

Abstract
Background and purpose:
Tobacco smoke represents a relevant risk factor for coronary heart disease (CHD). Although peroxisome proliferator-activated receptor (PPAR)γ activation reduces inflammation and atherosclerosis, expression of PPARγ in cells and its modulation by smoking are poorly investigated. We previously reported that monocyte/macrophages from healthy smokers exhibited an enhanced constitutive expression of PPARγ. Here, we evaluated PPARγ expression and basal cytokine release in monocytes and monocyte-derived macrophages (MDMs) from 85 CHD patients, classified by their smoking habit (smokers, non-smokers and ex-smokers), and assessed the role of PPARγ ligands in this context.
Experimental approach:
PPARγ protein was detected by Western blot and semi-quantified by PPARγ/β-actin ratio; cytokine release was measured by elisa and nuclear factor-kappaB (NF-κB) translocation by electrophoretic mobility shift assays.
Key results:
As compared to the other groups, MDMs from smoker CHD patients exhibited a reduced PPARγ/β-actin ratio and an increased spontaneous release of tumour necrosis factor-α (TNF-α) and interleukin-6, but with no major variations in monocytes. In cells from selected CHD patients, rosiglitazone inhibited TNF-α release and NF-κB translocation induced by phorbol-12-myristate 13-acetate. The selective PPARγ antagonist GW9662 reversed these effects, with some variations related to smoking habit.
Conclusions and implications:
In CHD patients, exposure to tobacco smoke profoundly affected PPARγ expression, and this was related to levels of secretion of pro-inflammatory cytokines. MDMs from CHD smokers showed the lowest PPARγ expression and released more inflammatory cytokines. Moreover, rosiglitazone's ability to inhibit cytokine release and its reversal by GW9662 clearly indicated PPARγ involvement in these changes in CHD patients.
Keywords: PPARγ, monocyte/macrophages, coronary heart diseases, tobacco smoke, cytokines, NF-κB
Introduction
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily and comprise three distinct subtypes: PPARα, β/δ and γ (nomenclature follows Alexander et al., 2008). Upon activation, they heterodimerize with the retinoid X receptor, and then bind to PPAR responsive elements in the promoter region of target genes, thus regulating their expression. PPARγ is a key mediator in adipogenesis and controls critical steps of glucose and lipid homeostasis (Straus and Glass, 2007). Besides being expressed at high levels in white adipose tissue, PPARγ has been demonstrated in a large variety of cells, including human monocyte/macrophages (Li and Palinski, 2006; Amoruso et al., 2007; Straus and Glass, 2007; Rigamonti et al., 2008; Szanto and Roszer, 2008). PPARγ can be activated by naturally occurring ligands [e.g. 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and oxidized low-density lipoproteins], as well as by synthetic agents, such as the thiazolidinedione (TZD) class of anti-diabetic drugs, and some selected non-steroidal anti-inflammatory drugs (Jiang et al., 1998; Ricote et al., 1998; Amoruso et al., 2007; Straus and Glass, 2007).
A large array of experimental results indicates that PPARγ regulates inflammatory processes and atherosclerosis, the anti-inflammatory potential largely residing in the ability of PPARγ agonists to inhibit monocyte/macrophage activation and expression of pro-inflammatory molecules, such as tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1β, inducible nitric oxide synthase, gelatinase B, COX-2 and nuclear factor-kappaB (NF-κB) (Jiang et al., 1998; Ricote et al., 1998; Li et al., 2000; Li and Palinski, 2006; Amoruso et al., 2007; Caito et al., 2008; Rigamonti et al., 2008).
We recently demonstrated that PPARγ protein is constitutively present in human monocytes and monocyte-derived macrophages (MDMs), its expression being up-regulated along with differentiation (Amoruso et al., 2007). We also reported that cells from healthy smokers present an enhanced expression of PPARγ, as compared to non-smokers, and that this effect is reproduced to some extent by in vitro challenge with nicotine (Amoruso et al., 2007).
