

Abstract
Elevated basal concentrations of glucagon and reduced postprandial glucagon suppression are partly responsible for the increased hepatic glucose production seen in type 2 diabetic patients. Recently, it was demonstrated that an antagonistic human monoclonal antibody (mAb) blocking glucagon receptor (GCGR) has profound glucose-lowering effects in various animal models. To further understand the effects on glucose homeostasis mediated by such an antibody, a pharmacokinetic-pharmacodynamic (PK-PD) study was conducted in a diabetic ob/ob mouse model. Four groups of ob/ob mice were randomized to receive single intraperitoneal administration of placebo, 0.6, 1, or 3 mg/kg of mAb GCGR, a fully human mAb against GCGR. The concentration-time data were used for noncompartmental and compartmental analysis. A semi-mechanistic PK-PD model incorporating the glucose-glucagon inter-regulation and the hypothesized inhibitory effect of mAb GCGR on GCGR signaling pathway via competitive inhibition was included to describe the disposition of glucose and glucagon over time. The pharmacokinetics of mAb GCGR was well characterized by a two-compartment model with parallel linear and nonlinear saturable eliminations. Single injection of mAb GCGR caused a rapid glucose-lowering effect with blood glucose concentrations returning to baseline by 4 to 18 days with increasing dose from 0.6 to 3 mg/kg. Elevation of glucagon concentrations was also observed in a dose-dependent manner. The results illustrated that the feedback relationship between glucose and glucagon in the presence of mAb GCGR could be quantitatively described by the developed model. The model may provide additional understanding in the underlying mechanism of GCGR antagonism by mAb.
Key words: anti-GCGR antibody, glucagon, hepatic glucose production, type 2 diabetes
INTRODUCTION
Glucagon, a 29-amino acid peptide, is secreted from pancreatic alpha cells in response to falling blood glucose. Under normal physiological conditions, glucagon signaling is important in glucose homeostasis and represents the principle counter-regulatory mechanism that opposes insulin action of glucose lowering (1). Glucagon stimulates glucose production by promoting hepatic gluconeogenesis and glycogenolysis preferentially in the liver and kidney (2) after binding and activating the glucagon receptor (GCGR), a seven-transmembrane G-protein-coupled receptor (3,4).
It is well recognized that elevation of hepatic glucose production contributes to the hyperglycemic state of type 2 diabetes (5,6). Accumulating evidences have shown that higher basal glucagon concentrations and lack of suppression of postprandial glucagon secretion are partially responsible for the increased hepatic glucose production observed in diabetic patients (7–11). In addition, preclinical studies performed in various diabetic animal models have suggested that inhibition of the GCGR signaling pathway represents one of the potential approaches for treating type 2 diabetes (12,13). For instance, reduction in GCGR expression using antisense oligonucleotides has been shown to lower glycemia in db/db mice and Zucker diabetic fatty rats (12,14,15).
Hormonal regulation of glucose is mainly controlled by insulin and glucagon, among others. Unlike well-published, mechanism-based models of insulin action on glucose metabolism (16–18), limited information is available for modeling the physiological effect of glucagon as well as the consequence of blocking GCGR pathway on glucose homeostasis. In addition, there have been no reports so far, to our knowledge, of modeling the glucose-lowering effects caused by long-lasting antagonistic monoclonal antibodies (mAbs). Preclinical data performed with a series of antagonistic GCGR mAbs was recently published (19). One of the antibodies, mAb B, demonstrated long-lasting, dose-dependent, glucose-lowering effect following single injections at 1 or 3 mg/kg in the leptin-deficient ob/ob mice. The ob/ob mouse is a commonly used mouse model of type 2 diabetes with moderate hyperglycemia and hyperinsulinemia (20). Ob/ob mice compensate for the extreme insulin resistance induced by their massive obesity and thus are able to maintain plasma glucose at concentrations that are only slightly elevated. Humans have a more progressive onset of type 2 diabetes than ob/ob mice.
In the present investigation, we studied the in vivo pharmacological response in ob/ob mice, following single intraperitoneal (i.p.) doses of mAb GCGR, an anti-GCGR mAb with similar potency as mAb B. The primary purpose was to quantitatively characterize the homeostatic regulation of glucose and glucagon, as well as the changes in their profiles over time evoked by acute blockage of GCGR signal by mAb GCGR. The proposed semi-mechanistic pharmacokinetic-pharmacodynamic (PK-PD) model was based on the concepts of the indirect response models (21,22) and incorporated regulatory mechanisms, specifically, glucose-glucagon feedback in both directions and the inhibitory effect of mAb GCGR on GCGR signaling via competitive binding with glucagon. We expected to see reduction in blood glucose concentrations accompanied with elevation of glucagon upon single-dose mAb GCGR treatment. The results demonstrated in this study could assist in understanding the mechanism underlying GCGR antagonism in general and support the clinical development of mAb GCGR for the treatment of type 2 diabetes.
MATERIALS AND METHODS
Test Article
mAb GCGR is a fully human IgG2 recombinantly expressed in Chinese hamster ovarian cells. From mAb GCGR, mAb B (19) was derived by changing a single amino acid to achieve the product homogeneity. Equal potency and efficacy were demonstrated in various in vitro assays and animal models (data not shown). mAb GCGR was supplied as a frozen liquid formulation containing 70 mg/mL mAb GCGR.
Animal Husbandry
The ob/ob mouse study was conducted at Amgen Inc. (Thousand Oaks, CA, USA) and approved by the Institutional Animal Care and Use Committee. Two hundred fifty 14-week-old male ob/ob mice (The Jackson Laboratory, Bar Harbor, ME, USA) weighing approximately 40–50 g were maintained on a 12-h light/dark cycle with free access to food and water.
