

Abstract
Fundamental to cell adhesion and migration, integrins are large heterodimeric membrane proteins that uniquely mediate inside-out signal transduction, whereby adhesion to the extracellular matrix is activated from within the cell by direct binding of talin to the cytoplasmic tail of the β integrin subunit. Here, we report the first structure of talin bound to an authentic full-length β integrin tail. Using biophysical and whole cell measurements, we show that a specific ionic interaction between the talin F3 domain and the membrane–proximal helix of the β tail disrupts an integrin α/β salt bridge that helps maintain the integrin inactive state. Second, we identify a positively charged surface on the talin F2 domain that precisely orients talin to disrupt the heterodimeric integrin transmembrane (TM) complex. These results show key structural features that explain the ability of talin to mediate inside-out TM signalling.
Keywords: cell adhesion, crystallography, integrin activation, NMR, talin
Introduction
Integrins are essential to biological functions that range from leukocyte trafficking to tissue integrity; these adhesion receptors are also therapeutic targets in thrombosis, inflammation, and cancer. Each α and β subunit of the integrin heterodimer consists of several linked globular extracellular domains, a single transmembrane (TM) helix, and a generally short cytoplasmic tail. Cellular modulation of integrin affinity (‘activation') has a pivotal role in the biological function of these receptors and is the subject of much current interest (Hynes, 2002; Calderwood, 2004; Campbell and Ginsberg, 2004; Ginsberg et al, 2005). There is some controversy about the exact structure of the high- and low-affinity states, but strong evidence indicates that activation is initiated by protein–protein interactions with the cytoplasmic domains, causing tail separation and propagation of conformational changes to the outside of the cell (Lu et al, 2001; Takagi et al, 2001; Kim et al, 2003; Partridge et al, 2005; Arnaout et al, 2007; Luo et al, 2007; Wegener and Campbell, 2008; Askari et al, 2009). A key event is binding to the integrin β cytoplasmic tail by talin (Tadokoro et al, 2003; Nieswandt et al, 2007; Petrich et al, 2007a, 2007b), a 270 kDa protein (capable of forming homodimers) with an N-terminal head domain (comprising F0, F1, F2, and F3 subdomains) and a C-terminal rod domain that binds to vinculin and actin (Critchley and Gingras, 2008) (Figure 1C). Binding of the F3 domain to integrin β tails is sufficient for integrin activation (Calderwood et al, 2002), although other head domains contribute to activation (Bouaouina et al, 2008).
Figure 1.

Integrin and talin sequence comparisons. (A) Sequence of the cytoplasmic tail of the β1A, β1D, and β3 integrins. Residues in β1D and β3 that differ from β1A are highlighted, and a key membrane–proximal aspartate residue is indicated with β1 numbering. Secondary structure (α helices in blue, 310 helices in green) is based on the β1D/talin2 complex structure. Residues embedded in the membrane (Lau et al, 2008b) are shaded in grey. Residues are coloured by chemical properties: acidic residues are shown in red, basic in blue, aliphatic in green, aromatic in cyan, polar in lavender, and others are given unique colouring. (B) Sequence of the F2–F3 domains of talin1 and talin2. Residues in talin2 that differ from talin1 are highlighted, and secondary structure was determined as in (A). The F2 domain is underlined in cyan and the F3 in yellow. (C) A schematic of the domain structure of talin. Talin homodimerization (not shown) occurs at the C-terminus.
Structural studies of integrin activation have been hampered by the weak nature of the talin/integrin interaction. Various strategies have been used to overcome this problem, beginning with a crystal structure of a short membrane–distal fragment of the β3 tail covalently tethered to the talin1 F2–F3 fragment (Garcia-Alvarez et al, 2003). This showed that the F3 domain interacts with the NPxY motif of the β3 tail in canonical PTB domain fashion (Calderwood et al, 2003), but this gave no information about the membrane–proximal region of the β3 tail, a region known to be essential for activation (Hughes et al, 1995; Vinogradova et al, 2002; Ulmer et al, 2003). A second structure, solved by NMR, elucidated the interface between the F3 domain and the β3 membrane–proximal helix (Wegener et al, 2007). This was made possible by constructing a chimaeric peptide of the β3 helix attached to a sequence from PIPK1γ that binds tightly to the talin NPxY binding pocket (Barsukov et al, 2003; de Pereda et al, 2005). These studies offered some insight into the β3/talin1 complex and the structural basis of integrin activation.
Prior structural studies of integrin activation by talin have involved a single integrin and talin isoform. However, mammals express two isoforms of talin (Figure 1B) and eight different β integrins, some of which exhibit additional splice variants (Figure 1A). In this study, we explore a wider range of talin/integrin interactions to identify a pair more suitable for structure determination. The resulting crystal structure, along with a multifaceted experimental approach, reveals a concerted series of evolutionarily conserved interactions that initiate inside-out signalling.
Results
The structure of the talin2/β1D complex
Through NMR studies of β tail/talin complexes (Table I; Supplementary Figure S1) we found that integrin β tails differ widely in their affinity for different isoforms of talin and that β1D binds to talin2 with a much higher affinity than any previously studied integrin/talin pair. Talin2 and β1D, a splice variant of β1 differing from β1A only in its C-terminus (Figure 1A), are the major isoforms found in striated muscle (Belkin et al, 1996; Senetar et al, 2007; Conti et al, 2008), and the formation of this higher affinity complex is consistent with the high forces that this talin/integrin complex is subjected to in myotendinous junctions (Belkin et al, 1996; Conti et al, 2008).
Table 1.