Despite the fact that PPARγ is mostly considered as an anti-inflammatory and anti-atherogenic factor, information on its quantitative expression is scarce, and very few investigations have evaluated the role of PPARγ in patients with coronary heart disease (CHD). The causal role of cigarette smoking in both heart and lung diseases is well established, and tobacco has been shown to modify macrophage responsiveness (Taylor et al., 1998; Bardelli et al., 2005; Gunella et al., 2006; Karimi et al., 2006; Benowitz, 2008; Caito et al., 2008).
Therefore, it would be of particular interest, to the clinical management of diseases as well, to know whether or not tobacco smoking affects the expression of PPARγ in CHD patients. Hence, this observational pilot study was aimed to evaluate the constitutive expression of PPARγ protein, as well as the basal release of pro-inflammatory cytokines, in monocytes and MDMs isolated from representative CHD patients of both sexes, according to smoking habit. To better define the PPARγ role in this context, we have also evaluated the ability of rosiglitazone, a clinically important PPARγ ligand, to affect cytokine release and NF-κB activation in cells from selected CHD patients.
Methods
Patient selection
The research protocol for this study was approved by the Ethical Committee of Ospedale Civile Maggiore, Verona, Italy, and informed written consent was obtained from all participants. In the period 1 February–31 May 2008, 85 CHD patients were consecutively recruited in this study. The patients enrolled were aged 45–90 years, were symptomatic for CHD (either stable or unstable angina) and had angiographic evidence of significant coronary artery disease (diameter stenosis >70%) in at least one major epicardial coronary vessel. All patients were on current medical therapy, none receiving TZDs (which increase PPARγ); the female population was represented by post-menopausal women, none of whom were receiving hormone replacement therapy. The smoking status of CHD patients was classified as: smokers (n= 21), non-smokers (n= 29) and ex-smokers (n= 35).
Before blood sampling, a cardiologist evaluated each patient; medical history, and clinical and laboratory data were registered in a dedicated database. Blood samples were withdrawn between 0800 and 0900 h; smokers refrained from smoking at least 1 h before phlebotomy.
Preparation of monocytes, partially differentiated macrophages (M4d) and MDMs
Human monocytes were isolated from heparinized venous blood (30–40 mL) of CHD patients by standard techniques of dextran sedimentation, histopaque (density =1.077 g·cm−3) gradient centrifugation (400×g, 30 min, room temperature) and recovered by thin suction at the interface, as described (Amoruso et al., 2007). Cells were resuspended in RPMI 1640 medium, supplemented with 5% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine and antibiotics; purified monocyte populations were obtained by adhesion. Cell viability (trypan blue dye exclusion) was usually >98%. MDMs were prepared from monocytes, by culture (8–10 days) in RPMI 1640 medium containing 20% FBS, glutamine and antibiotics. Monocytes, cultured as above for 4 days, were also assessed and are referred to as M4d (Amoruso et al., 2007).
MDMs were defined as macrophage-like cells by evaluating surface markers CD14, MHCII, CD1a and CD68 (Amoruso et al., 2007; 2008;). Briefly, adherent cells were detached by gentle scraping with a plastic scraper. After three washings with sterile phosphate-buffered saline (PBS), cells were resuspended at the final concentration of 1 × 105 cells·mL−1, and fluorescent dye-labelled antibodies against the different surface markers (anti-CD14 from Becton Dickinson, Oxford, UK; anti-CD68 and anti-MHCII from Dako, Milan, Italy; anti-CD1a from Bioscence, San Diego, CA, USA) were added for 30 min on ice. Incubation was performed in the dark, and expression of surface markers was analysed by flow cytometry.