In Vivo Study Design
In type 2 diabetes, postprandial hyperglucagonemia is an important contributor to failed suppression of hepatic glucose release after meal ingestion. In addition, deficit in β-cell mass and impaired postprandial insulin secretion contribute to the phenotype of the disease. Plasma glucagon concentrations in patients with diabetes are often comparable to those of nondiabetic individuals in fasted state. Insulin concentrations are also low. In our study, samples were collected in ob/ob mice that had been fed ad libitum.
On day 0 (time 0), ob/ob mice were sorted into treatment groups with similar distributions based on blood glucose and body weight. At pre-specified time points, animals were injected intraperitoneally with vehicle or mAb GCGR at 0.6, 1, or 3 mg/kg (injection dose volume of 2.0 mL/kg). A composite sampling approach (eight to ten mice per dose group per time point) was used to collect blood samples via cardiac puncture for measurements of serum concentration of mAb GCGR and plasma concentration of glucose and glucagon at various time points: at approximately 24, 48, 96, 168, 240, 336 (vehicle group and 1 and 3 mg/kg dose groups only), and 432 h (vehicle group and 3 mg/kg dose group only) post-dose.
Glucose and Glucagon Analysis
Dipeptidyl peptidase-4 inhibitor (100 μM) and aprotinin (85 μg/mL) were added to each blood sample, to avoid possible degradation of glucagon. Plasma glucagon concentrations were measured by a competitive enzyme immunoassay developed at Amgen Inc. (Thousand Oaks, CA, USA) using a combination of highly specific antibody to glucagon and a biotin-avidin affinity system as previously described. The lower limit of quantification of the assay was 80 pg/mL. Average assay accuracy ranged from −4% to 7%, and the total error for quality controls ranged from 4% to 12%.
All blood samples taken for blood glucose measurement were collected from the retro-orbital sinus of nonanesthetized mice and measured using a hand-held glucometer, One Touch® Profile (LifeScan, Inc., Milpitas, CA, USA), with within-run precision under 5% (23).
Measurement of mAb GCGR Concentration in Serum
The concentrations of mAb GCGR in serum samples were quantified by a sandwich enzyme-linked immunosorbent assay using anti-idiotype mAb. The capture reagent was a mouse anti-mAb GCGR mAb supplied by Amgen Inc. (Thousand Oaks, CA, USA). Briefly study samples were added to the coated plates after blocking nonspecific binding. Horseradish peroxides-conjugated mouse anti-mAb GCGR antibody was used as the detector antibody. Colorimetric determination of the horseradish peroxidase reaction with the tetramethylbenzidine peroxide substrate solution (KPL Inc., Gaithersburg, MD, USA) was measured by optical density at 450 to 650 nm. The conversion of optical density units to concentrations for mAb GCGR in the study samples was achieved through comparison with a standard curve assayed on the same plate. A five-parameter, logistic, auto-estimate regression model with a weighing factor of 1/Y using Watson Laboratory Information Management System (version 7.0.0.01, Thermo Scientific, Waltham, MA, USA) data reduction package was used. The lower limit of quantification of the assay was 0.02 µg/mL. The inter-assay coefficient of variation ranged from 2% to 7%. Average assay accuracy ranged from −3% to 5%, and the total error for quality controls ranged from 5% to 12%. Cross-reactivity was not observed with glucagon at concentration up to 100 ng/mL, with glucagon-like peptide 1 at concentration up to 1 ng/mL or with a structurally similar fully human IgG2 antibody at the concentration of 3 to 300 ng/mL. Interference was observed with anti-mAb GCGR neutralizing rabbit antibody at concentration greater than 0.02 µg/mL. However, anti-drug antibodies are not expected in a single-dose study.
Noncompartmental Data Analysis
Data from mice over the period of pre-dose to 432 h were combined to display the concentration-time profiles of mAb GCGR, glucose, and glucagon. The composite mean concentration-time data were subjected to noncompartmental analysis using WinNonlin Professional version 4.1e (Pharsight, Mountain View, CA, USA). PK parameters such as area under the concentration-time curve (AUC) were estimated using the linear/logarithmic trapezoidal method (for the up/down portions of the curve, respectively) up to the last measured concentration that was above the lower detection limit (AUClast). The maximum serum concentration (Cmax) and the time it occurred (tmax) after i.p. administration were recorded as observed.
Composite plasma concentrations of glucose and glucagon were expressed as a percentage change from the pre-dose baseline. The area under the effect-time curve for glucose (AUCglucose) and the area above the effect-time curve for glucagon (AUCglucagon) values were calculated using the linear trapezoidal method. The maximum percent change in glucose and glucagon from baseline and the time at which it occurred (ETmax) were recorded as observed.
Compartmental Data Analysis
Analysis of PK and PD data was performed with nonlinear mixed-effects modeling software (NONMEM version VI, Level 2, ICON, Ellicott City, MD, USA) with the ADVAN6 subroutine for estimation of the model parameters. The Fortran compiler g77 (version 2.95) was used for NONMEM installation, and NMQual (version 6.3.2, Metrum Institute, Augusta, ME, USA) was implemented to fix reported bugs. The first-order conditional estimation method implemented in NONMEM was used for both PK and PK-PD model development. The residual variability for both PK and PD was assumed to follow an exponential error model. A standard sequential modeling approach was used for PK-PD modeling. First, the PK model was fitted simultaneously across various treatments with mAb GCGR concentration data pooled from eight to ten mice per time point per dose group. All animals at 168 and 240 h post-dose in the 0.6 mg/kg group and all animals at 336 h post-dose in the 1 mg/kg group had mAb GCGR concentrations that were below quantitation limit and were omitted from the PK analyses. The predicted PK parameters of mAb GCGR were then fixed and used to drive the fitting of plasma glucose and glucagon concentration-time data from placebo and various mAb GCGR-treated dose groups simultaneously to generate one set of parameters to describe the effect of mAb GCGR on PD response. Various proposed PD models for glucose-glucagon regulation were fitted and compared. Inter-dose random effects were tried on parameters such as baseline glucose and glucagon concentrations. The final model selection criteria included reduction of objective function value, evaluation of precision of parameter estimates, visual inspection of predicted versus observed concentrations, and plots of weighted residuals versus time or predicted concentrations. Only results of the final model fitting are presented in this paper.
