

Abstract
Apoptosis rather than differentiation is a physiological process during myogenesis and muscle regeneration. When cultured myoblasts were induced to differentiate, we detected an increase in caspase 8 activity. Pharmacological inhibition of caspase 8 activity decreased apoptosis. Expression of a dominant-negative mutant of the adapter protein FADD also abrogated apoptosis, implicating a death ligand pathway. Treatment with TRAIL, but not Fas, induced apoptosis in these myoblasts. Accordingly, treatment with a soluble TRAIL decoy receptor or expression of a dominant-negative mutant of the TRAIL receptor DR5 abrogated apoptosis. While TRAIL expression levels remained unaltered in apoptotic myoblasts, DR5 expression levels increased. Finally, we also detected a reduction in FLIP, a death-receptor effector protein and caspase 8 competitive inhibitor, to undetectable levels in apoptotic myoblasts. Thus, our data demonstrate an important role for the TRAIL/DR5/FADD/caspase 8 pathway in the apoptosis associated with skeletal myoblast differentiation. Identifying the functional apoptotic pathways in skeletal myoblasts may prove useful in minimizing the myoblast apoptosis that contributes pathologically to a variety of diseases and in minimizing the apoptosis of transplanted myoblasts to treat these and other disease states.
Keywords: Myoblast, Apoptosis, FADD, DR5, FLIP
Introduction
Apoptosis is a common physiological process that occurs in all tissues and is critical to proper development and homeostasis [1]. In fact, apoptosis and differentiation are often coordinately regulated with apoptosis serving the critical function of removing excess or physiologically ineffective cells [1–3]. Skeletal myoblast apoptosis occurs during myogenesis [2] and muscle regeneration [3] and has been carefully documented in cultures of primary myoblasts [4, 5], in established myoblast cell lines [6, 7], and in an established muscle satellite cell line [8]. While the coordinate regulation of differentiation and apoptosis is clearly important under normal physiological conditions, it is likely detrimental to the use of myoblast transfer as a treatment for a variety of diseases [9, 10]. Further, inappropriate myoblast apoptosis contributes pathologically to a variety of diseases [11–13]. Thus, an understanding of the apoptotic process within skeletal myoblasts may identify targets for therapeutic manipulation relevant to disease states with associated muscle degeneration and to the use of myoblast transfer as a therapeutic approach. Further, such information may prove relevant to the use of apoptotic modifiers to treat a variety of other pathological conditions [14–18].
While analysis of the apoptotic process that occurs during the differentiation of skeletal myoblasts is only beginning [7, 19, 20], the apoptotic process in other systems has been extensively studied. Mitochondrial permeabilization, resulting in the release of several pro-apoptotic proteins as well as the ensuing dissipation of the mitochondrial membrane potential, is a critical component of the apoptotic process in most systems. The molecules directly responsible for this mitochondrial assault are the pro-apoptotic members of the Bcl2 family [21–23]. The signaling pathways through which particular pro-apoptotic Bcl2 family members are engaged are broadly categorized as either “intrinsic” or “extrinsic”. The “intrinsic” pathway is initiated through altered activity of either kinases or transcription factors while the “extrinsic” pathway is initiated by death ligand signaling [24]. Several of the proteins released from the mitochondria play either a direct or indirect role in caspase activation [22, 23]. Caspases are cysteine-aspartic acid specific proteases that play a crucial role during apoptosis [25]. These are categorized as either “initiator” caspases or “executioner” caspases. Activation of initiator caspase 9 is a result of the release of cytochrome C from the mitochondria while activation of initiator caspase 8 is a direct result of death ligand signaling [24, 26].
Herein, we report a role for caspase 8 activation during the apoptosis associated with skeletal myoblast differentiation. This finding prompted an investigation into the role of death ligand signaling and led us to discover that signaling by the death ligand TRAIL, through the TRAIL receptor DR5 and the adapter protein FADD, also plays a role in this apoptotic process. Further, we report that this death ligand pathway is engaged through increased expression of the TRAIL receptor DR5 and decreased expression of FLIP, a caspase 8 antagonist.
Materials and methods
Cells and cell culture
The growth and differentiation properties of 23A2 myoblasts have been reported previously [7, 19]. The 23A2 derivatives expressing dominant negative FADD (23A2:dnFADD myoblasts) and the 23A2 and C2C12 derivatives expressing dominant negative DR5 (23A2:dnDR5 myoblasts and C2C12:dnDR5 myoblasts) were generated by transfecting myoblasts with 300 ng of the pcDNA3:AU1:dnFADD construct or 300 ng of the pcDNA3:AU1:dnDR5 construct using Lipofectamine Plus (GibcoBRL) as specified by the manufacturer. Briefly, cells were plated at 105 in 100 mm dishes and transfected the next day with 300 ng of DNA. After 48 h, the culture was passaged and placed in selection media containing G418 (750 µg/ml). After 14 days of drug selection, individual G418 resistant colonies were isolated and propagated for further analysis. All cells were maintained on gelatin coated plates in growth medium (GM), which consists of basal modified Eagle's medium (BME), 10% fetal bovine serum (FBS), 100 I.U./ml penicillin and 100 µg/ml streptomycin (1% P/S). Differentiation was induced by switching cells from GM to differentiation medium (DM), which consists of BME, 1% P/S and no FBS.
Western blot analysis
Cells were lysed as previously described. The protein concentrations of all lysates were determined using Coomassie Protein Assay Reagent from Pierce as per manufacturer's instructions. Following protein determination, lysates (200 µg for AU1 detection and 100 µg for Bid, FLAG, TRAIL, DR5 and FLIP detection) were denatured in sample buffer (final concentration in lysates of 2% SDS, 10% glycerol, 2% 2-mercaptoethanol, pH 6.8) and electrophoresed through denaturing polyacrylamide gels (15% for Bid and AU1 detection, 12% for FLIP detection, 10% for FLAG and TRAIL detection, and 8% for DR5 detection. Following SDS polyacrylamide gel electrophoresis (SDS-PAGE), samples were transferred electrophoretically for four amp hours to Hybond-P polyvinylidene difluoride membranes in transfer buffer containing 80% methanol and 1 g/L SDS. Membranes were blocked for one hour in 1 × TBS/0.1% NP40 with 10% newborn calf serum and 5% drymilk. The following primary antibodies were incubated with the appropriate membranes at a dilution of 1:500 overnight: anti-Bid antibody (Santa Cruz), anti-AU1 antibody (Covance), anti-FLAG (M5, Sigma), anti-TRAIL (H-257, Santa Cruz), anti-FLIP (Dave-2, Alexis) and anti-DR5 (TRAIL-R2, R&D Systems). The actin primary antibody (Sigma) was used at a dilution of 1:30,000 for one hour. Appropriate HRP-conjugated secondary antibodies, each diluted 1:1000, were incubated with the membranes for one hour. After each incubation with antibody and prior to the addition of chemiluminescent substrate, membranes were washed five times in 1 × TBS (tris-buffered saline pH 7.4) with 1% NP-40. Membranes were then incubated with SuperSignal West Pico Chemiluminescent Substrate (Pierce) for 60 s and bands were visualized using either Kodak Scientific Imaging Film or a Bio-Rad Fluor-S Multi-imager.