Kd values of talin F3/β integrin tail interactions
| Talin1 | Talin2 | |
|---|---|---|
| β1A | 490±10 | 652±20 |
| β1D | 95±4 | 36±2 |
| β3 | 273±6 | 438±15 |
| Kd values are given in μM±s.e. | ||
This tighter complex crystallized, thus enabling us to solve the structure of the β1D integrin tail/talin2 F2–F3 complex at 2.2 Å resolution (Figure 2A; Table II). This is the first structure of talin bound to an authentic β integrin tail and the first involving either of these two striated muscle-specific isoforms. Each asymmetric unit in the crystal contained two integrin/talin heterodimers (Supplementary Figure S2A) with distinct electron density visible for the N-terminal 37 residues of the integrin tail (Supplementary Figure S3). The last 13 residues were not observed, suggesting that they remain unstructured. The talin2 F2–F3 domains exhibit similar folds and relative orientations to those seen in previous talin1 F2–F3 structures (Supplementary Figure S2B; Supplementary Table I), and the interface of the membrane–proximal integrin helix with the talin F3 activation loop is similar to that observed for the β3/PIPK1γ chimaera (Supplementary Figure S2C). This new structure allows detailed comparisons of the complexes formed between different talin and integrin isoforms; further analysis to define the structural basis of differing integrin/talin affinities—focusing on the more membrane–distal portions of this interface—will be an important area of future study. However, here we show that the β1D integrin tail/talin2 F2–F3 structure also reveals several novel features that permit the formulation of a new comprehensive structural model of integrin activation.
Figure 2.

The talin2/β1D structure. (A) One heterodimer from the crystal structure of talin2 F2–F3 bound to the β1D integrin tail. Labelling is for talin2/β1D with talin1/β3 numbering in parentheses. Highlighted residues interact with the membrane or form a key integrin/talin salt bridge. All structure images were generated with MOLMOL (Koradi et al, 1996). (B) The talin2/β1D structure was merged with the β3 transmembrane segment (PDB ) (Lau et al, 2008b) and aligned to the calculated membrane tilt angle of 25°. The electrostatic potential is mapped on talin, illustrating the juxtaposition of several positively charged residues next to the membrane surface.
Table 2.
Data collection and refinement statistics
| Data collection | |
| Beam line | ESRF ID23.EH2 |
| Space group | P212121 |
| Cell parameters | a=53.26 Å, b=108.72 Å, c=131.85 Å |
| α=β=γ=90° | |
| Wavelength (Å) | 0.8726 |
| Resolution (Å) | 41.95–2.17 (2.28–2.17)a |
| Total reflections | 154 133 (21 979) |
| Unique reflections | 41 362 (5982) |
| Rmerge | 0.114 (0.338) |
| Completeness (%) | 99.6 (99.9) |
| Multiplicity | 3.7 (3.7) |
| I/σ(I) | 8.3 (3.5) |
| Refinement | |
| Rwork (%) | 21.3 |
| Rfree (%) | 24.9 |
| Overall mean B values (Å2) | 32.3 |
| Number of amino acid residues per asymmetric unit | 500 |
| Number of water molecules | 382 |
| Matthews coefficient | 3.05 (water content, 59.7%) |
| RMSD from ideal values | |
| Bonds/angles (Å/deg) | 0.005/0.874 |
| Estimated error based on maximum likelihood | |
| Coordinate/phase (Å/deg) | 0.32/25.1 |
| aHighest resolution shell is shown in parenthesis. | |
A positively charged patch on the talin F2 domain binds to the cell membrane
The talin2/β1D structure exhibits a well-defined N-terminal β tail helix extending from K752 to A773 (corresponding to K716-A737 in β3). This helix overlaps with a recent NMR structure of the β3 TM domain that exhibits a membrane-embedded helix extending to I721 and tilted by about 25° to the membrane bilayer (Lau et al, 2008b). Thus, β1D residues K752-I757 can be overlaid and merged with the membrane-embedded residues K716-I721 of the β3 TM structure, a procedure that could not be performed with the β3/PIPK1γ structure because of fraying of the integrin N-terminus in solution. The predicted orientation of the talin2/β1D structure with respect to the membrane (Figure 2B) results in the striking juxtaposition of a positively charged patch in the F2 domain (residues K258, K274, R276, and K280 in talin2; K256, K272, K274, and R277 in talin1) with the membrane (Figure 2A). These residues are conserved across species (Supplementary Figure S4), and we thus hypothesized that the residues forming this membrane orientation patch (MOP) have a key role in integrin activation.
This hypothesis was tested using a well-established αIIbβ3 integrin activation assay in CHO cells (Han et al, 2006) (Supplementary Figure S5). Expression of wild-type (wt) talin1 F3 slightly increased integrin activation, but expression of a longer talin1 construct with additional N-terminal domains (F1–F2–F3) caused a much more pronounced increase in integrin activation (Figure 3B). This activation was partially or fully abrogated by mutating the four MOP residues singly, doubly, or quadruply (4E) to glutamate (Figure 3A). To ensure that the observed effects were not because of protein instability, decreased integrin binding, or major structural changes, NMR studies were conducted on the 4E mutant. The mutations did not significantly affect integrin binding or the NMR spectrum of talin1 F2–F3, indicating that the 4E construct was folded and stable (Supplementary Figure S6). To test the generalizability of these findings, these experiments were repeated in CHO cells expressing a chimaeric integrin consisting of the extracellular and TM domains of αIIbβ3 and the intracellular domains of α5β1A (O'Toole et al, 1994) (Figure 3C). Consistent with previous findings (O'Toole et al, 1994; Bouaouina et al, 2008; Hato et al, 2008), the effect of talin on this integrin was reduced in comparison to αIIbβ3, owing to both higher basal activation and decreased maximal activation in response to talin. However, talin1 F1–F2–F3 did increase activation of this integrin, and this increase was abrogated by mutating MOP residues.
Figure 3.