Pharmacokinetic Model
A two-compartment model with parallel linear and saturable nonlinear elimination characterized by a Michaelis–Menten (VMAX, KM) component from the central compartment was used to describe the PK of mAb GCGR (Fig. 1), shown by the following differential equations:
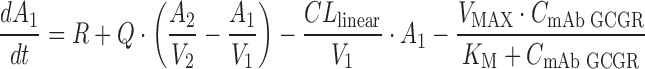 |
1 |
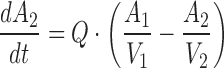 |
2 |
where A1 and A2 represent the amounts of mAb GCGR in the central and peripheral compartments, respectively; CmAb GCGR is the serum concentration of mAb GCGR; CLlinear is the linear clearance; Q is the intercompartmental clearance between the central and peripheral compartments; VMAX is the maximum velocity for the saturable elimination from central compartment; and KM is the Michaelis–Menten constant for the saturable elimination from the central compartment. A zero-order drug input rate (R) was included following i.p. dosing to better capture the absorption process. The choice of the PK model structure was based on preliminary evaluation of one- and two-compartment models with first-order elimination versus saturable elimination. The PK parameters estimated from fitting to the above-mentioned PK model were fixed in the following PD analysis.
Fig. 1.

Pharmacokinetic model of mAb GCGR disposition in ob/ob mice. Elimination occurs from the central compartment. The Michaelis–Menten constant (K M) and maximum velocity (V MAX) describe the saturable component of the total clearance, and CLlinear describes the linear component of the total clearance. Absorption occurred through a zero-order process following i.p. dosing with drug input rate R. Transfer of mAb GCGR between central and peripheral compartments is quantified by the intercompartmental clearance, Q
Pharmacodynamic Model
Given the known stimulatory effect of glucagon on glucose production in response to decreasing glucose concentration (1,2) and the inhibitory effect of glucose on glucagon secretion in response to increasing glucose concentration (24,25), a feedback model incorporating the glucose-glucagon inter-regulation and mAb GCGR effect on inhibiting GCGR pathway via competitive interaction was proposed and depicted in Fig. 2. Glucose and glucagon plasma concentrations across treatment groups were fitted simultaneously using the proposed model described by the following equations:
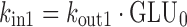 |
3 |
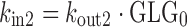 |
4 |
 |
5 |
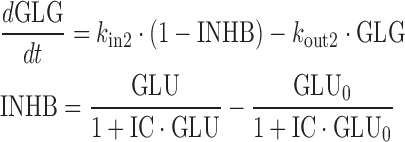 |
6 |
where GLU and GLG represent the glucose and glucagon concentrations over time. These equations represent a natural extension from the traditional indirect response model of one PD variable to a system of first-order differential equations with two interactive variables. Glucose is produced at a constant (zero-order) rate, kin1. Its removal is described by the first-order degradation rate, kout1. Similarly, glucagon concentration is controlled by production and degradation rate constants (kin2 and kout2). At time zero, the glucose and glucagon systems are assumed to be at their physiological steady state, represented by the baseline equations (Eqs. 3 and 4). As glucagon binds to GCGR, a stimulatory factor, STIM, is generated (Eq. 5). The form of the STIM function was selected based on the requirement that STIM approaches zero if GLG approaches GLG0 when there is no inhibitory effect of mAb GCGR on GCGR (i.e., CmAb GCGR = 0). This STIM term describes the glucagon capability for driving the physiological process that increases glucose production. S is a model parameter that describes the slope effect on glucose production from stimulation by glucagon, and EC describes the antagonistic effect of mAb GCGR that modifies S. The assumption of mAb GCGR (competitively) inhibiting the stimulatory effect of glucagon on glucose production was supported by Schild analysis using recombinant human GCGR cell lines in which increasing concentrations of an equally potent anti-GCGR agent, mAb B, induced parallel rightward shifts of the glucagon dose–response curve as measured by glucagon-stimulated cAMP accumulation (19), suggesting a classical inhibition model of Gaddum and Schild (4,26,27). As a feedback, glucose controls glucagon concentrations by inhibiting the production of glucagon (Eq. 6) with the term, INHB. The form of INHB function was selected based on the requirement that INHB approaches zero if GLU approaches GLU0. IC represents the slope factor of glucose on glucagon production. All parameters are greater than zero.
Fig. 2.

Pharmacodynamic model of mAb GCGR disposition in ob/ob mice. Disposition of glucose and glucagon after administration of mAb GCGR according to the feedback pharmacokinetic-pharmacodynamic model. GLU and GLG represent the glucose and glucagon concentrations; C mAb GCGR, mAb GCGR concentration; k in1, glucose production rate; k out1, glucose degradation rate; k in2, glucagon production rate; k out2, glucagon degradation rate; S, slope constant for the effect of glucagon on glucose production; EC, slope constant for the effect of mAb GCGR concentration on reducing glucagon stimulation of glucose production; IC, slope constant for the effect of glucose on reducing glucagon production. The open bar represents stimulation; the solid bar represents inhibition
To demonstrate the applicability of the developed PD model, simulations were conducted to predict glucose-time profiles after multiple i.p. dosing (every 5 days for 5 weeks) with a surrogate chimeric mAb in diet-induced obesity (DIO) mice, a model that demonstrates glucose lowering effect after chronic treatment. Parameter estimates derived from the modeling were used in the simulations. The threefold lower in vitro potency of the surrogate mAb relative to mAb GCGR was factored into the EC value. The baseline glucose value was adjusted to approximately 160 mg/dL to reflect the glucose range in DIO mice. To validate the model, simulation results were compared to the observed data.