Cytosolic nucleosome ELISA
Myoblasts were plated at equal density and the next day switched to fresh GM or DM for eight hours. This is the optimal time point for this assay as DNA fragmentation ismaximal yet apoptotic myoblasts have not yet detached from the plate [7]. Cytosolic nucleosomes were measured using the Cell Death Detection ELISA Plus Kit (Roche Diagnostics) per manufacturer's instructions and as previously described [7] with the following change. Absorbance at 405 nm was measured at 60 s intervals using a spectrophotometer. Values were taken from the time point where all samples were within the linear range of the assay for each individual experiment and normalized to cell number from parallel plates.
Caspase 8 assay
Caspase 8 colorimetric assays were performed using the caspase 8 Colorimetric Assay Kit from R&D Systems per manufacturer's instructions and as previously described [7]. Experiments were performed within the linear range of the assay and absorbance was normalized to the protein concentration of each lysate.
Statistics
Statistical analysis was performed using KaleidaGraph data analysis per the manufacturer's instructions (version 3.6). Briefly, for analysis of the data in Fig. 1, a single sample t-test was used where the test value was the value assigned to that obtained in GM for (A) and to that obtained with the solvent control for (B). A paired t-test was use for the analysis of data in Figs. 2–6.
Fig. 1.
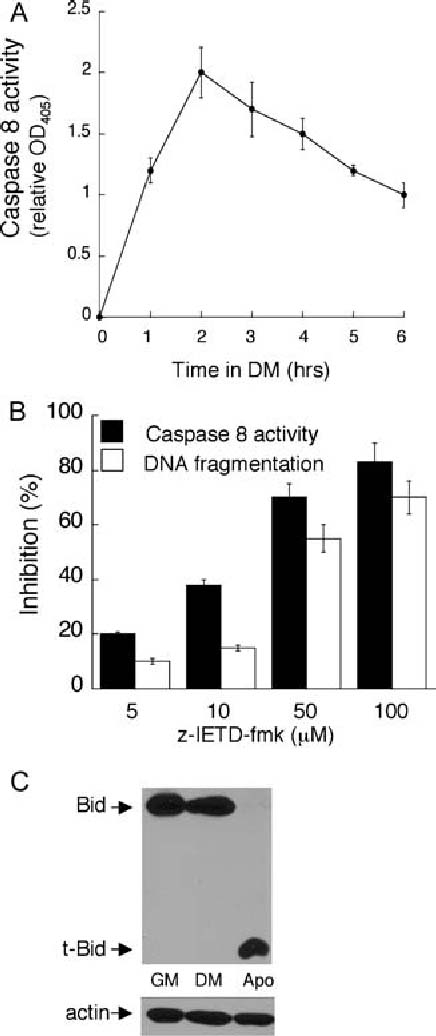
Caspase 8 activation is required for the apoptosis associated with skeletal myoblast differentiation. In (A) equal cell numbers were plated and the next day switched to DM in for the indicated time prior to the determination of caspase 8 activity. Error bars represent the standard deviation of the mean from three values from three independent experiments. Each value obtained for caspase 8 activity in DM is significantly different than that obtained in GM (P < 0.02). In (B) equal cell numbers were plated and the next day switched to DM in the presence of the indicated concentration of IETD-fmk or solvent for two hours prior to the assessment of caspase 8 activity or for eight hours prior to the assessment of DNA fragmentation. Caspase 8 activity was measured using the caspase 8 colorimetric kit (R&D Systems) and DNA fragmentation was measured using the Cell Death Detection ELISA Plus Kit (Roche Diagnostics) as described in Materials and Methods. In (B) per cent inhibition was calculated for each sample relative to values obtained from lysates of cells treated with solvent. Error bars represent the standard deviation of the mean from three values from three independent experiments. Statistical analysis was performed as described in “Materials and methods”. Each value obtained after treatment with z-IETD-fmk is significantly different from the corresponding value obtained after treatment with solvent (P < 0.003). In (C), equal cell numbers were plated and the next day switched to either fresh GM or DM for 18 h. Apoptotic myoblasts were pelleted from the DM and lysates were prepared from the apoptotic myoblasts, from the surviving myoblasts cultured in DM and from the myoblasts cultured in GM. Western analyses, with actin serving as a protein loading and transfer control, were performed as described in Materials and Methods. Shown are results from one experiment that are representative of 2 independent experiments
Fig. 2.
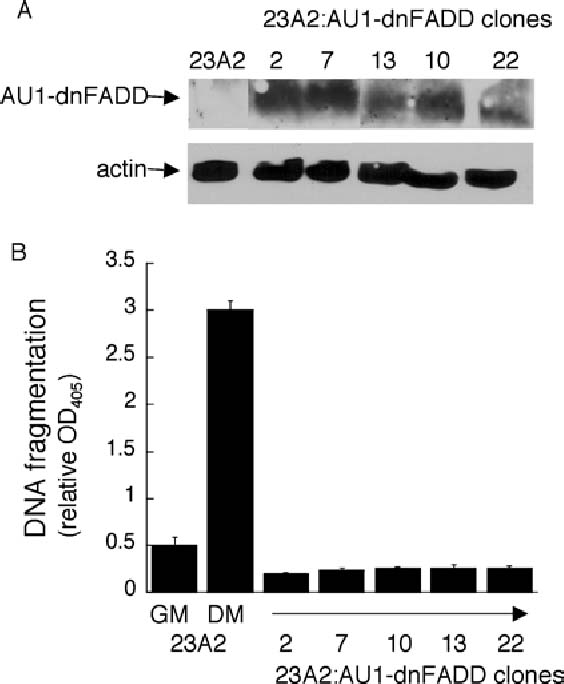
Expression of dominant negative FADD abrogates the apoptosis associated with skeletal myoblast differentiation. In (A) equal cell numbers were plated and the next day lysates were prepared. Western analyses, with actin serving as a protein loading and transfer control, were performed as described in Materials and Methods. In (B) equal cell numbers were plated and the next day switched to either fresh GM or DM for eight hours prior to the assessment of DNA fragmentation as described in Fig. 1. Error bars represent mean ± SEM from triplicate samples. Statistical analysis was performed as described in Materials and methods. The value obtained for 23A2 myoblasts cultured in DM is significantly different than the value obtained in GM (P < 0.002) and the values obtained for each clone cultured in DM are significantly different than the value obtained for 23A2 myoblasts cultured in DM (P < 0.003)
Fig. 6.
