in vivo toxicity study of solid lipid microparticles as carrier for pulmonary administration. AAPS PharmSciTech. 2004;5(2):e27. doi: 10.1208/pt050227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pople P, Singh KK. Glyceryl behanate-monograph. In: Rowe RC, Sheskey PJ, Quinn ME, editors. Handbook of pharmaceutical excipients. 6. London: Pharmaceutical Press; 2009. pp. 286–288. [Google Scholar]
- 16.Dalpiaz A, Mezzena M, Scatturin A, Scalia S. Solid lipid microparticles for the stability enhancement of the polar drug N-6-cyclopentyladenosine. Int J Pharm. 2008;355(1–2):81–86. doi: 10.1016/j.ijpharm.2007.11.044. [DOI] [PubMed] [Google Scholar]
- 17.Appendix XII B. Dissolution guidance on dissolution testing. British Pharmacopoeia Volume 2009. Norwich, UK: TSO; 2009.
- 18.Salama RO, Traini D, Chan HK, Young PM. Preparation and characterisation of controlled release co-spray dried drug-polymer microparticles for inhalation 2: evaluation of in vitro release profiling methodologies for controlled release respiratory aerosols. Eur J Pharm Biopharm. 2008;70(1):145–152. doi: 10.1016/j.ejpb.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Moss OR. Simulants of lung interstitial fluid. Health Phys. 1979;36(3):447–448. [PubMed] [Google Scholar]
- 20.Davies NM, Feddah MR. A novel method for assessing dissolution of aerosol inhaler products. Int J Pharm. 2003;255(1–2):175–187. doi: 10.1016/S0378-5173(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 21.Section 2.9.18—appendix XII C. Consistency of formulated preparations for inhalation. British Pharmacopoeia, 2009.
- 22.Tursilli R, Piel G, Delattre L, Scalia S. Solid lipid microparticles containing the sunscreen agent, octyl-dimethylaminobenzoate: effect of the vehicle. Eur J Pharm Biopharm. 2007;66(3):483–487. doi: 10.1016/j.ejpb.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Tajber L, Corrigan DO, Corrigan OI, Healy AM. Spray drying of budesonide, formoterol fumarate and their composites—I. Physicochemical characterisation. Int J Pharm. 2009;367(1–2):79–85. doi: 10.1016/j.ijpharm.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 24.Velaga SP, Berger R, Carlfors J. Supercritical fluids crystallization of budesonide and flunisolide. Pharm Res. 2002;19(10):1564–1571. doi: 10.1023/A:1020477204512. [DOI] [PubMed] [Google Scholar]
- 25.Brubach JB, Jannin V, Mahler B, Bourgaux C, Lessieur P, Roy P, et al. Structural and thermal characterization of glyceryl behenate by X-ray diffraction coupled to differential calorimetry and infrared spectroscopy. Int J Pharm. 2007;336(2):248–256. doi: 10.1016/j.ijpharm.2006.11.057. [DOI] [PubMed] [Google Scholar]
- 26.Souto EB, Mehnert W, Muller RH. Polymorphic behaviour of Compritol888 ATO as bulk lipid and as SLN and NLC. J Microencapsul. 2006;23(4):417–433. doi: 10.1080/02652040600612439. [DOI] [PubMed] [Google Scholar]
- 27.Eldem T, Speiser P, Altorfer H. Polymorphic behavior of sprayed lipid micropellets and its evaluation by differential scanning calorimetry and scanning electron microscopy. Pharm Res. 1991;8(2):178–184. doi: 10.1023/A:1015831801813. [DOI] [PubMed] [Google Scholar]
- 28.Rao M, Ranpise A, Borate S, Thanki K. Mechanistic evaluation of the effect of sintering on Compritol 888 ATO matrices. AAPS PharmSciTech. 2009;10(2):355–360. doi: 10.1208/s12249-009-9211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pham S, Wiedmann TS. Note: dissolution of aerosol particles of budesonide in Survanta, a model lung surfactant. J Pharm Sci. 2001;90(1):98–104. doi: 10.1002/1520-6017(200101)90:1<98::AID-JPS11>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 30.Moore JW, Flanner HH. Mathematical comparison of dissolution profiles. Pharm Tech. 1996;20:64–75. [Google Scholar]
- 31.Guidence for industry; Dissolution testing of immediate release solid dosage forms. In: Administration FaD, editor. August 1997.
- 32.Zhang J, Ebbens S, Chen X, Jin Z, Luk S, Madden C, et al. Determination of the surface free energy of crystalline and amorphous lactose by atomic force microscopy adhesion measurement. Pharm Res. 2006;23(2):401–407. doi: 10.1007/s11095-005-9144-1. [DOI] [PubMed] [Google Scholar]
- 33.Newell HE, Buckton G, Butler DA, Thielmann F, Williams DR. The use of inverse phase gas chromatography to measure the surface energy of crystalline, amorphous, and recently milled lactose. Pharmaceut Res. 2001;18(5):662–666. doi: 10.1023/A:1011089511959. [DOI] [PubMed] [Google Scholar]
- 34.Smith IJ, Parry-Billings M. The inhalers of the future? A review of dry powder devices on the market today. Pulm Pharmacol Ther. 2003;16(2):79–95. doi: 10.1016/S1094-5539(02)00147-5. [DOI] [PubMed] [Google Scholar]