RESULTS
Noncompartmental PK and PD Analysis
Table I summarizes the noncompartmental PK and PD parameters in mAb GCGR-treated ob/ob mice. For the dose range studied (0.6–3 mg/kg), mAb GCGR exhibits nonlinear PK profile with greater than dose-proportional increase in exposure with increasing doses following single i.p. administration (see Fig. 3). The Cmax and AUClast values increased by 19.6- and 46.1-fold, respectively, for a fivefold increase in dose, and tmax shows a slight rightward shift from 1 to 2 days with increasing dose levels.
Table I.
Noncompartmental Pharmacokinetic and Pharmacodynamic Parameters after Single i.p. Administration of 0.6, 1, or 3 mg/kg mAb GCGR to Male ob/ob Mice
| Dose (mg/kg) | mAb GCGR | Glucose | Glucagon | ||||||
|---|---|---|---|---|---|---|---|---|---|
| t max (day) | C max (μg/mL) | AUClast (μg·day/mL) | ETmax (day) | MI (%) | AUCglucose (%·day) | ETmax (day) | MS (%) | AUCglucagon (%·day) | |
| 0.6 | 1 | 1.61 | 3.04 | 1 | 30.9 | 90.4 | 1 | 50.6 | 55.0 |
| 1 | 1 | 3.87 | 11.6 | 1 | 37.1 | 177 | 2 | 90.5 | 393 |
| 3 | 2 | 31.6 | 140 | 4 | 46.8 | 378 | 4 | 221 | 1,540 |
C max maximum observed serum concentration, t max time of Cmax, AUC last area under the serum concentration-time curve from time zero to the time of the last quantifiable concentration, ET max time of maximum observed effect, MI maximum inhibition (% change from baseline), MS maximum stimulation (% change from baseline), AUC glucose area under the effect-time curve for glucose, AUC glucagon area above the effect-time curve for glucagon
Fig. 3.

Composite mean (+SD) serum concentration-time profiles of mAb GCGR after single i.p. administration of 0.6, 1, or 3 mg/kg mAb GCGR to male ob/ob mice
The mean glucose and glucagon profiles after single i.p. administration of mAb GCGR are shown in Fig. 4. As demonstrated in Fig. 4a, single i.p. administration of mAb GCGR induced a dose-dependent decrease in glucose concentrations. A rapid decrease in glucose concentrations within 1 day after mAb GCGR treatment was observed. The ETmax was observed to occur between 1 and 4 days post-dose as doses increased from 0.6 to 3 mg/kg. Average value of maximum inhibition of glucose increased from 30.9% to 46.8% as dose increased from 0.6 to 3 mg/kg. The cumulative effect of mAb GCGR, as measured by AUCglucose, increased by approximately fourfold as dose increased from 0.6 to 3 mg/kg. Average glucose concentrations returned to baseline by 4 days following a dose of 0.6 mg/kg, and by 7 and 18 days following a dose of 1 and 3 mg/kg, respectively. As demonstrated in Fig. 4b, single i.p. administration of mAb GCGR elevated glucagon concentrations in a dose-dependent manner. The ETmax was observed to occur between 1 and 4 days post-dose as doses increased from 0.6 to 3 mg/kg. Average value of maximum stimulation of glucagon increased from 50.6% to 221% above baseline as dose increased from 0.6 to 3 mg/kg. The cumulative effect of mAb GCGR, as measured by AUCglucagon, increased by approximately 28-fold as dose increased from 0.6 to 3 mg/kg.
Fig. 4.

Composite mean (±SEM) concentration-time profiles of glucose (left) and glucagon (right) after single i.p. administration of 0.6, 1, or 3 mg/kg mAb GCGR to male ob/ob mice
Pharmacokinetics-Pharmacodynamics Modeling
The observed serum concentration-time profiles of mAb GCGR data in ob/ob mice after single i.p. doses of mAb GCGR were compared with the results fitted by the two-compartment PK model with linear and nonlinear elimination from the central compartment (see Fig. 5). The PK model adequately captured the concentration-time data at all dose levels. The estimated values of the PK parameters are presented in Table II. Model fitting yielded the Michaelis–Menten constant (KM) of 1.58 ± 0.07 μg/mL. In general, the model provided reasonable description of the data suitable for the subsequent PK-PD modeling.
Fig. 5.

Observed and predicted serum concentration-time profiles of mAb GCGR after single i.p. administration of 0.6, 1, or 3 mg/kg mAb GCGR to male ob/ob mice. The symbols represent observed values, and the lines are predicted by the pharmacokinetic model
Table II.