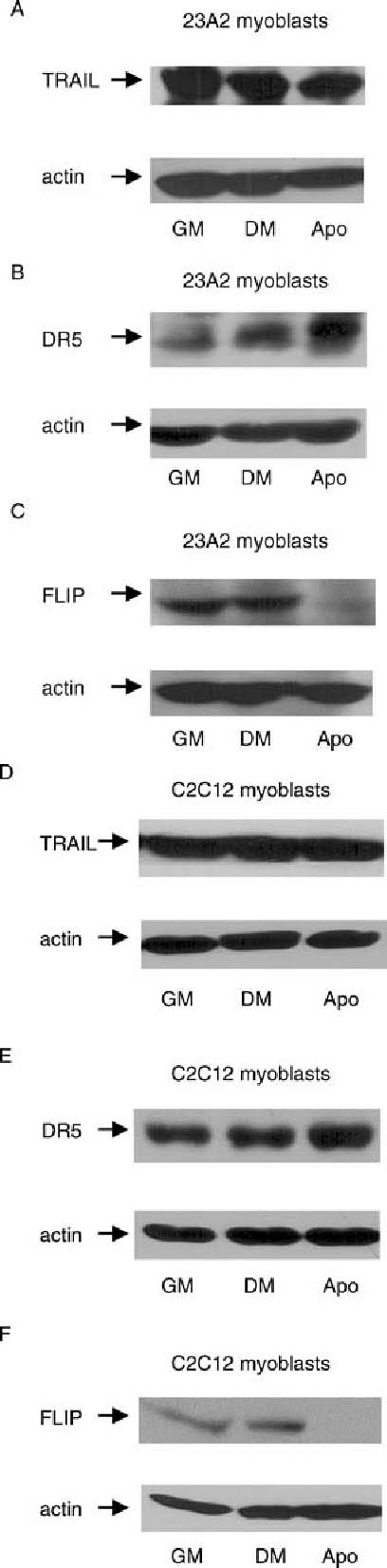
Apoptotic myoblasts possess increased DR5 levels and decreased FLIP levels. Equal cell numbers were plated and the next day switched to either fresh GM or DM for 18 h. Apoptotic myoblasts were pelleted from the DM and lysates were prepared from the apoptotic myoblasts, from the surviving myoblasts cultured in DM and from the myoblasts cultured in GM. Western analyses, with actin serving as a protein loading and transfer control, were performed as described in Materials and Methods. Shown are results from one experiment that are representative of at least 3 independent experiments for each
Results
Inhibition of caspase 8 and FADD signaling abrogates the apoptosis associated with skeletal myoblast differentiation We have previously reported a requirement for signaling by the executioner caspase 3 during the apoptosis associated with 23A2 myoblast differentiation [7, 19]. To determine if caspase 8 was an initiator caspase activated during this process, we measured the activation of caspase 8 when skeletal myoblasts were switched from growth media (GM) to differentiation media (DM). We determined that caspase 8 activity increases in a time-dependent manner with a 2-fold maximal activation after roughly two hours of culture in DM (Fig. 1(A)). To determine the significance of caspase 8 activation to the apoptosis of skeletal myoblasts cultured in DM, myoblasts were cultured in the presence of IETD-fmk, a selective pharmacological inhibitor of caspase 8. Cytosolic nucleosomes are the result of DNA fragmentation and nuclear envelope breakdown, hallmarks of the apoptotic process. We have carefully correlated cell density and time in DM with the level of cytosolic nucleosomes and the percentage of myoblasts undergoing apoptosis [7, 19]. Through this analysis we determined that, when myoblasts are plated at a density of 4 × 105 per 100 mm dish, the level cytosolic nucleosomes present after eight hours of culture in DM is representative of the 30% of myoblasts that finally undergo apoptosis [7, 19]. Thus, analysis of cytosolic nucleosomes is generally performed under these conditions. Treatment with IETD-fmk provided a dose-dependent inhibition of caspase 8 activity and a corresponding inhibition in the level of cytosolic nucleosomes (Fig. 1(B)).
Since activation of caspase 8 often results in the cleavage of Bid (a BH3-only pro-apoptotic member of the Bcl2 family) to truncated Bid (tBid) [27, 28], we also analyzed Bid protein status as a consequence of culture in DM. The apoptotic process associated with differentiation, however, typically removes only 30% of the myoblasts. This process is continual and finally complete within 12–18 h of culture in DM [6, 7]. Further, cell cycle position is implicated in this process [6, 29, 30] as induction of the cyclin-dependent kinase (CDK) inhibitor p21(CIP1) [31–34] correlates with myoblast survival. This protection conferred by p21(CIP1) requires the retinoblastoma protein [6, 29] suggesting that this protection is a consequence of the ensuing cell cycle exit. In an asynchronous population, therefore, only a fraction of those cells destined to under apoptosis might be expected to possess altered levels of Bid at any time point. As such, we had difficulty detecting reproducible changes in the expression level of Bid in a population consisting of both those myoblasts able to survive (roughly 70% presumably as a consequence of cell cycle exit) and those undergoing apoptosis. We, therefore, compared Bid protein status in lysates prepared from the apoptotic myoblasts that had detached from the plate after 18 h of culture in DM, from the corresponding surviving myoblasts cultured in DM and from myoblasts cultured in GM. We found that the level of full length Bid detected in myoblasts cultured in GM was similar to that observed in corresponding surviving myoblasts cultured in DM, while t-Bid was not detected in either (Fig. 1(C)). In contrast, only t-Bid was detectable in the apoptotic myoblasts (Fig. 1(C)).
A role for caspase 8 in the apoptosis associated with differentiation suggested that a death ligand pathway might be involved. All known death ligand receptors signal through the adapter protein FADD to induce the activation of caspase 8 [35]. Truncated FADD lacking the death effector domain (DED) responsible for interaction with the DED of caspase 8 functions as a dominant negative mutant [36]. This mutant protein has been used successfully to implicate a role for death ligand signaling [36, 37]. 23A2 myoblasts were transfected with pCDNA3-FADD-DN and G418 resistant clones were screened for the expression of AU1-tagged FADD-DN (Fig. 2(A)). The ability of each clone expressing AU1-tagged FADD-DN to undergo apoptosis as a consequence of culture in DM was then compared to control 23A2 myoblasts. DNA fragmentation is induced 3-fold when 23A2 myoblasts are switched from GM to DM [7, 38] (Fig. 2(B)). The DNA fragmentation detected in each of the FADD-DN clones, however, is below even that detected in control 23A2 myoblasts cultured in GM (Fig. 2(B)).