A key role for a talin/membrane interaction in integrin activation. (A) GFP-talin1 F1–F2–F3 wild type (F1–F2–F3) or F1–F2–F3 with various mutations in the F2 domain were transfected into αIIbβ3-expressing CHO cells. Activated integrins were detected with PAC1 antibody and analysed by FACS 24 h after transfection. Integrin activity was normalized against GFP-F1–F2–F3 wt-transfected cells. Error bars represent standard errors of three independent experiments. 4E corresponds to all four membrane orientation patch (MOP) residues mutated to glutamate, and 4A corresponds to all four mutated to alanine. (B) As in (A), but with GFP-talin1 F3 wt or F1–F2–F3 wt. Error bars (barely visible because of small size) represent standard errors of two independent experiments. F3 caused a statistically significant increase in integrin activation (*) of P=0.0388 by one tail test. (C) As in (A) and (B), but GFP-talin1 F3 wt, F1–F2-F3 wt, or F1–F2–F3 mutants were transfected into CHO cells expressing a chimaeric integrin containing the intracellular domains of α5β1A, as described in the text. Error bars represent standard errors of four independent experiments.
To show a direct interaction between the talin MOP and the membrane, we performed vesicle cosedimentation assays (Figure 4). A solution containing protein and vesicles was separated by centrifugation into a pellet consisting of the vesicles plus bound protein and a supernatant containing unbound material. In the presence of neutral phosphatidylcholine vesicles, talin1 F2–F3 remained in the unbound supernatant fraction. However, addition of 20% negatively charged phosphatidylserine to these vesicles caused 40% of talin1 F2–F3 to precipitate with the vesicles, and this binding was fully abrogated by the 4E mutation. Increasing the negatively charged content of the vesicles to 100% phosphatidylserine caused wt talin1 F2–F3 to be fully bound; the 4E mutation significantly decreased this binding, although some residual binding was still detected. Similar results were achieved with the talin1 F2 domain, although binding was only detected in 100% phosphatidylserine vesicles, and the 4E mutation fully abrogated vesicle binding. Thus, we have identified a specific new talin/membrane interaction site that is essential for full integrin activation and is sensitive to the presence of negatively charged moieties in the membrane.
Figure 4.

The talin F2 domain membrane orientation patch (MOP) interacts with negatively charged membrane phospholipids. Talin1 F2–F3 and F2 (0.15 mg/ml), either wt or 4E, were mixed with vesicles (0.5 mg/ml) consisting of phosphatidylcholine (PC), phosphatidylserine (PS), or a 4:1 ratio of PC:PS and then centrifuged. (A) Coomassie-stained gel of one representative experiment. Unbound protein was located in the supernatant (S) and bound protein in the pellet (P). (B) Graphical representation of the percentage of protein bound to lipid vesicles (average of three independent experiments±standard error).
The talin F3 domain forms a key membrane–proximal salt bridge with the β integrin tail
The β1D/talin2 structure reveals a salt bridge between β1D D759 and talin2 K327 that caps the membrane–proximal portion of the interaction (Figure 2A). The distance between the aspartate side chain carboxyl oxygen atom and lysine side chain nitrogen atom is 3.7 Å (Supplementary Figure S3C). We note that this is a relatively large distance, but it is within the commonly used 4.0 Å limit (Barlow and Thornton, 1983; Kumar and Nussinov, 1999). This geometry is consistent between the two dimers in the asymmetric unit, and the electron density of these side chains is well defined (B values ranging from an average of 23 Å2 for both Cβ atoms, to an average of 31 Å2 for the aspartate side chain carboxyl oxygen atoms, and 45 Å2 for the lysine nitrogen atom). An analysis of the β1D/talin2 by PISA (Krissinel and Henrick, 2007) predicts that β1D D759 and talin2 K327 contribute favourably to the overall interaction (data not shown).
These two residues are conserved in other paralogues of talin and integrins (Figure 1, D723 in β3 and K324 in talin1). NMR experiments show that swapping the charge of these residues in the β3/talin1 pair (D/R in β3 and K/D in talin1) abrogates the membrane–proximal interaction (Figure 5A–C) and decreases the affinity of the overall interaction (Supplementary Table II). A more conservative D/A mutation in β3 yields the same result, indicating that this is primarily an electrostatic interaction (Supplementary Figure 7A). The effect of these mutations is virtually identical to that of a FF727/730AA mutation in the β3 membrane–proximal helix, which also abrogates integrin activation (Wegener et al, 2007) (Supplementary Figure S7A). A similar effect was observed with the β1A/talin1 pair (Supplementary Figures S7B and S8).
Figure 5.

A key salt bridge between talin and the β integrin tail. (A) Weighted shift maps of perturbations observed in 1H–15N HSQC spectra of the β3 tail on the addition of talin1 F3. Experiments were performed on β3 wt with talin1 wt, β3 D723R with talin1 wt, and β3 wt with talin1 K324D. Grey bars correspond to residues that could not be tracked because of exchange broadening. (B) Chemical shift perturbations in β3 on binding to talin1 F3 wt domain mapped onto the β1D/talin2 structure (largest shifts in blue, smallest in red). (C) As in (B) but with β3 D723R. (D) As in Figure 3A, but exploring the effect of talin1 F0–F1–F2–F3 wt or K324D on activation of αIIbβ3 expressed in CHO cells.
This same β3 residue, D723, has been shown earlier, by integrin activation assays (Hughes et al, 1996), α/β TM association studies (Kim et al, 2009), and NMR (Lau et al, 2009), to stabilize the integrin inactive state by interacting with R995 in αIIb. Talin1 K324 would thus compete with αIIb R995 for an electrostatic interaction with β3 D723, thereby weakening any αIIb R995–β3 D723 interaction (Figure 6A). This hypothesis was tested in αIIbβ3-expressing CHO cells. The addition of wt talin1 F0–F1–F2–F3 markedly increased integrin activation, but this increase was fully abrogated when talin1 K324D was introduced instead (Figure 5D). β3 D723 thus constrains bidirectional integrin signalling through an interaction with αIIb R995 in the absence of talin, but also participates in the activation process through an interaction with talin1 K324.