Pharmacokinetic (PK) Parameters Obtained from Fitting the Serum mAb GCGR Data with a Two-compartment PK Model with Parallel Linear and Nonlinear Clearance
| Parameter | Estimate | RSE (%) |
|---|---|---|
| CLlinear (mL/day/kg) | 14.9 | 0.896 |
| Q (mL/day/kg) | 2.83 | 68.1 |
| V 1 (mL/kg) | 895 | 7.08 |
| V 2 (mL/kg) | 6.94 | 39.8 |
| K M (μg/mL) | 1.58 | 4.27 |
| V MAX (μg/day/kg) | 1,020 | 6.96 |
| R (μg/day/kg) | 24100 | 4.68 |
| σ 2 | 0.0942 | 9.61 |
CL linear linear clearance, Q intercompartmental clearance, V 1 volume of central compartment, V 2 volume of peripheral compartment, K M Michaelis–Menten constant, V MAX maximum velocity for the saturable elimination from central compartment, R infusion rate, σ 2 residual variance, RSE relative standard error
The observed plasma concentration-time profiles of glucose and glucagon data in ob/ob mice after single i.p. doses of vehicle or mAb GCGR were compared with the results fitted by the final feedback PK-PD model as described by Eqs. 5 and 6 (see Fig. 6). In control mice, glucose and glucagon concentrations remained unchanged before and after vehicle injection with estimated initial values of 101 ± 1 mg/dL and 533 ± 48 pg/mL, respectively (Fig. 6a, b, top left; Table III). The concentrations of these two biomarkers were controlled by their respective production rates (kin1 and kin2) balanced by their degradation rates (kout1 and kout2) as shown in Eqs. 3 and 4. Baseline glucagon concentration was the only parameter that was included with a random effect to account for the inter-dose difference. The difference in objective function values for models with and without this random effect was 6.48. As expected, this parameter of random effect is poorly estimated, due to the composite sampling nature of concentration data with three dose groups. As shown in Fig. 6b, glucagon concentrations were over-predicted in the placebo group but, to a lesser extent, under-predicted in the treated groups. The slight under-prediction was greater in the 1-mg/kg group compared to the 0.6- and 3-mg/kg groups. However, the model appeared to provide a reasonable description of the data, which shows consistency with the physiological situation in the ob/ob mouse model.
Fig. 6.

Observed and predicted plasma concentration of glucose a and glucagon b after single i.p. administration of 0.6 to 3 mg/kg mAb GCGR to male ob/ob mice. The symbols represent observed values, and the lines are predicted by the feedback pharmacokinetic-pharmacodynamic model. The solid line represents population prediction; the dotted line represents individual (dose-group) prediction
Table III.
Pharmacodynamic (PD) Parameters Obtained from Fitting the Plasma Glucose and Glucagon Data Simultaneously with the Final Pharmacokinetic-PD Model with Feedback Mechanism
| Parameter | Estimate | RSE (%) |
|---|---|---|
| GLU0 (mg/dL) | 101 | 1.19 |
| GLG0 (pg/mL) | 533 | 8.91 |
| ω 2 GLG0 | 0.0138 | 102 |
| k in1 (mg/dL/day) | 266 | 12.8 |
| k in2 (pg/mL/day) | 439 | 21.5 |
| S (pg/mL−1) | 0.001 | 5.44 |
| EC (μg/mL−1) | 0.606 | 23.3 |
| IC (mg/dL−1) | 0.0507 | 9.82 |
| σ 2 glucose | 0.245 | 30.9 |
| σ 2 glucagon | 0.0278 | 5.22 |
ω 2 GLG0 inter-dose variance for baseline glucagon, S slope constant for the effect of glucagon on glucose production, EC slope constant for the effect of monoclonal antibody glucagon receptor concentration on reducing glucagon stimulation of glucose production, IC slope constant for the effect of glucose on reducing glucagon production, k in1 glucose production rate, k in2 glucagon production rate, GLU 0 baseline glucose, GLG 0 baseline glucagon, σ 2 glucose residual variance for glucose, σ 2 glucagon residual variance for glucagon
The stimulatory component (STIM) in Eq. 5 reflects the ability of glucagon to stimulate glucose production with the dampening effect from mAb GCGR. S and EC are estimated to be 0.001 ± 0.00005 pg/mL−1 and 0.606 ± 0.141 μg/mL−1, respectively. Inhibitory effect (INHB) in Eq. 6 reflects the ability of glucose to suppress glucagon secretion. IC is estimated to be 0.051 ± 0.005 mg/dL−1. When mAb GCGR concentrations are maintained at the predicted maxima of the three dose levels (0.6, 1, and 3 mg/kg) with the current sampling time points, the maximum glucose reductions from the current typical baseline (101 mg/dL) are predicted to be approximately 13.1%, 25.0%, and 44.7%, respectively (see Fig. 7).
Fig. 7.

Constant mAb GCGR concentration versus steady-state glucose concentration. Any point (x, y) on the curve shows that the steady-state glucose is eventually reached at a new concentration y if mAb GCGR concentration is maintained at a constant concentration of x
As evidenced by the reasonably close agreement between the observed and predicted glucose and glucagon profiles, the feedback model generally described the PD data well and successfully captured the reversibility of both biomarkers back to baseline after single-dose treatment of mAb GCGR, suggesting homeostasis is maintained. The PD parameters of fixed effects were determined with good precision, with the highest percent standard error being 23.3%. Residual variability for the two biomarkers was reasonable. The diagnostic plots did not show apparent deficiency for the data fit (see Fig. 8).
Fig. 8.

Goodness-of-fit plots for the feedback pharmacodynamic model. a Predicted versus observed values for glucose, b weighted residuals versus time for glucose, c predicted versus observed values for glucagon, and d weighted residuals versus time for glucagon
The simulated glucose concentration-time profile following multiple dosing of the surrogate mAb is shown in Fig. 9. Simulation using established parameters well captured the glucose change over time following chronic treatment of mAb given every 5 days at 3 mg/kg.
Fig. 9.

Simulated glucose-time profiles in diet-induced obesity mice after i.p. administration of surrogate monoclonal antibody at 3 mg/kg every 5 days. The symbols represent observed values, the solid line is the simulated median, and the dotted lines are the simulated percentiles 5, 95. The variability, which is described by the 5% and 95% percentiles, originates exclusively from the inter-dose variance for baseline glucagon
DISCUSSION
Following single i.p. administration, mAb GCGR exhibits nonlinear PK profiles, and Cmax and AUClast increased more than dose proportionally (Table I and Fig. 3). This finding is consistent with available data of mAb GCGR studied in multiple preclinical species (data not shown). In mAb GCGR-treated animals, dramatic and long-lasting reduction in blood glucose concentrations was associated with marked elevated glucagon, whereas placebo-treated group shows no major fluctuations in both glucose and glucagon concentrations throughout the experiment. Based on the dose-dependent, glucose-lowering effect as seen in Fig. 4, it is believed that mAb GCGR has an indirect effect on glucose concentration, as exemplified by the time delay of maximal reduction of glucose relative to the observed maximal mAb GCGR concentration (Table I). The indirect effect on glucagon concentration is even more apparent in that the time of maximum observed concentration of glucagon has a 1–2-day delay relative to that of mAb GCGR.