TRAIL receptor DR5 signaling plays a role in the apoptosis associated with skeletal myoblast differentiation
The discovery that expression of FADD-DN blocked the apoptosis associated with differentiation further implicated a death ligand pathway in this process. The first death ligand pathway we investigated was the Fas ligand (FasL)/Fas pathway. This pathway can be activated either by treatment with soluble FasL or by treatment with an activating Fas antibody [39]. Treatment with concentrations of the activating Fas monoclonal antibody Jo2 (BD-Pharmingen), sufficient to induce apoptosis in other cell lines [40], did not induce apoptosis in myoblasts cultured in GM or increase apoptosis in myoblasts cultured in DM (Fig. 3(A)). Treatment with soluble FasL, genetically modified to form active complexes (SUPERFas Ligand, Alexis) also did not induce or increase apoptosis in 23A2 myoblasts cultured in either GM or DM, respectively (Fig. 3(B)). Further, full length FasL presented as membranous vesicles (Upstate) did not affect apoptosis in myoblasts cultured in either GM or DM (data not shown). We next determined if the death ligand TRAIL could induce apoptosis in skeletal myoblasts. We found that treatment with recombinant human TRAIL could induce apoptosis in a dose-dependent (Fig. 4(A)) and time dependent (Fig. 4(B)) manner in myoblasts cultured in GM and could increase apoptosis in a dose-dependent (Fig. 4(A)) and time dependent (Fig. 4(B)) manner in myoblasts cultured in DM. To determine if endogenous TRAIL plays a role in the apoptotic process that occurs as a consequence of culture in DM, we utilized a soluble human TRAIL decoy receptor chimera (TRAIL:R4:Fc) which functions by binding endogenous TRAIL and preventing its association with the endogenous TRAIL receptor. In support of a role for endogenous TRAIL in this apoptotic process, the level of cytosolic nucleosomes in myoblasts cultured in DM supplemented with the TRAIL:R4:Fc decoy was inhibited by more than 50% when compared to the level of cytosolic nucleosomes in myoblasts cultured in DM (Fig. 4(C)).
Fig. 3.
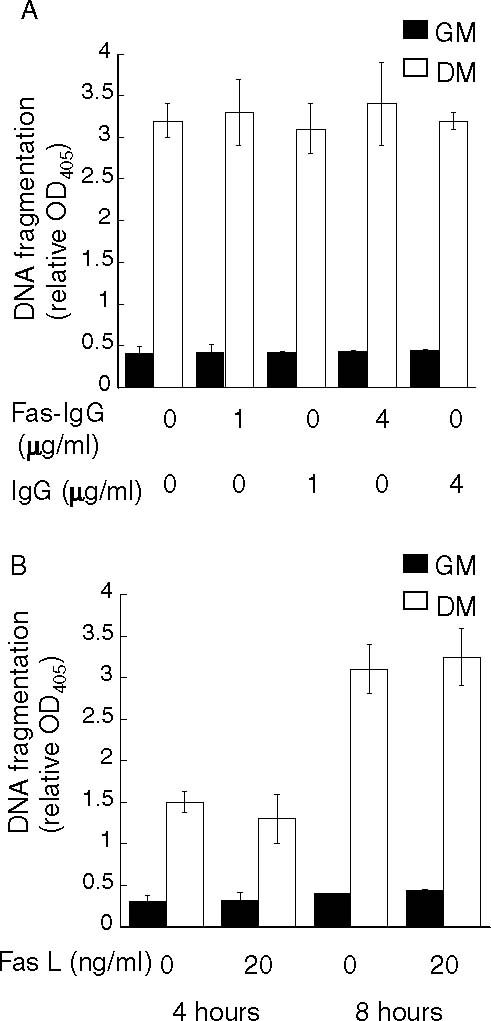
Treatment of 23A2 myoblasts with either an activating anti-Fas antibody or with FasL does not induce apoptosis. In (A) equal cell numbers were plated and the next day switched to either fresh GM or DM with the indicated concentration of activating anti-mouse Fas antibody (clone Jo2, form NA/LE; BD Pharmingen) permanufacturer's instructions or control IgG for eight hours prior to the assessment of DNA fragmentation as described in Fig. 1. In (B) equal cell numbers were plated and the next day switched to either fresh GM or DM with the indicated concentration of SUPERFas Ligand for either four or eight hours prior to the assessment of DNA fragmentation as described in Fig. 1. In each, error bars represent mean ± SEM from triplicate samples. Statistical analysis was performed as described in Materials and methods. Values obtained in the presence of Fas-IgG or FasL are not significantly different from values obtained in the presence of IgG control or solvent (P > 0.7 and P > 0.2, respectively)
Fig. 4.
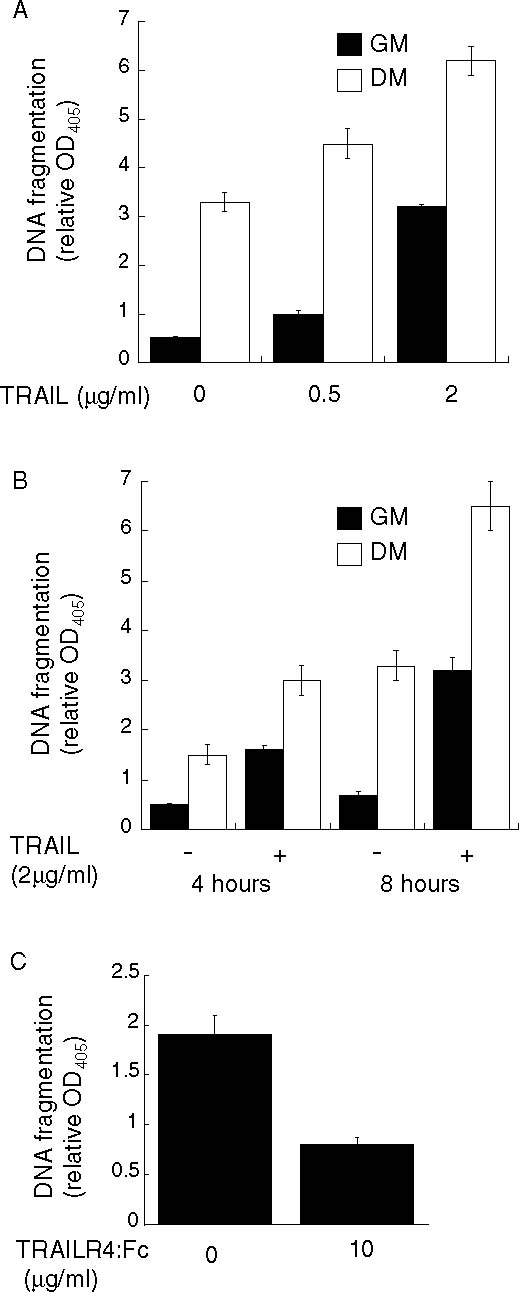
Treatment of 23A2 myoblasts with recombinant TRAIL induces apoptosis while a soluble TRAIL decoy receptor inhibits apoptosis. In (A) and (B), equal cell numbers were plated and the next day switched to either fresh GM or DM with the indicated concentration of recombinant TRAIL for eight hours in (A), or for the indicated time in (B), prior to the assessment of DNA fragmentation as described in Fig. 1. In (C), equal cell numbers were plated and the next day switched to DM with the indicated concentration of the soluble TRAIL decoy receptor, TRAILR4:Fc (R&D Systems) for four hours. In each, error bars represent mean ± SEM from triplicate samples. Statistical analysis was performed as described in Materials and methods. Each value obtained after treatment with TRAIL is significantly different from the corresponding value obtained after treatment with solvent (P < 0.03) and the value obtained in the presence of TRAILR4:Fc is significantly different from the value obtained after treatment with solvent (P < 0.05)
DR5 is the only TRAIL receptor in murine cells [41]. To further confirm that the TRAIL pathway was actually responsible for the apoptosis associated with differentiation, 23A2 myoblasts were transfected with pCDNA3-dnDR5 [42] and G418 resistant clones were screened for the expression of FLAG-tagged dnDR5 (Fig. 5(A)). The ability of each clone expressing FLAG-tagged dnDR5 to undergo apoptosis as a consequence of culture in DM was then compared to control 23A2 myoblasts. As before, DNA fragmentation is induced roughly 3-fold when 23A2 myoblasts are switched from GM to DM (Figs. 2(B) and 5(B)). The DNA fragmentation detected in each of the dnDR5 clones is even less than that routinely observed in myoblasts cultured in GM (Fig. 5(B)). Further, while treatment with recombinant TRAIL increased the appearance of cytosolic nucleosomes roughly two-fold (Figs. 4 and 5(B)) in control 23A2 myoblasts, treatment of the dnDR5 clones with recombinant TRAIL did not elevate the level of cytosolic nucleosomes (Fig. 5(B)). To determine if the TRAIL/DR5 pathway contributed to the apoptosis associated with differentiation in another established skeletal myoblast cell line, C2C12 myoblasts were transfected with pCDNA3-dnDR5 and G418 resistant clones were screened for the expression of FLAG-tagged dnDR5 (Fig. 5(C)). As observed in 23A2 myoblasts, culture in DM results in roughly a 3-fold increase in the appearance of cytosolic nucleosomes and this is further elevated when these myoblasts are treated with recombinant TRAIL. Further, expression of dnDR5 abrogates both the apoptosis associated with C2C12 differentiation and the enhanced apoptosis induced by treatment with recombinant TRAIL (Fig. 5(D)).