Figure 6.

Disruption of the α/β integrin dimer by talin. (A) Overlay of the talin2/β1D structure (plus β3 TM) with the αIIbβ3 TM structure (PDB ) (Lau et al, 2009). Talin is shown in yellow, αIIb in blue, β3/β1D bound to talin in red, and β3/β1D bound to αIIb in magenta. Inset shows inner membrane clasp competition. (B) The talin2/β1D structure (plus β3 TM) has been reoriented by 20° so that maximal contact is achieved between the membrane and the talin F2 membrane orientation patch (MOP). Membrane-targeting residues in F2 are highlighted in blue, and talin2 K325 (talin1 K322) in the F3 domain is highlighted in green. (C) The structure in (B) shown in an orthogonal ‘back' view. The inactive αIIbβ3 transmembrane domain complex has been added to illustrate the change in β tilt angle on activation. The β3 TM structure has been extended into the cytoplasm by combining it with the β1D tail structure. Talin2 K327 (talin1 K324) is highlighted in cyan. (D) The same view as (C), but with only the two β integrin tails shown to highlight the 20° change in tilt angle.
Discussion
By exploring a wide range of talin/integrin interactions, we identified a talin/integrin pair (talin2/β1D) that binds much more tightly than any previously studied pair and was more amenable to crystallization. The resulting crystal structure hinted at novel interactions between the talin F2 domain and the cell membrane and between the talin F3 domain and a membrane–proximal aspartate residue in the β tail. We subsequently validated these interactions by additional biophysical methods and showed their biological relevance through in-cell experiments. These findings can now be incorporated into a new structural model of integrin activation by talin.
A recent NMR structure revealed that the αIIbβ3 integrin TM domains form a dimer of unique structure stabilized by two interactions: an outer membrane embrace that involves glycine-mediated packing of the two TM helices, and an inner membrane interface that includes the D723/R995 salt bridge and a conserved GFF motif in the α subunit (Lau et al, 2009). A structure generated by disulfide-based distance restraints and molecular modelling revealed a similar arrangement (Zhu et al, 2009). An overlay of the talin2/β1D structure with the αIIbβ3 NMR structure reveals steric clashes between αIIb and talin2 located around the integrin inner membrane clasp (Figure 6A). Thus, the replacement of the interaction between β3 D723 (or β1 D759) and the α subunit with a β3 D723/talin interaction seems to be a key event in integrin activation. In addition, analysis of the talin2/β1D structure and mutation of the MOP residues showed the importance of this positively charge patch on F2. Closer inspection reveals that contact between the phospholipid headgroups of the membrane and the talin F2 MOP is better achieved if the tilt of the β TM domain changes by about 20° in a plane perpendicular to the plane of the membrane and to that of the initial β tilt (Figure 6B–D). The α subunit would be expected to remain in a vertical orientation, as it is constrained by tryptophan residues at each membrane interface (Yau et al, 1998; Lau et al, 2008a). An induced change in the β TM orientation would, however, disrupt the precise packing of the β subunit against the α TM domain. This 20° reorientation also brings K325 in the talin2 F3 domain (K322 in talin1) adjacent to the membrane, implying that the F2 MOP is a component of a larger membrane-interacting-charged surface, spanning multiple domains of talin. This is consistent with a previous report showing that mutation of this residue in talin1 disrupts integrin activation (Wegener et al, 2007); it could also explain why talin1 F2–F3 4E exhibits residual binding to membrane lipids (Figure 4).
Our results are therefore consistent with a model of integrin activation in which membrane-based reorientation, together with the weakened electrostatic interaction at the α/β cytoplasmic face, results in tail separation (Figure 7; Supplementary Movies S1 and S2). Indeed, this model explains why disruption of the αIIb R995–β3 D723 interaction is insufficient to activate integrins in the absence of talin binding (Tadokoro et al, 2003; Wegener et al, 2007) and is compatible with the observation that mutation of αIIb R995 or β3 D723 weakens, but does not eliminate, α/β TM association (Kim et al, 2009; Lau et al, 2009). The α/β TM domains interact only weakly (Kim et al, 2009; Lau et al, 2009), and the ability of talin to induce α/β TM separation was recently shown directly (Kim et al, 2009), thus adding plausibility to the mechanism of activation reported here.
Figure 7.

Model of integrin activation by talin, shown in three orientations. When talin binds to the β integrin tail it forms an extensive interface with the tail, including a membrane–proximal salt bridge, disrupting the salt bridge between the α and β subunits. To maximize contacts between the membrane and the positively charged membrane orientation patch (MOP) on the talin F2 domain, the β integrin must be reoriented with respect to the membrane by approximately 20°. Through these actions talin causes α/β separation, inducing the active state in the extracellular region.
The fact that the relevant talin and integrin residues are highly conserved implies that this mechanism of integrin activation is generalizable across different isoforms. The sequences of the membrane–proximal regions of the β1 and β3 tails are remarkably similar (Figure 1A), and previous studies have showed that the D759A mutant in β1 increases integrin affinity for fibronectin (Sakai et al, 1998; Millon-Fremillon et al, 2008). Interestingly, mice with this knockin mutation in β1 do not display a pronounced phenotype (Czuchra et al, 2006). This could relate to the observation that though β3 integrins have more distinct ‘on' and ‘off' states, the activation of β1 integrins is more dynamically regulated, and β1 integrins exist in a more default ‘on' state (Hynes, 2002; Hato et al, 2008). Thus, the combined effects of decreasing both α/β association and β/talin association by mutating D759 could compensate for one another in the case of β1 integrins. In contrast, the result of mutating D723 in β3 would be dominated by the effect of breaking its interaction with the α subunit. Even in β3 integrins, though, the effect of decreased talin interaction may partially counteract the effect of weakened α/β association, as αIIb R995D has a greater activating effect than β3 D723R (Hughes et al, 1996). Regardless, experiments with a chimaeric integrin containing the α5β1A cytoplasmic domains did reveal an essential role for the talin MOP in β1 integrin activation (Figure 3C). Compared with activation of αIIbβ3, talin caused a smaller increase in activation of the chimaeric α5β1A integrin—consistent with experiments reported on native α5β1A (Bouaouina et al, 2008)—but the effect of mutating MOP residues was the same as in αIIbβ3, largely abrogating talin-induced integrin activation.