To elucidate the potential underlying mechanism of GCGR-mediated hepatic glucose production initiated by glucagon and the antagonizing effects of mAb GCGR, a semi-mechanistic PK-PD model was developed. This model quantitatively characterized the homeostatic regulation between glucose and glucagon as well as the changes in the profiles over time caused by acute treatment of mAb GCGR in ob/ob mice. The dose range used in this study (0.6–3 mg/kg) was appropriately chosen to differentiate the dose-dependent glucose-lowering effects upon GCGR blockage. Based on results from a pilot dose-finding study (data not shown), the dose of 3 mg/kg was sufficient to produce full inhibition of GCGR-mediated glucose production. The PK of mAb GCGR in ob/ob mice was well characterized by a two-compartment model with linear and nonlinear elimination (Fig. 5). The saturable nonlinear elimination mechanism was modeled using a standard Michaelis–Menten component. The estimated KM value of 1.58 ± 0.07 μg/mL (Table II) is similar to the in vitro IC50 of 1.43 ± 0.67 μg/mL (unpublished data) determined based on functional assay evaluating inhibition of glucagon-induced cAMP accumulation in murine GCGR-expressed cell line, which suggest that the saturable nonlinear elimination of mAb GCGR is associated with its interaction with GCGR. A mechanistic PK model for drugs exhibiting target-mediated drug disposition (TMDD) (28,29,30) could also be used to describe the kinetics of mAb GCGR. However, the binding equations used to describe the TMDD model with quasi-steady-state approximation resemble Michaelis–Menten kinetics (30). We believe that Michaelis–Menten model, as a simplification of the full TMDD model, provides reasonable description of the mAb GCGR concentration-time data, suitable for the subsequent PK-PD analysis.
A standard sequential modeling approach was used to reduce the model fitting time and to obtain a reasonable description of the data. Since only one sample per mouse was obtained during sacrifice, a composite sampling approach was used. Thus, the data analysis adopted a naïve pooling method, which ignores the variability of PK parameters from each animal and regard data as if they came from a single animal per dose group. Due to this data limitation, simultaneous model fitting was not considered to have additional value. Future work with more extensive sampling should consider including simultaneous fitting of the mAb GCGR, glucose, and glucagon data with a TMDD model and a component of the glucose-glucagon feedback loop.
The estimated PK parameters listed under Table II appear to deviate from the values of a typical IgG2 antibody in mice, with large central volume and fast linear clearance for instance. These apparent deviations may be related to the sparseness of the PK data such as the lack of data available during the initial absorption phase and the lack of concentrations measurable at the terminal phase for the low dose group. Considering the above-mentioned limitations, the established PK model did manage to provide a reasonable phenomenological description of the observed trend.
The choice of the PK-PD model was based on the mechanism of action of mAb GCGR as well as the known biological fact of glucagon and GCGR on glucose homeostasis. It is known that glucagon stimulates glucose production via GCGR signaling pathway in response to hypoglycemic state (1,2) and, on the other hand, glucose suppresses glucagon secretion in response to hyperglycemic state, although the exact mechanisms for such regulations are not fully understood (24,25,31). The current proposed model combines the form of indirect response model with stimulation and inhibition of input as proposed by Dayneka et al. (21) and included the effect of glucagon on glucose disposition and vice versa, as well as the antagonistic property of mAb GCGR on GCGR signaling.
The stimulation of glucagon on glucose input rate and competitive antagonistic interaction between mAb GCGR and glucagon on binding to GCGR was reflected in the stimulation term (STIM) in Eq. 5. The stimulation on glucose production is driven by the change of glucagon. The STIM term also has a competitive-antagonist type of dependence on mAb GCGR concentration as described by 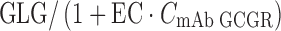 in which GLG concentration is modulated by mAb GCGR. In the absence of mAb GCGR, STIM term goes to zero; hence, glucose concentration at steady state was controlled only by kin1 and kout1.
in which GLG concentration is modulated by mAb GCGR. In the absence of mAb GCGR, STIM term goes to zero; hence, glucose concentration at steady state was controlled only by kin1 and kout1.
To model glucose inhibitory effect (INHB) on glucagon production, the change of glucose 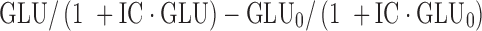 was used as an inhibitor on glucagon production rate, kin2, as demonstrated in Eq. 6. If GLU is smaller than GLU0, then glucagon and, in turn, glucose will be stimulated until the steady state is restored. Similarly, when GLU is larger than GLU0, both glucagon and glucose will be inhibited. According to Eq. 6, when GLU level is close to, or greater than, 160 mg/dL, the change of GLG over time is negative, and Eq. 6 and represents the removal of GLG from the system. Lack of glucagon suppression contributes to hyperglycemia. Given the age of the ob/ob mice used in the study, glucose concentration measured was never greater than 160 mg/dL throughout the experiment and was unlikely to be at such high level from a physiological point of view. The equation is reasonable in a sense that it captures the potential regulation of glucagon suppression.
was used as an inhibitor on glucagon production rate, kin2, as demonstrated in Eq. 6. If GLU is smaller than GLU0, then glucagon and, in turn, glucose will be stimulated until the steady state is restored. Similarly, when GLU is larger than GLU0, both glucagon and glucose will be inhibited. According to Eq. 6, when GLU level is close to, or greater than, 160 mg/dL, the change of GLG over time is negative, and Eq. 6 and represents the removal of GLG from the system. Lack of glucagon suppression contributes to hyperglycemia. Given the age of the ob/ob mice used in the study, glucose concentration measured was never greater than 160 mg/dL throughout the experiment and was unlikely to be at such high level from a physiological point of view. The equation is reasonable in a sense that it captures the potential regulation of glucagon suppression.