Fig. 5.
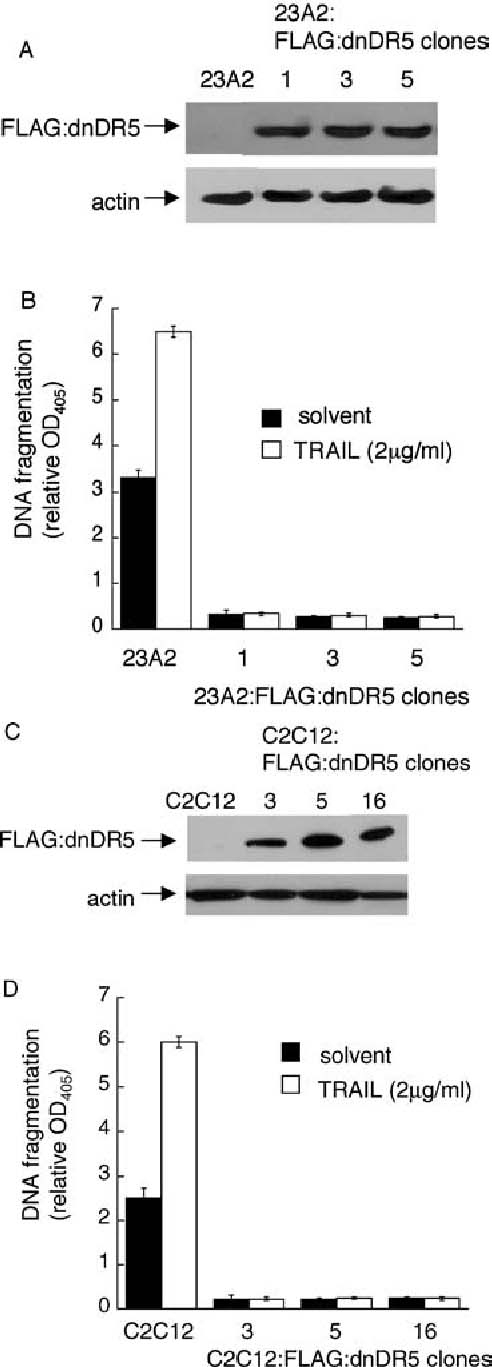
Expression of dominant negative DR5 abrogates the apoptosis associated with 23A2 and C2C12 skeletal myoblast differentiation. In (A) and (C) equal cell numbers were plated and the next day lysates were prepared. Western analyses, with actin serving as a protein loading and transfer control, were performed as described in Materials and Methods. In (B) and (D) equal cell numbers were plated and the next day switched to either fresh GM or DM, including either solvent or recombinant TRAIL, for eight hours prior to the assessment of DNA fragmentation as described in Fig. 1. Error bars represent mean ± SEM from triplicate samples. Statistical analysis was performed as described in Materials and methods. The values obtained for each clone cultured in DM are significantly different than the value obtained for the respective parental control cells cultured in DM (P < 0.0004 for 23A2 myoblasts and P < 0.0001 for C2C12 myoblasts). Further, the values obtained for each clone cultured in DM with TRAIL are significantly different than the value obtained for the respective parental control cells cultured in DM with TRAIL (P < 0.0006 for 23A2 myoblasts and P <. 0001 for C2C12 myoblasts)
Increased DR5 levels and decreased FLIP levels are detected in apoptotic myoblasts
We next sought to determine the mechanism through which the DR5/FADD pathway was engaged during the apoptotic process associated with skeletal myoblast differentiation. In general, signaling through death ligand pathways can be initiated either through increased expression of the death ligand or the death receptor or through decreased expression of a naturally occurring inhibitory protein such as FLIP (FADD-like IL-1-converting enzyme-inhibitory protein) [41, 43]. Similar to our analysis of Bid protein levels, we had difficulty detecting reproducible changes in the expression levels of these proteins in a population consisting of both those myoblasts able to survive and those undergoing apoptosis. We, therefore, compared the expression levels of these proteins in lysates prepared from the apoptotic myoblasts that had detached from the plate after 18 h of culture in DM, from the corresponding surviving myoblasts cultured in DM and from myoblasts cultured in GM. We determined that the expression level of DR5, but not TRAIL, was elevated in apoptotic myoblasts when compared to that observed in corresponding surviving myoblasts cultured in DM or to myoblasts cultured in GM (Fig. 6(A), (B), (D) and (E)). The increase in DR5 expression in apoptotic 23A2 myoblasts is greater than that in apoptotic C2C12 myoblasts when each is compared to their non-apoptotic counterparts (Fig. 6(B) and (E)). As a consequence of alternative splicing, two isoforms of FLIP, FLIPL and FLIPS, can be expressed in other systems [41, 43]. Although the antibody we used recognizes an epitope found in both FLIPL and FLIPS, FLIPS is not detected in our cells under any condition examined. We have determined, however, that FLIPL is expressed in surviving 23A2 and C2C12 myoblasts, but that both apoptotic 23A2 and C2C12 myoblasts display a reduction in FLIP expression to levels that are below detection (Fig. 6(C) and (F)).