PPARγ protein expression and semi-quantitative analysis
Monocytes, partially differentiated (M4d) and fully differentiated macrophages (MDMs) from CHD patients were used to evaluate their constitutive expression of PPARγ protein. Cells (2 × 106), seeded in six-well plates, were washed twice with ice-cold PBS and scraped off the wells in lysis buffer containing 3% SDS, 0.25 M Tris and 1 mM phenylmethyl sulphonyl fluoride, and lysed by sonication; when necessary, cell lysates were stored at −80°C. The determination of protein concentration was done with a Bradford-based assay. Protein samples (20 µg) were analysed by SDS–PAGE (10% acrylamide) and electro-blotted on nitrocellulose membrane (Protran, Perkin Elmer Life Sciences, Boston, MA, USA). Immunoblots were performed using monoclonal mouse anti-human PPARγ (Santa Cruz, CA, USA) and anti-humanβ-actin (Sigma, St Louis, MO, USA) antibodies; anti-mouse secondary antibody was coupled to horseradish peroxidase. Chemiluminescence signals were analysed under non-saturating conditions with an image densitometer (Versadoc, Bio-Rad, Hercules, CA, USA). Semi-quantitative evaluation of PPARγ protein was performed as described (Amoruso et al., 2007), by calculating the ratio between its expression and the expression of the reference housekeeping protein, β-actin.
Electrophoretic mobility shift assay for NF-κB
Nuclear extracts (5 µg) from monocytes and MDMs were incubated with 2 µg poly(dI-dC) and [32P]ATP-labelled oligonucleotide probe (100 000–150 000 cpm; Promega, Milan, Italy) in binding buffer for 30 min at room temperature, as described (Bardelli et al., 2005; Gunella et al., 2006). The NF-κB consensus oligonucleotide (5'-AGTTGAGGGGACTTTCCCAGGC–3') was from Promega. The nucleotide–protein complex was separated on a polyacrylamide gel, the gel was dried and radioactive bands were detected by autoradiography (Bardelli et al., 2005; Gunella et al., 2006).
Cytokine release
Cells (1 × 106), seeded in six-well plates, were maintained in a 5% CO2 incubator at 37°C for 6 h; supernatants were collected and stored. TNF-α, IL-6 and IL-10 (the latter was evaluated as the most relevant anti-inflammatory cytokine) were estimated by elisa (PeliKine Compact human elisa kit), following the manufacturer's instructions (CLB/Sanquin, Amsterdam, The Netherlands). Results are expressed in pg·mL−1. In some experiments, we also evaluated the ability of the selective PPARγ agonist rosiglitazone and the selective antagonist GW9662 (Alexander et al., 2008) to affect cytokine release in monocytes/macrophages stimulated by phorbol-12 myristate 13-acetate (PMA). To perform these experiments, cells were pretreated with increasing concentrations of rosiglitazone (10−10 to 10−5 M), with or without GW9662 (10−6 M), for 1 h and then challenged with PMA 10−7 M for 24 h. As previously shown, the 24 h challenge ensured maximal cytokine release (Bardelli et al., 2005; Amoruso et al., 2008). In these experiments, GW9662 was used at 10−6 M as this concentration was known to block PPARγ-induced effects in previous experiments (Amoruso et al., 2008).
Statistical analysis
All statistical analyses were performed using SPSS statistical software (version 15.0, SPSS Inc., Chicago, IL, USA). Data are mean ± SEM of ‘n’ independent experiments; cytokine determinations were performed in duplicate. Concentration–effect curves for rosiglitazone were constructed, and its IC50 values (on PMA-induced cytokine release) were interpolated from curves of best fit. Statistical evaluation was performed by analysis of variance with Bonferroni corrections. Differences were considered statistically significant when P < 0.05.
Materials
FBS was from Gibco (Paisley, UK). Rosiglitazone was from Cayman Chemicals (Milan, Italy); the selective antagonist GW9662 was from Biomol (Exeter, UK). Histopaque, RPMI 1640 medium, glutamine, HEPES, streptomycin, penicillin, amphotericin B, protease inhibitors, PMA and monoclonal mouse anti-humanβ-actin antibodies were obtained from Sigma (Milwaukee, WI, USA). The monoclonal mouse anti-human PPARγ (E-8) antibody was from Santa Cruz. Tissue culture plates were from Nunc Ltd (Roskilde, Denmark); all cell culture reagents, with the exception of FBS, were endotoxin free according to details provided by the manufacturer.