When there is no change in glucose concentration from its initial value, the INHB term goes to zero, and glucagon concentration at steady state is only controlled by kin2 and kout2. Consequently, in the absence of mAb GCGR, euglycemic condition is maintained, and no glucose reduction or glucagon elevation was observed as depicted in Fig. 6a (top left) and Fig. 6b (top left), respectively. On the other hand, the glucose-glucagon system is indeed perturbed by positive mAb GCGR concentration, as shown in this study. Assume a constant mAb GCGR is maintained, then a new steady-state concentration of glucose (and of glucagon) will eventually be reached based on the current model. The dependence of the new glucose steady-state concentration on mAb GCGR concentration is displayed in Fig. 7. The decrease of glucose is about 45% at 31.6 μg/mL (which is the Cmax of 3 mg/kg dose), beyond which, there is essentially no additional glucose lowering.
Models with added complexities were also tested. These models either did not converge successfully or the precision of the PD parameters were compromised or there was no significant drop in the objective function value.
Besides the mAb GCGR mediated glucagon elevation described above, it is plausible that a compensatory mechanism such as increased secretion of glucagon from pancreatic alpha cells due to reduced GCGR signaling could also contribute to the observed glucagon elevation in our single-dose treatment. However, the fact that the glucagon concentrations returned to baseline in a dose-dependent manner and the adequacy of the developed PD model in describing the glucose and glucagon data suggests that the competitive interaction hypothesis is sufficient to explain the observed transient elevation of glucagon after single-dose treatment.
To apply the developed model for predicting PD in different study scenarios, we simulated the glucose-lowering effect of a surrogate mAb under chronic treatment and compared it to the observed data. The simulation was for a study conducted in a different animal model (DIO mice) with a different compound having lower in vitro potency compared to mAb GCGR. Overall, the model was predictive of the sustained glycemic control observed in DIO mice after adjustment of EC value and glucose baseline to reflect the mAb property and the nature of the animal model (Fig. 9). Simulation of glucagon was not conducted, as glucagon was not monitored in the study. It should be noted that chronic administration of GCGR antagonists could lead to alpha cell hypertrophy or hyperplasia, as resulted in hypersecretion of glucagon (15). Hence, when the data from chronic treatment of mAb GCGR become available, a mechanism of increased glucagon secretion caused by prolonged GCGR blockage could be incorporated into the feedback PK-PD model. In addition, with more data, this model will be further evaluated, and other possible mechanism such as the initial glucose effect on its own elimination or insulin action on glucose uptake could be investigated.
Although our model successfully described the glucose and glucagon dynamics in the absence or presence of mAb GCGR, it is important to point out that the model is not fully mechanistic. One deficiency of the model is the negligence of other metabolic hormones that play a role in the glucose homeostatic system. Hormonal regulation of glucose is controlled by both insulin and glucagon, among others. Insulin has broad glucose-lowering effects including inhibition of hepatic glucose production via gluconeogenesis and glycogenolysis as well as stimulation of glucose output via peripheral glucose uptake (32,33). In the current study, insulin was not collected due to limitation on maximum volume of blood that could be drawn from each ob/ob mouse. The physiological action of insulin on inhibiting hepatic glucose output or on promoting glucose disposal was therefore omitted during the model building process. mAb GCGR did not seem to have any direct effect on the activities related to the insulin pathway, although theoretically, blockage of GCGR pathway might alleviate the body’s demand on glucose-lowering insulin and changes the insulin production rate to a certain extent. To better understand the complexities and the role of metabolic hormones in glucose homeostasis, a more physiologically based PK-PD model considering all the crucial hormones needs to be explored in the future. Other drug-related phenomenon upon chronic treatment of mAb GCGR such as drug tolerance caused by depletion of precursors or rebound effect following treatment cessation in the presence of marked elevation of glucagon also needs to be taken into account.
CONCLUSION
A semi-mechanistic feedback PK-PD model incorporating the glucose-glucagon inter-regulation and the hypothesized mechanism of mAb GCGR was successfully developed. It can describe the observed mAb GCGR PK, as well as glucose and glucagon profiles reasonably well. As a first attempt for modeling the glucose-glucagon system, such a model should allow additional insights on glucose homeostasis and have future applicability in the prediction of efficacy of anti-GCGR antibody based therapy in type 2 diabetes.