In summary, this high-resolution structure of talin in complex with a full-length authentic integrin β cytoplasmic domain reveals that separation of the heterodimeric membrane-spanning helices is caused by the combined effects of talin-induced destabilization of the α/β inner membrane clasp and reorientation of the β TM domain. This model illustrates how localized membrane-specific protein interactions within the cell can lead to disruption of an interaction between TM helices in a large membrane-spanning receptor, effecting structural changes of great biological significance outside of the cell.
Materials and methods
Preparation and purification of proteins
Full-length β integrin tails were prepared as reported earlier for the β3 tail (Oxley et al, 2008). Specifically, using the In-Fusion cloning system (Clontech), β3 K716-T762, β1A K752-K798, and β1D K752-L801 were cloned into pET16b vectors modified with a 3C protease cleavage site between the N-terminal polyhistidine tag and the protein sequence. An additional construct of β1D K752-L801 was cloned into pET30b to produce the β1D integrin tail with a C-terminal polyhistidine tag (β1D-His6). 15N-labelled β integrin tails were expressed into inclusion bodies in Escherichia coli grown in M9 minimal media containing 15NH4Cl. Integrin tails were purified under denaturing conditions (50 mM sodium phosphate, 300 mM NaCl, 8 M urea, 0.035% β-mercaptoethanol, pH 7.0) by Talon immobilized metal affinity chromatography (Clontech), eluting the polyhistidine-tagged integrin tail in 200 mM imidazole. Further purification was performed by C4 reverse phase HPLC. For constructs produced in pET16b, the polyhistidine tag was removed by cleavage with 3C protease, and the integrin tails were further purified by HPLC. Mutations in the β integrin tails and other constructs were introduced using the QuikChange kit (Stratagene).
The talin1 F2 (K196-G309), talin1 F3 (G309-S405), and talin2 F3 (G311-S408) domains were expressed using the pGEX-6P-2 vector in a manner similar to that reported earlier (de Pereda et al, 2005). In this case, E. coli was grown in Luria broth (LB) and was lysed in 50 mM Tris, 200 mM NaCl, 0.035% 2-mercaptoethanol, 0.4% Triton-X, pH 7.0. After glutathione sepharose chromatography and 3C cleavage, the protein was further purified by gel filtration chromatography into pH 7.0 NMR buffer (50 mM sodium phosphate, 100 mM NaCl, 1 mM DTT). The talin1 F2–F3 domains (K196-S405) and talin2 F2–F3 domains (K198-S408) were cloned into pET151 using traditional methods. They were expressed in E. coli grown in LB and lysed as for the F3 domain constructs, although in a different buffer (50 mM sodium phosphate, 300 mM NaCl, 0.035% β-mercaptoethanol, pH 7.0). Fusion proteins were purified with Talon resin, polyhistidine tags were removed by cleavage with TEV protease, and the proteins were further purified by gel filtration chromatography.
NMR spectroscopy
All NMR experiments were performed on spectrometers equipped with Oxford Instruments superconducting magnets (500 and 600 MHz 1H operating frequencies) and GE/Omega computers. Unless otherwise indicated, samples were prepared in pH 6.1 NMR buffer with 5% D2O and Complete protease inhibitors (Roche). Experiments were performed at 25°C. The 1H and 15N resonances of 15N-labelled β integrin tails were assigned using a 1 mM sample in 20 mM sodium acetate pH 5.0 and using 3D NOESY-HSQC and 3D TOCSY-HSQC spectra. Resonance assignments were then transferred to pH 6.1 through pH titrations. Spectra were referenced in the direct dimension to DSS at 0 p.p.m., with indirect referencing in the 15N dimension using an 15N/1H frequency ratio of 0.101329118 (Wishart et al, 1995). Data were processed using NMRPipe (Delaglio et al, 1995) and spectra were visualized using the program SPARKY (www.cgl.ucsf.edu/home/sparky) or CCPN Analysis (Vranken et al, 2005). Resonance assignments for β1D were first performed on β1D-His6, because of higher expression levels, and assignments were transferred to untagged β1D.
1H–15N HSQC titrations were performed with 0.05 mM 15N-labelled integrin tail and increasing amounts of unlabelled talin, from 0 to 1 mM. Weighted combined 1H and 15N amide shifts (Δ(H,N)) were calculated using the equation 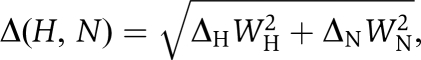 where WH and WN are weighting factors for the 1H and 15N amide shifts, respectively (WH=1, WN=0.154) (Ayed et al, 2001) and Δ=δbound−δfree. Dissociation constants were determined by fitting changes in backbone chemical shift with concentration to the following equation:
where WH and WN are weighting factors for the 1H and 15N amide shifts, respectively (WH=1, WN=0.154) (Ayed et al, 2001) and Δ=δbound−δfree. Dissociation constants were determined by fitting changes in backbone chemical shift with concentration to the following equation: 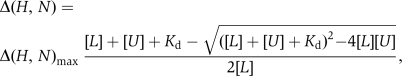 where Kd is the dissociation constant, Δ(H,N) is the weighted shift change, Δ(H,N)max is the shift change at saturation, and [L] and [U] are the concentrations of the labelled and unlabelled proteins, respectively. Data from peaks that were well resolved, had a significant change in position, and were discernable throughout the titration were fit simultaneously to this equation with the program OriginPro 8, extracting a single Kd and multiple Δ(H,N)max values.
where Kd is the dissociation constant, Δ(H,N) is the weighted shift change, Δ(H,N)max is the shift change at saturation, and [L] and [U] are the concentrations of the labelled and unlabelled proteins, respectively. Data from peaks that were well resolved, had a significant change in position, and were discernable throughout the titration were fit simultaneously to this equation with the program OriginPro 8, extracting a single Kd and multiple Δ(H,N)max values.