Results
Baseline patient characteristics
This study enrolled 85 consecutive CHD patients who were admitted to the Division of Cardiology in the period 1 February–31 May 2008, and gave their informed consent. Thereafter, the CHD patients were stratified according to their lifestyle smoking behaviour, so that the study population comprised smoker (n= 21), non-smoker (n= 29) and ex-smoker (n= 35) patients of both sexes.
Baseline demographic, clinical and laboratory characteristics of the three subgroups are summarized in Tables 1 and 2. The study population was primarily composed of male patients (n= 65), with a minority (n= 20) of post-menopausal female patients, none receiving hormonal replacement therapy. The smoker group (n= 21) was represented by current heavy tobacco smokers, that is, consumers of at least 20 cigarettes per day for more than 10 years, and included 16 men and 5 women. Sixteen men and 13 women who had never smoked during their life composed the non-smoker CHD group, whereas the ex-smoker group included 33 men and 2 women. All the ex-smoker CHD patients were former heavy smokers and had given up cigarettes at least 1 year before the enrolment. In particular, 10 patients had stopped smoking 1–2 years before, five patients had stopped 3–5 years before, 15 patients stopped smoking 6–10 years before and five patients had given up >10 years before.
Table 1.
Characteristics of coronary heart disease patients and their medication
| Ex-smokers (n =35) | Non-smokers (n =29) | Smokers (n =21) | |
|---|---|---|---|
| Sex (M/F; % female) | 33/2 (6%) | 16/13 (45%) | 16/5 (24%) |
| Age (year) | 64.6 ± 1.5 | 66.9 ± 1.9 | 63 ± 1.5 |
| BMI (kg·m−2) | 26.4 ± 1.5 | 27.1 ± 0.7 | 26.3 ± 0.7 |
| Multi-vessel disease (n; %) | 12 (34%) | 10 (34%) | 6 (29%) |
| Unstable angina (n; %) | 18 (51%) | 17 (59%) | 11 (52%) |
| Hypertension (n; %) | 23 (66%) | 21 (73%) | 11 (52%) |
| Dyslipidemia (n; %) | 26 (74%) | 19 (66%) | 13 (62%) |
| Diabetes (n; %) | 14 (40%) | 13 (45%) | 5 (24%) |
| Family history (n; %) | 7 (20%) | 10 (34%) | 9 (43%) |
| Nitrates (n; %) | 20 (57%) | 23 (80%) | 18 (86%) |
| Ca-antagonists (n; %) | 5 (14%) | 5 (17%) | 4 (19%) |
| ARBs (n; %) | 7 (20%) | 8 (27%) | 2 (10%) |
| ACE-inhibitors (n; %) | 15 (43%) | 15 (52%) | 9 (43%) |
| Beta-blockers (n; %) | 27 (77%) | 19 (66%) | 13 (62%) |
| Statins (n; %) | 23 (66%) | 16 (55%) | 12 (57%) |
| Anti-diabetics (n; %) | 14 (40%) | 10 (34%) | 5 (24%) |
| ASA/anti-platelet (n; %) | 35 (100%) | 29 (100%) | 21 (100%) |
Values are mean ± SEM.
BMI, body mass index; ARBs, angiotensin II receptor antagonists; ACE, angiotensin converting enzyme; ASA: aspirin.
Table 2.
Serum glucose and lipid profile of coronary heart disease patients
| Ex-smokers (n =35) | Non-smokers (n =29) | Smokers (n = 21) | |
|---|---|---|---|
| Glucose (mM) | 6.78 ± 0.37 | 7.43 ± 0.4 | 5.94 ± 0.4* |
| Total cholesterol (mM) | 4.78 ± 0.2 | 4.78 ± 0.22 | 4.94 ± 0.26 |
| HDL-cholesterol (mM) | 1.12 ± 0.04 | 1.06 ± 0.05 | 1.14 ± 0.07 |
| LDL-cholesterol (mM) | 3.06 ± 0.2 | 3.01 ± 0.2 | 13.13 ± 0.2 |
| Triglycerides (mM) | 1.61 ± 0.14 | 1.26 ± 0.1 | 1.47 ± 0.26 |
Values are mean ± SEM.
P < 0.02 versus non-smokers.