Discussion
Little has been reported regarding the molecular initiation of apoptosis in skeletal myoblasts. We have reported a role for the executioner caspase 3 [7, 19], and others have suggested a role for caspase 12 and endoplasmic reticulum (ER) stress signaling through activation of the ATF6 transcription factor [20]. We, however, do not detect activation of ATF6 (personal observation). Furthermore, ER stress-mediated apoptosis in other systems involves calcium efflux initiated by the pro-apoptotic Bcl2 family members Bak and Bax. Calcium release mediates activation of the protease m-calpain that cleaves and activates caspase 12 and initates the caspase cascade [44].We have determined, however, that calpain activity does not play a role in the apoptotis associated with skeletal myoblast apoptosis (Shaltouki and Weyman, manuscript in preparation). Herein, we document a role for the initiator caspase 8 and the TRAIL/DR5/FADD pathway in the apoptosis associated with skeletal myoblast differentiation. Specifically, this apoptosis is inhibited by pharmacological abrogation of caspase 8 activity, by expression of a dominant negative mutant of the death receptor adapter protein FADD, or by expression of a dominant negative mutant of DR5.
We have further determined that, relative to both proliferating myoblasts cultured in growth media (GM) and surviving, differentiating myoblasts cultured in differentiation media (DM), those myoblasts induced to undergo apoptosis in DM possess an increased level of the TRAIL receptor DR5 and a decreased level of the death-receptor effector protein and caspase 8 competitive inhibitor FLIP. TRAIL expression is easily observed and similar levels are detected in proliferating myoblasts cultured in GM as well as those myoblasts either undergoing differentiation or apoptosis in DM. Since soluble TRAIL decoy receptor did inhibit apoptosis, and since skeletal myoblasts are exceptionally migratory (personal observation), we hypothesize that those myoblasts with increased levels of DR5 and decreased levels of FLIP as a consequence of culture in DM are sensitized to undergo apoptosis when constitutively expressed TRAIL ligand is encountered. Future experiments are focused on determining the mechanism through which DR5 levels are elevated in myoblasts undergoing differentiation-associated apoptosis. DR5 was originally identified as target of the p53 transcription factor and increased expression of DR5 sensitizes cells to TRAIL mediated apoptosis [45]. DR5 induction by p53 is cell type specific occurring in only spleen, small intestine and thymus [46]. Consistent with this, p53 plays a role in the differentiation of skeletal myoblasts but not in the apoptosis of skeletal myoblasts [47] (Freer/Weyman, manuscript in preparation). Thus, future experiments are focused on determining the mechanism through which DR5 levels are elevated in myoblasts undergoing differentiation-associated apoptosis.
Cells with high levels of FLIP are resistant to apoptosis while cells with low levels of FLIP are sensitive to apoptosis [41]. An important role for FLIP expression on the survival of differentiating skeletal myoblasts is suggested by the finding that skeletal muscle, along with heart, lymphoid tissue and kidney, posess high levels of FLIP protein [43]. Consistent with this, we have determined that FLIP levels in myoblasts undergoing apoptosis as a consequence of culture in DM are reduced to undetectable levels. FLIP expression levels are regulated by cell cycle position, by the transcription factor p53, and by the PI3K/Akt/FOXO31 pathway in other systems. Firstly, FLIP levels decrease during S-phase [48], but the molecular mechanism responsible for this decrease has not been reported. Since cell cycle proteins have been implicated in the regulation of the differentiation-associated apoptosis of skeletal myoblasts [6, 30, 49, 50] the role of cell cycle progression in this process is currently under investigation. Secondly, decreased FLIP levels can result as a consequence of increased proteolysis mediated by the ubiquitin pathway. This mechanism is employed during p53-mediated apoptosis [51]. While we, and others [47] have eliminated a role for p53 in the differentiation-associated apoptosis of skeletal myoblasts, we have not excluded the possibility that decreased levels of FLIP during this process are a consequence of accelerated degradation. These experiments are more complicated than employing pharmacological inhibitors of the proteosome since many apoptotic molecules involved in TRAIL-induced apoptosis would be affected [52]. Thirdly, FLIP levels are decreased in the absence of growth factor mediated signaling through the PI3K/Akt pathway [53]. Decreased signaling through this pathway allows nuclear localization of the FOXO3a transcription factor and FLIP levels are repressed by FOXO3a. Decreased FLIP mRNA levels are associated with Foxo3A-mediated repression [54], but the molecular details of this repression remain to be determined. PI3K/Akt signaling does play a role in myoblast survival during differentiation [32] and culture in differentiation media does induce a decrease in PI3K/Akt signaling [55]. However, abrogation of PI3K/Akt signaling, or constitutively active FOXO3a signaling, for at least 24 h is required to detect decreased levels of FLIP [54, 56]. We have determined that the decrease in PI3K/Akt signaling as a consequence of culture in differentiation media is transient, with signaling levels rising again after 6 h [55]. Further, this same pathway is responsible for increased expression of TRAIL and the pro-apoptotic Bcl2 family member Bim [57, 58] and the expression level of neither of these proteins is increased during the apoptosis associated with differentiation (herein and Freer/Weyman, manuscript in preparation). DR5 expression is not controlled by the PI3K/Akt pathway [57]. Taken together, these data do not suggest that the PI3K/Akt/Foxo3a pathway is responsible for the decreased levels of FLIP detected in myoblasts undergoing differentiation-associated apoptosis. Thus, future experiments are focused on determining the mechanism through which FLIP levels are reduced in myoblasts undergoing differentiation-associated apoptosis.
While our studies performed on cultured myoblasts clearly demonstrate a role for the TRAIL /DR5 /FADD / caspase 8 pathway in the apoptosis associated with skeletal myoblast differentiation, no obvious muscle related phenotype has been reported for TRAIL−/− [59], DR5−/− [60], FADD−/− [61, 62], FLIP−/− [63] or caspase 8−/− mice [64]. In FADD−/−, FLIP−/− or caspase 8−/− mice, this lack of muscle phenotype may be attributed to the fact that these mice are not viable [61–64]. TRAIL−/− and DR5−/− mice, however, develop normally. A defective apoptotic phenotype in any cell type in the TRAIL−/− and DR5−/− mice is, however, observed in response to DNA damaging agents such as chemical carcinogen or ionizing radiation [59, 60]. Thus, we speculate that lack of a developmental muscle phenotype could indicate either that this pathway has no role during developmental myogenesis or that compensatory signaling negates detection of such a role. Further, a reduced apoptotic phenotype in either TRAIL−/− and DR5−/− skeletal myoblasts might only be observed in response to specific stimuli such as conditions that lead to muscle wasting or regeneration, or in the context of myoblast transplantation [1–3, 9, 10, 12, 13, 65–71]. These future studies, combined with further analysis to understand the molecular mechanisms mediating the increased level of the TRAIL receptor DR5 and the decreased level of FLIP in apoptotic myoblasts, might identify additional therapeutic targets for manipulation to modify skeletal myoblast apoptosis relevant to the amelioration of disease states with associated muscle degeneration as well as to the use of myoblast transfer as a therapeutic approach.
Conclusion
Inhibition of TRAIL, DR5, FADD or caspase 8 signaling inhibits the apoptosis associated with skeletal myoblast differentiation. Myoblasts undergoing apoptosis rather than differentiation possess increased levels of the TRAIL receptor DR5 and decreased levels of the caspase 8 antagonist FLIP.
Acknowledgment
We thank A. Almasan (Learner Research Institute, Cleveland Clinic), V. Dixit (Genetech, Inc.) and E.S.Alnemri (Thomas Jefferson University) for generously providing the dnFADD and dnDR5 constructs. This work was supported by an NIH RO1 CA84212 awarded to C.M. Weyman.