Crystallization
Samples for crystallization contained 6 mg/ml (250 μM) talin2 F2–F3 and 3 mg/ml (500 μM) β1D integrin tail in crystallization buffer (10 mM Tris, 100 mM NaCl, pH 7.0). Crystals were grown by the sitting drop method at 4°C in 0.1 M ammonium acetate, 0.02 M magnesium chloride, 0.05 M HEPES (pH 7.0), and 5% PEG 8k. For data collection, crystals were soaked in the same buffer plus 30% glycerol and then flash frozen in liquid nitrogen.
Data were collected at the ESRF on beamline ID23.EH2 at a wavelength of 0.8726 Å. The crystal diffracted to 2.17 Å resolution. Data were indexed and integrated using MOSFLM, and scaled and merged using SCALA from the CCP4 program suite (Collaborative Computational Project Number 4, 1994). The structure was phased by molecular replacement using the talin1 F2–F3 domains from PDB entries , , and (Garcia-Alvarez et al, 2003) as search models and using the program Phaser (Read, 2001). The crystal indexed to the space group P212121 and contained two integrin/talin dimers in the asymmetric unit. Model building was performed in Coot (Emsley and Cowtan, 2004), and refinement in Refmac (Winn et al, 2003) and PHENIX Refine (Adams et al, 2002). Noncrystallographic symmetry restraints were included only in the initial stages of refinement. The integrin tail was not included in the original molecular replacement model, but it could be built into electron density early in the refinement process. The structure refined to Rwork=21.30% and Rfree=24.85%. In a Ramachandran plot, 91.2% of residues lie in favoured regions, 8.6% in allowed regions, 0.2% in generously allowed regions, and 0.0% in disallowed regions.
Integrin activation assays
PAC1 binding was measured by two-colour flow cytometry as described earlier (Han et al, 2006). In brief, A5 cells (CHO cells expressing integrin αIIbβ3) were transfected with N-terminal GFP fused talin1 F3 or F1–F2–F3 constructs, or co-transfected with GFP and Talin F0–F1–F2–F3 constructs. Experiments were similarly performed on CHO cells expressing a chimaeric integrin consisting of the extracellular and TM domains of αIIbβ3 and the intracellular domains of α5β1A, which has been described earlier (O'Toole et al, 1994). Twenty-four hours after talin transfection, cells were collected, incubated with activation-specific anti-αIIbβ3 antibody PAC1 (Shattil et al, 1985), and then stained by R-phycoerythrin-conjugated anti-IgM antibody. Five minutes before analysis, propidium iodide (PI) was added, and PAC1 binding was measured with FACSCalibur (BD Bioscience). Only GFP-positive and PI-negative cells (live cells) were analysed to calculate the level of integrin activation. The ability of talin constructs containing various mutations to activate integrins was presented as percent of maximal integrin activation, and was calculated as (Fo−Fr)/(Fmax−Fr), where Fo is the mean fluorescence intensity (MFI) of PAC1 binding of various mutant-transfected cells, Fr is the MFI of PAC1 binding in the presence of competitive inhibitor eptifibatide (Scarborough et al, 1993), and Fmax is the MFI of PAC1 binding of wt F1–F2–F3 or F0–F1–F2–F3-transfected cells.
Phospholipid cosedimentation assays
Large multilamellar vesicles were prepared essentially as described earlier (Niggli et al, 1994). Briefly, films of dried phospholipids (Sigma) were swollen at 5 mg/ml in 20 mM Hepes (pH 7.4), 0.2 mM EGTA for 3 h at 42°C. The vesicles were then centrifuged (20 000 g for 20 min at 4°C), and the pellet was resuspended in the same buffer at 5 mg/ml. Protein samples were diluted into 20 mM Tris/HCl (pH 7.4), 0.1 mM EDTA, 15 mM β-mercaptoethanol. After centrifugation (20 000 g for 20 min at 4°C) proteins (0.15 mg/ml) were incubated (30 min, 25°C) in the absence or presence of phospholipid vesicles (0.5 mg/ml), 200 μl total volume, followed by centrifugation (25 000 g for 20 min at 4°C). Pellet and supernatant fractions were subjected to SDS–PAGE and proteins detected by Coomassie-blue staining. The percentage of protein bound (protein in pellet/total protein) was calculated by measuring band density in ImageJ (Abramoff et al, 1994).
Accession codes
Atomic coordinates for the talin2/β1D complex have been deposited in the Protein Data Bank under accession number . Chemical shift resonance assignments have been deposited in the Biological Magnetic Resonance Bank (BMRB) under the following accession numbers: 16159 (β1A), 16158 (β1D), and 16162 (β1D-His6). Assignments for β3 have been reported earlier (Oxley et al, 2008) and deposited in the BMRB under accession number 15552.
Supplementary Material
Supplementary Movie 1
Supplementary Movie 2
Supplementary Data
Review Process File
Acknowledgments
We thank Tobias Ulmer for providing the coordinates of the αIIbβ3 transmembrane structure. This work was supported by funding from the Wellcome Trust (IDC and DRC), the NIH (grants HL078784 and AR27214 to MHG), Cancer Research UK (DRC), the Rhodes Trust (NJA), the NIH Cell Migration Consortium (IDC, MHG, and KLW), the Marie Curie Fellowship program (IV), and Trinity College Oxford (IV).