Although not homogeneous regarding the male/female ratio, the three subgroups were similar for baseline characteristics and disease severity, except for a reduced prevalence of diabetes and hypertension, and a higher proportion with a family history of CHD, in the smoker group (Table 1). All patients were on current medical therapy, aspirin and/or other anti-platelet drugs being administered to all patients; CHD diabetic patients were also treated with insulin and/or oral anti-diabetics, except TZD (Table 1). As reported in Table 1, only 57% of the CHD ex-smokers received nitrates, as compared to 80% and more in the two other groups. On the contrary, the percentage of CHD ex-smokers treated with β-adrenoceptor antagonists (beta-blockers) and statins was higher than the two other groups (Table 1).
Serum glucose, triglycerides, total cholesterol and HDL- and LDL-cholesterol values are summarized in Table 2. These values were in a normal or near-normal range, and, apart from a minor serum glucose value in the smoker group (including fewer diabetic patients than the two others), no major changes were observed among the three subgroups (Table 2).
Therefore, the three study groups are rather similar, so that the eventual variations in PPARγ expression cannot be ascribed to different disease characteristics, abnormal parameters and/or therapy.
Characterization of monocytes and MDM from CHD patients
Phenotype evaluation of monocytes and MDM among the three CHD groups (smokers, non-smokers and ex-smokers) was performed according to Amoruso et al. (2008). Cell surface expression of CD14, CD68 and MHCII was about 90, 65 and 98%, respectively; in monocytes, no major variation in the percentage of positive cells being observed in relation to smoking habit. In MDM, cell surface expression of CD14, CD68 and MHCII was about 40, 93 and 70%, respectively, with no statistical difference among the three groups (data not shown). As previously reported (Amoruso et al., 2008), the absence of CD1a expression demonstrated that no differentiation towards dendritic cells occurred in our MDM preparations (data not shown).
Expression of PPARγ protein in monocytes, M4d and MDM from CHD patients
As shown in Figure 1A, PPARγ expression was up-regulated during differentiation to MDM, as previously reported (Amoruso et al., 2007). Expression of PPARγ varied with smoking habit in partially differentiated (M4d) and fully differentiated macrophages (MDMs), but not in monocytes (Figure 1B). MDMs from the smoker CHD group expressed the lowest PPARγ protein content, while MDMs from ex-smoker patients constitutively expressed higher amounts of PPARγ protein, compared to the two other groups (P < 0.05 vs. non-smokers; P < 0.001 vs. smokers) (Figure 1B). Similar results were also observed in M4d, cells from CHD smokers demonstrating the lowest PPARγ expression (Figure 1B).
Figure 1.
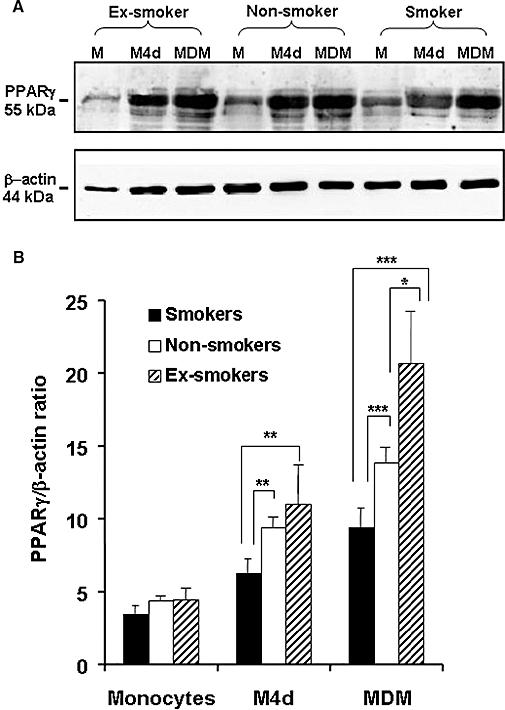
Constitutive peroxisome proliferator-activated receptor (PPAR)γ protein expression in monocytes, partially differentiated macrophages (M4d) and fully differentiated macrophages from coronary heart disease (CHD) patients, related to smoking status. In (A): representative Western blot for constitutive PPARγ protein and β-actin expression for individual CHD patients. In (B): semi-quantitative analysis of PPARγ protein expression in CHD patients (21 smokers, 29 non-smokers and 35 ex-smokers). Results are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
The enhanced PPARγ expression, detected in MDMs from CHD ex-smokers, appeared to be correlated to the length of time from smoking cessation: in fact, it was maximal 1–2 years after quitting and then declined in the following years, paralleling the amount observed in CHD non-smokers (Figure 2).