References
- 1.Jiang G, Zhang BB. Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab. 2003;284:E671–E678. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- 2.Cherrington AD. Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48:1198–1214. doi: 10.2337/diabetes.48.5.1198. [DOI] [PubMed] [Google Scholar]
- 3.Jelinek LJ, Lok S, Rosenberg GB, Smith RA, Grant FJ, Biggs S, Bensch PA, Kuijper JL, Sheppard PO, Sprecher CA, et al. Expression cloning and signaling properties of the rat glucagon receptor. Science. 1993;259:1614–1616. doi: 10.1126/science.8384375. [DOI] [PubMed] [Google Scholar]
- 4.Mayo KE, Miller LJ, Bataille D, Dalle S, Goke B, Thorens B, et al. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol Rev. 2003;55:167–194. doi: 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- 5.DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non-insulin-dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism. 1989;38:387–395. doi: 10.1016/0026-0495(89)90129-7. [DOI] [PubMed] [Google Scholar]
- 6.Staehr P, Hother-Nielsen O, Levin K, Holst JJ, Beck-Nielsen H. Assessment of hepatic insulin action in obese type 2 diabetic patients. Diabetes. 2001;50:1363–1370. doi: 10.2337/diabetes.50.6.1363. [DOI] [PubMed] [Google Scholar]
- 7.Muller WA, Faloona GR, Aguilar-Parada E, Unger RH. Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med. 1970;283:109–115. doi: 10.1056/NEJM197007162830301. [DOI] [PubMed] [Google Scholar]
- 8.Unger RH, Orci L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet. 1975;1:14–16. doi: 10.1016/S0140-6736(75)92375-2. [DOI] [PubMed] [Google Scholar]
- 9.Lins PE, Wajngot A, Adamson U, Vranic M, Efendic S. Minimal increases in glucagon levels enhance glucose production in man with partial hypoinsulinemia. Diabetes. 1983;32:633–636. doi: 10.2337/diabetes.32.7.633. [DOI] [PubMed] [Google Scholar]
- 10.Consoli A, Nurjhan N, Capani F, Gerich J. Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes. 1989;38:550–557. doi: 10.2337/diabetes.38.5.550. [DOI] [PubMed] [Google Scholar]
- 11.Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev. 2007;28:253–283. doi: 10.1210/er.2006-0026. [DOI] [PubMed] [Google Scholar]
- 12.Sloop KW, Michael MD, Moyers JS. Glucagon as a target for the treatment of type 2 diabetes. Expert Opin Ther Targets. 2005;9:593–600. doi: 10.1517/14728222.9.3.593. [DOI] [PubMed] [Google Scholar]
- 13.Burcelin R, Katz EB, Charron MJ. Molecular and cellular aspects of the glucagon receptor: role in diabetes and metabolism. Diabetes Metab. 1996;22:373–396. [PubMed] [Google Scholar]
- 14.Liang Y, Osborne MC, Monia BP, Bhanot S, Gaarde WA, Reed C, She P, Jetton TL, Demarest KT. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes. 2004;53:410–417. doi: 10.2337/diabetes.53.2.410. [DOI] [PubMed] [Google Scholar]
- 15.Sloop KW, Cao JX, Siesky AM, Zhang HY, Bodenmiller DM, Cox AL, et al. Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest. 2004;113:1571–1581. doi: 10.1172/JCI20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lima JJ, Matsushima N, Kissoon N, Wang J, Sylvester JE, Jusko WJ. Modeling the metabolic effects of terbutaline in beta2-adrenergic receptor diplotypes. Clin Pharmacol Ther. 2004;76:27–37. doi: 10.1016/j.clpt.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Landersdorfer CB, Jusko WJ. Pharmacokinetic/pharmacodynamic modelling in diabetes mellitus. Clin Pharmacokinet. 2008;47:417–448. doi: 10.2165/00003088-200847070-00001. [DOI] [PubMed] [Google Scholar]
- 18.Kirchheiner J, Bauer S, Meineke I, Rohde W, Prang V, Meisel C, et al. Impact of CYP2C9 and CYP2C19 polymorphisms on tolbutamide kinetics and the insulin and glucose response in healthy volunteers. Pharmacogenetics. 2002;12:101–109. doi: 10.1097/00008571-200203000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Yan H, Gu W, Yang J, Bi V, Shen Y, Lee E, et al. Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J Pharmacol Exp Ther. 2009;329:102–111. doi: 10.1124/jpet.108.147009. [DOI] [PubMed] [Google Scholar]
- 20.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 21.Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21:457–478. doi: 10.1007/BF01061691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabrielsson J, Jusko WJ, Alari L. Modeling of dose-response-time data: four examples of estimating the turnover parameters and generating kinetic functions from response profiles. Biopharm Drug Dispos. 2000;21:41–52. doi: 10.1002/1099-081X(200003)21:2<41::AID-BDD217>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 23.Solnica B, Naskalski JW, Sieradzki J. Analytical performance of glucometers used for routine glucose self-monitoring of diabetic patients. Clin Chim Acta. 2003;331:29–35. doi: 10.1016/S0009-8981(03)00079-2. [DOI] [PubMed] [Google Scholar]
- 24.Gerich JE, Charles MA, Grodsky GM. Regulation of pancreatic insulin and glucagon secretion. Annu Rev Physiol. 1976;38:353–388. doi: 10.1146/annurev.ph.38.030176.002033. [DOI] [PubMed] [Google Scholar]
- 25.Gerich JE, Charles MA, Grodsky GM. Characterization of the effects of arginine and glucose on glucagon and insulin release from the perfused rat pancreas. J Clin Invest. 1974;54:833–841. doi: 10.1172/JCI107823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaddum JH. Theories of drug antagonism. Pharmacol Rev. 1957;9:211–218. [PubMed] [Google Scholar]
- 27.Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy G. Mechanism-based pharmacodynamic modeling. Clin Pharmacol Ther. 1994;56:356–358. doi: 10.1038/clpt.1994.134. [DOI] [PubMed] [Google Scholar]
- 29.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28:507–532. doi: 10.1023/A:1014414520282. [DOI] [PubMed] [Google Scholar]
- 30.Gibiansky L, Gibiansky E, Kakkar T, Ma P. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn. 2008;35:573–591. doi: 10.1007/s10928-008-9102-8. [DOI] [PubMed] [Google Scholar]
- 31.Vieira E, Salehi A, Gylfe E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia. 2007;50:370–379. doi: 10.1007/s00125-006-0511-1. [DOI] [PubMed] [Google Scholar]
- 32.Flakoll PJ, Carlson MG, Cherrington AD. Physiologic action of insulin, Chapter 14. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes mellitus. A fundamental and clinical text. 2. Philadelphia: Lippincott Williams & Wilkins; 2000. pp. 148–161. [Google Scholar]
- 33.Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, Ford EL, et al. Insulin signaling in α cells modulates glucagon secretion in vivo. Cell Metab. 2009;9:350–361. doi: 10.1016/j.cmet.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]