Footnotes
The authors declare that they have no conflict of interest.
References
- Abramoff MD, Magelhaes PJ, Ram SJ (1994) Image processing with ImageJ. Biophoton Int 11: 36–42 [Google Scholar]
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58(Part 11): 1948–1954 [DOI] [PubMed] [Google Scholar]
- Arnaout MA, Goodman SL, Xiong JP (2007) Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol 19: 495–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askari JA, Buckley PA, Mould AP, Humphries MJ (2009) Linking integrin conformation to function. J Cell Sci 122 (Part 2): 165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayed A, Mulder FA, Yi GS, Lu Y, Kay LE, Arrowsmith CH (2001) Latent and active p53 are identical in conformation. Nat Struct Biol 8: 756–760 [DOI] [PubMed] [Google Scholar]
- Barlow DJ, Thornton JM (1983) Ion-pairs in proteins. J Mol Biol 168: 867–885 [DOI] [PubMed] [Google Scholar]
- Barsukov IL, Prescot A, Bate N, Patel B, Floyd DN, Bhanji N, Bagshaw CR, Letinic K, Di Paolo G, De Camilli P, Roberts GC, Critchley DR (2003) Phosphatidylinositol phosphate kinase type 1gamma and beta1-integrin cytoplasmic domain bind to the same region in the talin FERM domain. J Biol Chem 278: 31202–31209 [DOI] [PubMed] [Google Scholar]
- Belkin AM, Zhidkova NI, Balzac F, Altruda F, Tomatis D, Maier A, Tarone G, Koteliansky VE, Burridge K (1996) Beta 1D integrin displaces the beta 1A isoform in striated muscles: localization at junctional structures and signaling potential in nonmuscle cells. J Cell Biol 132: 211–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaouina M, Lad Y, Calderwood DA (2008) The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate beta1 and beta3 integrins. J Biol Chem 283: 6118–6125 [DOI] [PubMed] [Google Scholar]
- Calderwood DA (2004) Integrin activation. J Cell Sci 117 (Part 5): 657–666 [DOI] [PubMed] [Google Scholar]
- Calderwood DA, Fujioka Y, de Pereda JM, Garcia-Alvarez B, Nakamoto T, Margolis B, McGlade CJ, Liddington RC, Ginsberg MH (2003) Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: a structural prototype for diversity in integrin signaling. Proc Natl Acad Sci USA 100: 2272–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood DA, Yan B, de Pereda JM, Alvarez BG, Fujioka Y, Liddington RC, Ginsberg MH (2002) The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem 277: 21749–21758 [DOI] [PubMed] [Google Scholar]
- Campbell ID, Ginsberg MH (2004) The talin-tail interaction places integrin activation on FERM ground. Trends Biochem Sci 29: 429–435 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50(Part 5): 760–763 [DOI] [PubMed] [Google Scholar]
- Conti FJ, Felder A, Monkley S, Schwander M, Wood MR, Lieber R, Critchley D, Muller U (2008) Progressive myopathy and defects in the maintenance of myotendinous junctions in mice that lack talin 1 in skeletal muscle. Development 135: 2043–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley DR, Gingras AR (2008) Talin at a glance. J Cell Sci 121 (Part 9): 1345–1347 [DOI] [PubMed] [Google Scholar]
- Czuchra A, Meyer H, Legate KR, Brakebusch C, Fassler R (2006) Genetic analysis of beta1 integrin ‘activation motifs' in mice. J Cell Biol 174: 889–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pereda JM, Wegener KL, Santelli E, Bate N, Ginsberg MH, Critchley DR, Campbell ID, Liddington RC (2005) Structural basis for phosphatidylinositol phosphate kinase type Igamma binding to talin at focal adhesions. J Biol Chem 280: 8381–8386 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60(Part 12 Part 1): 2126–2132 [DOI] [PubMed] [Google Scholar]
- Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, Liddington RC (2003) Structural determinants of integrin recognition by talin. Mol Cell 11: 49–58 [DOI] [PubMed] [Google Scholar]
- Ginsberg MH, Partridge A, Shattil SJ (2005) Integrin regulation. Curr Opin Cell Biol 17: 509–516 [DOI] [PubMed] [Google Scholar]
- Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, Puzon-McLaughlin W, Lafuente EM, Boussiotis VA, Shattil SJ, Ginsberg MH (2006) Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr Biol 16: 1796–1806 [DOI] [PubMed] [Google Scholar]
- Hato T, Yamanouchi J, Tamura T, Yakushijin Y, Sakai I, Yasukawa M (2008) Cooperative role of the membrane-proximal and -distal residues of the integrin beta3 cytoplasmic domain in regulation of talin-mediated alpha IIb beta3 activation. J Biol Chem 283: 5662–5668 [DOI] [PubMed] [Google Scholar]
- Hughes PE, Diaz-Gonzalez F, Leong L, Wu C, McDonald JA, Shattil SJ, Ginsberg MH (1996) Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J Biol Chem 271: 6571–6574 [DOI] [PubMed] [Google Scholar]
- Hughes PE, O'Toole TE, Ylanne J, Shattil SJ, Ginsberg MH (1995) The conserved membrane-proximal region of an integrin cytoplasmic domain specifies ligand binding affinity. J Biol Chem 270: 12411–12417 [DOI] [PubMed] [Google Scholar]
- Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687 [DOI] [PubMed] [Google Scholar]
- Kim C, Lau TL, Ulmer TS, Ginsberg MH (2009) Interactions of platelet integrin alphaIIb and beta3 transmembrane domains in mammalian cell membranes and their role in integrin activation. Blood 113: 4747–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Carman CV, Springer TA (2003) Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301: 1720–1725 [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14: 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Kumar S, Nussinov R (1999) Salt bridge stability in monomeric proteins. J Mol Biol 293: 1241–1255 [DOI] [PubMed] [Google Scholar]