Figure 2.
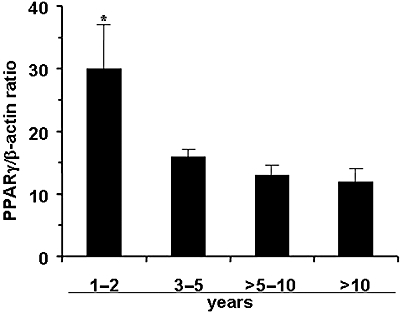
Constitutive peroxisome proliferator-activated receptor γ protein expression in monocyte-derived macrophages from 35 ex-smoker CHD patients. This group was subdivided according to the length of time they had stopped smoking (shown on the X-axis). Ten patients had given up smoking for 1–2 years, five for 3–5 years, 15 for 6–10 years and five had stopped for >10 years. Results shown are means (±SEM) from each subgroup. *P < 0.05.
Basal cytokine release in monocytes and MDMs from CHD patients
Because PPARγ is known to affect cytokine release (Jiang et al., 1998; Ricote et al., 1998; Amoruso et al., 2007), and monocyte/macrophages spontaneously release measurable amounts of inflammatory cytokines (Amoruso et al., 2007), we evaluated whether or not the smoking-related difference in PPARγ expression could be correlated with the amount of cytokine secreted.
As shown in Figure 3, untreated monocytes and MDMs from CHD patients release high amounts of TNF-α and IL-6; on the contrary, there was a minimal release of the anti-inflammatory cytokine IL-10. A similar cytokine release occurred in monocytes from smoker, non-smoker and ex-smoker CHD patients (Figure 3A), while MDMs from smoker CHD patients, presenting the lowest PPARγ expression (see Figure 1), released significantly higher amounts of TNF-α (P < 0.002) and IL-6 (P < 0.001), as compared to the other groups (Figure 3B). Interestingly, MDMs from ex-smoker CHD patients, with the highest PPARγ expression, released significantly less TNF-α than the two other groups (Figure 3B). There was no statistical difference between non-smoker and ex-smoker CHD patients regarding basal IL-6 release from MDMs. Conversely, on evaluating IL-10 secretion, no differences were observed between smoker, non-smoker and ex-smoker CHD patients (Figure 3).
Figure 3.
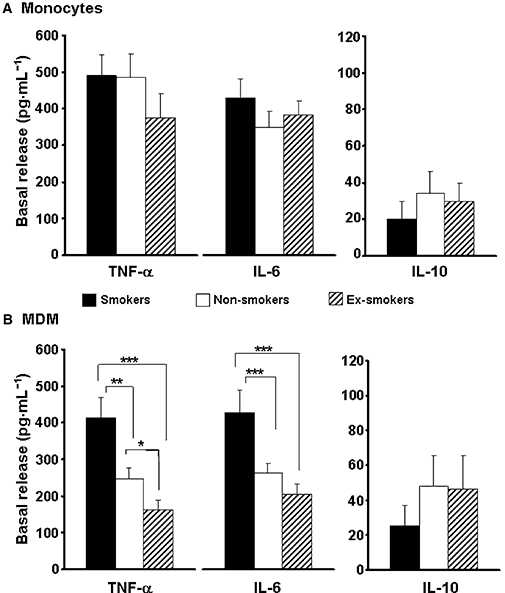
Basal release of tumour necrosis factor-α, interleukin-6 and interleukin-10 in monocytes (A) and monocyte-derived macrophages (B) from coronary heart disease patients. Twenty-one smokers, 29 non-smokers and 35 ex-smokers were evaluated. Results are mean ± SEM, and are expressed as pg·mL−1. *P < 0.05; **P < 0.002; ***P < 0.001.