Abbreviations
- FADD
Fas-associated death domain
- TRAIL
Tumor necrosis factor related apoptosis-inducing ligand
- FLIP
FADD-like IL-1-converting enzyme-inhibitory protein
References
- 1.Raff MC (1992) Social controls on cell survival and cell death. Nature 356:397–00 [DOI] [PubMed]
- 2.Fidzianska A, Goebel HH (1991) Human ontogenesis 3. Cell death in fetal muscle. Acta Neuropathol (Berl) 81:572–77 [DOI] [PubMed]
- 3.Miller JB, Stockdale FE (1986) Developmental regulation of the multiple myogenic cell lineages of the avian embryo. J Cell Biol 103:2197–208 [DOI] [PMC free article] [PubMed]
- 4.Sandri MC, Massimino ML, Geromel V, Arslan P (1996) Myoblasts and myotubes in primary cultures deprived of growth factors undergo apoptosis. Basic Appl Myol 6:257–60
- 5.Chinni C, de Niese MR, Tew DJ, Jenkins AL, Bottomley SP, Mackie EJ (1999) Thrombin, a survival factor for cultured myoblasts. J Biol Chem 274:9169–174 [DOI] [PubMed]
- 6.Wang J, Walsh K (1996) Resistance to apoptosis conferred by Cdk inhibitors during myocyte differentiation. Science 273:359–61 [DOI] [PMC free article] [PubMed]
- 7.Dee K, Freer M, Mei Y, Weyman CM (2002) Apoptosis coincident with the differentiation of skeletal myoblasts is delayed by caspase 3 inhibition and abrogated by MEK-independent constitutive Ras signaling. Cell Death Differ 9:209–18 [DOI] [PubMed]
- 8.Mampuru LJ, Chen SJ, Kalenik JL, Bradley ME, Lee TC (1996) Analysis of events associated with serum deprivation-induced apoptosis in C3H/Sol8 muscle satellite cells. Exp Cell Res 226:372–80 [DOI] [PubMed]
- 9.Skuk D, Tremblay JP (2000) Progress in myoblast transplantation: a potential treatment of dystrophies. Microsc Res Tech 48:213–22 [DOI] [PubMed]
- 10.Bouchentouf M, Benabdallah BF, Tremblay JP (2004) Myoblast survival enhancement and transplantation success improvement by heat-shock treatment in mdx mice. Transplantation 77:1349–356 [DOI] [PubMed]
- 11.Olson EN (1992) Interplay between proliferation and differentiation within the myogenic lineage. Dev Biol 154:261–72 [DOI] [PubMed]
- 12.Sandri M, Carraro U (1999) Apoptosis of skeletal muscles during development and disease. Int J Biochem Cell Biol 31:1373–390 [DOI] [PubMed]
- 13.Sandri M, El Meslemani AH, Sandri C et al (2001) Caspase 3 expression correlates with skeletal muscle apoptosis in Duchenne and facioscapulo human muscular dystrophy. A potential target for pharmacological treatment? J Neuropathol Exp Neurol 60:302–12 [DOI] [PubMed]
- 14.Yang Y, Kaul S, Zhang D, Anantharam V, Kanthasamy AG (2004) Suppression of caspase-3-dependent proteolytic activation of protein kinase C delta by small interfering RNA prevents MPP+-induced dopaminergic degeneration. Mol Cell Neurosci 25:406–21 [DOI] [PubMed]
- 15.Nagy Z, Simon L (2004) Neuroprotection in ischemic/hypoxic disorders: from the preclinical to the clinical testing. Adv Exp Med Biol 541:39–4 [PubMed]
- 16.Przedborski S (2004) Programmed cell death in amyotrophic lateral sclerosis: a mechanism of pathogenic and therapeutic importance. Neurologist 10:1– [DOI] [PubMed]
- 17.Sheikh MS, Huang Y (2004) Death receptors as targets of cancer therapeutics. Curr Cancer Drug Targets 4:97–04 [DOI] [PubMed]
- 18.Debatin KM, Krammer PH (2004) Death receptors in chemotherapy and cancer. Oncogene 23:2950–966 [DOI] [PubMed]
- 19.DeChant AK, Dee K, Weyman CM (2002) Raf-induced effects on the differentiation and apoptosis of skeletal myoblasts are determined by the level of Raf signaling: abrogation of apoptosis by Raf is downstream of caspase 3 activation. Oncogene 21:5268–279 [DOI] [PubMed]
- 20.Nakanishi K, Sudo T, Morishima N (2005) Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol 169:555–60 [DOI] [PMC free article] [PubMed]
- 21.Kroemer G (1999) Mitochondrial control of apoptosis: an overview. Biochem Soc Symp 66:1–5 [DOI] [PubMed]
- 22.Kuwana T, Newmeyer DD (2003) Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol 15:691–99 [DOI] [PubMed]
- 23.Orrenius S (2004) Mitochondrial regulation of apoptotic cell death. Toxicol Lett 149:19–3 [DOI] [PubMed]
- 24.Thorburn A (2004) Death receptor-induced cell killing. Cell Signal 16:139–44 [DOI] [PubMed]
- 25.Salvesen GS, Dixit VM (1997) Caspases: intracellular signaling by proteolysis. Cell 91:443–46 [DOI] [PubMed]
- 26.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 15:269–90 [DOI] [PubMed]
- 27.Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–01 [DOI] [PubMed]
- 28.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481–90 [DOI] [PubMed]
- 29.Wang J, Guo K, Wills KN, Walsh K (1997) Rb functions to inhibit apoptosis during myocyte differentiation. Cancer Res 57:351–54 [PubMed]
- 30.Guo K, Walsh K (1997) Inhibition of myogenesis by multiple cyclin-Cdk complexes. Coordinate regulation of myogenesis and cell cycle activity at the level of E2F. J Biol Chem 272:791–97 [DOI] [PubMed]
- 31.Lawlor MA, Rotwein P (2000) Coordinate control of muscle cell survival by distinct insulin-like growth factor activated signaling pathways. J Cell Biol 151:1131–140 [DOI] [PMC free article] [PubMed]
- 32.Lawlor MA, Rotwein P (2000) Insulin-like growth factor-mediated muscle cell survival: central roles for Akt and cyclin-dependent kinase inhibitor p21. Mol Cell Biol 20:8983–995 [DOI] [PMC free article] [PubMed]
- 33.Lawlor MA, Feng X, Everding DR, Sieger K, Stewart CE, Rotwein P (2000) Dual control of muscle cell survival by distinct growth factor-regulated signaling pathways. Mol Cell Biol 20:3256–265 [DOI] [PMC free article] [PubMed]
- 34.Reuveny M, Heller H, Bengal E (2004) RhoA controls myoblast survival by inducing the phosphatidylinositol 3-kinase-Akt signaling pathway. FEBS Lett 569:129–34 [DOI] [PubMed]
- 35.Jin Z, El-Deiry WS (2005) Overview of cell death signaling pathways. Cancer Biol Ther 4:139–63 [DOI] [PubMed]
- 36.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81:505–12 [DOI] [PubMed]