- Lau TL, Dua V, Ulmer TS (2008a) Structure of the integrin alphaIIb transmembrane segment. J Biol Chem 283: 16162–16168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau TL, Kim C, Ginsberg MH, Ulmer TS (2009) The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J 28: 1351–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau TL, Partridge AW, Ginsberg MH, Ulmer TS (2008b) Structure of the integrin beta3 transmembrane segment in phospholipid bicelles and detergent micelles. Biochemistry 47: 4008–4016 [DOI] [PubMed] [Google Scholar]
- Lu C, Takagi J, Springer TA (2001) Association of the membrane proximal regions of the alpha and beta subunit cytoplasmic domains constrains an integrin in the inactive state. J Biol Chem 276: 14642–14648 [DOI] [PubMed] [Google Scholar]
- Luo BH, Carman CV, Springer TA (2007) Structural basis of integrin regulation and signaling. Annu Rev Immunol 25: 619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millon-Fremillon A, Bouvard D, Grichine A, Manet-Dupe S, Block MR, Albiges-Rizo C (2008) Cell adaptive response to extracellular matrix density is controlled by ICAP-1-dependent beta1-integrin affinity. J Cell Biol 180: 427–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieswandt B, Moser M, Pleines I, Varga-Szabo D, Monkley S, Critchley D, Fassler R (2007) Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J Exp Med 204: 3113–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggli V, Kaufmann S, Goldmann WH, Weber T, Isenberg G (1994) Identification of functional domains in the cytoskeletal protein talin. Eur J Biochem 224: 951–957 [DOI] [PubMed] [Google Scholar]
- O'Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH (1994) Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol 124: 1047–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxley CL, Anthis NJ, Lowe ED, Vakonakis I, Campbell ID, Wegener KL (2008) An integrin phosphorylation switch: the effect of beta3 integrin tail phosphorylation on Dok1 and talin binding. J Biol Chem 283: 5420–5426 [DOI] [PubMed] [Google Scholar]
- Partridge AW, Liu S, Kim S, Bowie JU, Ginsberg MH (2005) Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J Biol Chem 280: 7294–7300 [DOI] [PubMed] [Google Scholar]
- Petrich BG, Fogelstrand P, Partridge AW, Yousefi N, Ablooglu AJ, Shattil SJ, Ginsberg MH (2007a) The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J Clin Invest 117: 2250–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrich BG, Marchese P, Ruggeri ZM, Spiess S, Weichert RA, Ye F, Tiedt R, Skoda RC, Monkley SJ, Critchley DR, Ginsberg MH (2007b) Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J Exp Med 204: 3103–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RJ (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr 57(Part 10): 1373–1382 [DOI] [PubMed] [Google Scholar]
- Sakai T, Zhang Q, Fassler R, Mosher DF (1998) Modulation of beta1A integrin functions by tyrosine residues in the beta1 cytoplasmic domain. J Cell Biol 141: 527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarborough RM, Naughton MA, Teng W, Rose JW, Phillips DR, Nannizzi L, Arfsten A, Campbell AM, Charo IF (1993) Design of potent and specific integrin antagonists. Peptide antagonists with high specificity for glycoprotein IIb-IIIa. J Biol Chem 268: 1066–1073 [PubMed] [Google Scholar]
- Senetar MA, Moncman CL, McCann RO (2007) Talin2 is induced during striated muscle differentiation and is targeted to stable adhesion complexes in mature muscle. Cell Motil Cytoskeleton 64: 157–173 [DOI] [PubMed] [Google Scholar]
- Shattil SJ, Hoxie JA, Cunningham M, Brass LF (1985) Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J Biol Chem 260: 11107–11114 [PubMed] [Google Scholar]
- Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA (2003) Talin binding to integrin beta tails: a final common step in integrin activation. Science 302: 103–106 [DOI] [PubMed] [Google Scholar]
- Takagi J, Erickson HP, Springer TA (2001) C-terminal opening mimics ‘inside-out' activation of integrin alpha5beta1. Nat Struct Biol 8: 412–416 [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Calderwood DA, Ginsberg MH, Campbell ID (2003) Domain-specific interactions of talin with the membrane-proximal region of the integrin beta3 subunit. Biochemistry 42: 8307–8312 [DOI] [PubMed] [Google Scholar]
- Vinogradova O, Velyvis A, Velyviene A, Hu B, Haas T, Plow E, Qin J (2002) A structural mechanism of integrin alpha(IIb)beta(3) ‘inside-out' activation as regulated by its cytoplasmic face. Cell 110: 587–597 [DOI] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59: 687–696 [DOI] [PubMed] [Google Scholar]
- Wegener KL, Campbell ID (2008) Transmembrane and cytoplasmic domains in integrin activation and protein-protein interactions (review). Mol Membr Biol 25: 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID (2007) Structural basis of integrin activation by talin. Cell 128: 171–182 [DOI] [PubMed] [Google Scholar]
- Winn MD, Murshudov GN, Papiz MZ (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol 374: 300–321 [DOI] [PubMed] [Google Scholar]
- Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 6: 135–140 [DOI] [PubMed] [Google Scholar]
- Yau WM, Wimley WC, Gawrisch K, White SH (1998) The preference of tryptophan for membrane interfaces. Biochemistry 37: 14713–14718 [DOI] [PubMed] [Google Scholar]
- Zhu J, Luo BH, Barth P, Schonbrun J, Baker D, Springer TA (2009) The structure of a receptor with two associating transmembrane domains on the cell surface: integrin alphaIIbbeta3. Mol Cell 34: 234–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1
Supplementary Movie 2
Supplementary Data
Review Process File