- 37.Ray S, Almasan A (2003) Apoptosis induction in prostate cancer cells and xenografts by combined treatment with Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand and CPT-11. Cancer Res 63:4713–723 [PubMed]
- 38.Dee K, DeChant A, Weyman CM (2003) Differential signaling through NFkappaB does not ameliorate skeletal myoblast apoptosis during differentiation. FEBS Lett 545:246–52 [DOI] [PubMed]
- 39.Kawahara A, Enari M, Talanian RV, Wong WW, Nagata S (1998) Fas-induced DNA fragmentation and proteolysis of nuclear proteins. Genes Cells 3:297–06 [DOI] [PubMed]
- 40.Wolfman JC, Palmby T, Der CJ, Wolfman A (2002) Cellular N-Ras promotes cell survival by downregulation of Jun N-terminal protein kinase and p38. Mol Cell Biol 22:1589–606 [DOI] [PMC free article] [PubMed]
- 41.Almasan A, Ashkenazi A (2003) Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev 14:337–48 [DOI] [PubMed]
- 42.MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES (1997) Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem 272:25417–5420 [DOI] [PubMed]
- 43.Roth W, Reed JC (2004) FLIP protein and TRAIL-induced apoptosis. Vitam Horm 67:189–06 [DOI] [PubMed]
- 44.Zhang K, Kaufman RJ (2006) The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66:S102–S109 [DOI] [PubMed]
- 45.Wu GS, Burns TF, McDonald ER 3rd et al (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17:141–43 [DOI] [PubMed]
- 46.Burns TF, Bernhard EJ, El-Deiry WS (2001) Tissue specific expression of p53 target genes suggests a key role for KILLER/DR5 in p53-dependent apoptosis in vivo. Oncogene 20:4601–612 [DOI] [PubMed]
- 47.Cerone MA, Marchetti A, Bossi G, Blandino G, Sacchi A, Soddu S (2000) p53 is involved in the differentiation but not in the differentiation-associated apoptosis of myoblasts. Cell Death Differ 7:506–08 [DOI] [PubMed]
- 48.Algeciras-Schimnich A, Griffith TS, Lynch DH, Paya CV (1999) Cell cycle-dependent regulation of FLIP levels and susceptibility to Fas-mediated apoptosis. J Immunol 162:5205–211 [PubMed]
- 49.Mercer SE, Ewton DZ, Deng X, Lim S, Mazur TR, Friedman E (2005) Mirk/Dyrk1B mediates survival during the differentiation of C2C12 myoblasts. J Biol Chem 280:25788–5801 [DOI] [PMC free article] [PubMed]
- 50.Peschiaroli A, Figliola R, Coltella L et al (2002) MyoD induces apoptosis in the absence of RB function through a p21(WAF1)-dependent re-localization of cyclin/cdk complexes to the nucleus. Oncogene 21:8114–127 [DOI] [PubMed]
- 51.Fukazawa T, Fujiwara T, Uno F et al (2001) Accelerated degradation of cellular FLIP protein through the ubiquitin-proteasome pathway in p53-mediated apoptosis of human cancer cells. Oncogene 20:5225–231 [DOI] [PubMed]
- 52.Zhang HG, Wang J, Yang X, Hsu HC, Mountz JD (2004) Regulation of apoptosis proteins in cancer cells by ubiquitin. Oncogene 23:2009–015 [DOI] [PubMed]
- 53.Suhara T, Mano T, Oliveira BE, Walsh K (2001) Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP). Circ Res 89:13–9 [DOI] [PubMed]
- 54.Skurk C, Maatz H, Kim HS et al (2004) The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem 279:1513–525 [DOI] [PubMed]
- 55.Karasarides M, Dee K, Schulman D, Wolfman A, Weyman CM (2006) Active Ras-induced effects on skeletal myoblast differentiation and apoptosis are independent of constitutive PI3-kinase activity. Cell Biol Int [DOI] [PubMed]
- 56.Panka DJ, Mano T, Suhara T, Walsh K, Mier JW (2001) Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP expression in tumor cells. J Biol Chem 276:6893–896 [DOI] [PubMed]
- 57.Wang Q, Wang X, Hernandez A, Hellmich MR, Gatalica Z, Evers BM (2002) Regulation of TRAIL expression by the phosphatidylinositol 3-kinase/Akt/GSK-3 pathway in human colon cancer cells. J Biol Chem 277:36602–6610 [DOI] [PubMed]
- 58.Gilley J, Coffer PJ, Ham J (2003) FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol 162:613–22 [DOI] [PMC free article] [PubMed]
- 59.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ (2002) Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol 168:1356–361 [DOI] [PubMed]
- 60.Finnberg N, Gruber JJ, Fei P et al (2005) DR5 knockout mice are compromised in radiation-induced apoptosis. Mol Cell Biol 25:2000–013 [DOI] [PMC free article] [PubMed]
- 61.Zhang J, Cado D, Chen A, Kabra NH, Winoto A (1998) Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392:296–300 [DOI] [PubMed]
- 62.Yeh WC, Pompa JL, McCurrach ME et al (1998) FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279:1954–958 [DOI] [PubMed]
- 63.Yeh WC, Itie A, Elia AJ et al (2000) Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12:633–42 [DOI] [PubMed]
- 64.Varfolomeev EE, Schuchmann M, Luria V et al (1998) Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9:267–76 [DOI] [PubMed]
- 65.Ellis RE, Yuan JY, Horvitz HR (1991) Mechanisms and functions of cell death. Annu Rev Cell Biol 7:663–98 [DOI] [PubMed]
- 66.Taylor DA, Atkins BZ, Hungspreugs P et al (1998) Regenerating functional myocardium: improved performance after skeletal myoblast transplantation. Nat Med 4:929–33 [DOI] [PubMed]
- 67.Scorsin M, Hagege A, Vilquin JT et al (2000) Comparison of the effects of fetal cardiomyocyte and skeletal myoblast transplantation on postinfarction left ventricular function. J Thorac Cardiovasc Surg 119:1169–175 [DOI] [PubMed]
- 68.Suzuki K, Murtuza B, Smolenski RT et al (2001) Cell transplantation for the treatment of acute myocardial infarction using vascular endothelial growth factor-expressing skeletal myoblasts. Circulation 104:I207–I212 [DOI] [PubMed]
- 69.Skuk D, Tremblay JP (2003) Cell therapies for inherited myopathies. Curr Opin Rheumatol 15:723–29 [DOI] [PubMed]
- 70.Skuk D, Tremblay JP (2003) Myoblast transplantation: the current status of a potential therapeutic tool for myopathies. J Muscle Res Cell Motil 24:285–00 [DOI] [PubMed]
- 71.Skuk D, Caron NJ, Goulet M, Roy B, Tremblay JP (2003) Resetting the problem of cell death following muscle-derived cell transplantation: detection, dynamics and mechanisms. J Neuropathol Exp Neurol 62:951–67 [DOI] [PubMed]