Effects of PPARγ ligands on cytokine release and NF-κB activation
To explore further the correlations between PPARγ expression level and cytokine release, we evaluated the effects of the selective PPARγ agonist rosiglitazone, which is currently used as a treatment for type 2 diabetes, and the PPARγ antagonist GW9662. We performed the experiments on monocytes and MDMs from selected CHD patients (n= 4), smokers and non-smokers; cells were pretreated with the PPARγ ligands for 1 h and then challenged with PMA (10−7 M) for 24 h. In keeping with our previous results (Bardelli et al., 2005; Amoruso et al., 2008), this 24 h challenge induced a maximal cytokine release from cells. Rosiglitazone inhibited, in a concentration-dependent (10−10 to 10−5 M) manner, PMA-induced TNF-α release in monocytes (Figure 4A) and MDMs (Figure 4B) from both groups of CHD patients. It was more effective in MDMs from non-smokers than smokers, but no major variations were observed in monocytes. The calculated IC50 values were as follows: 9 and 33 nM in MDMs (non-smokers and smokers, respectively), 17 and 28 nM in monocytes (non-smokers and smokers respectively). Similar results were obtained by evaluating IL-6 release (data not shown). As depicted in Figure 4, the selective antagonist GW9662, used at 10−6 M, reversed the agonist effect, providing evidence that cytokine inhibition by rosiglitazone was mediated by PPARγ activation, and that there was a correlation between the level of PPARγ expression and the release of cytokines. Indeed, GW9662 completely reversed rosiglitazone inhibition in cells from non-smoker CHD patients, but was less effective in cells from smoker CHD patients.
Figure 4.
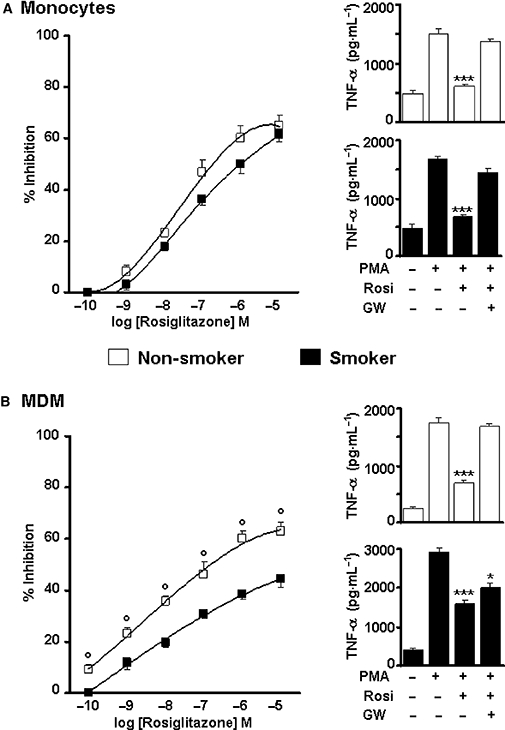
Effects of rosiglitazone on phorbol-12-myristate 13-acetate (PMA)-stimulated tumour necrosis factor-α (TNF-α) release from monocytes (A) and monocyte-derived macrophages (B) of selected non-smoker and smoker coronary heart disease patients. Cells were challenged with increasing concentrations of rosiglitazone (10−10 to 10−5 M) for 1 h and then stimulated by PMA (10−7 M) for 24 h. On the left: % inhibition by rosiglitazone; on the right: reversal by GW9662, at 10−6 M. Results are mean ± SEM; n= 4. °P < 0.01 versus smokers; *P < 0.05 versus PMA; ***P < 0.001 versus PMA. Note the different scale for TNF-α release in B.
Considering the pivotal role of the NF-κB pathway on cytokine release, we also evaluated the activation of NF-κB in cells from smoker and non-smoker CHD patients, and the possible effects of PPARγ ligands on its translocation. As demonstrated in Figure 5A, both monocytes and MDMs from smoker CHD patients showed a stronger constitutive nuclear NF-κB translocation, when compared to those from non-smokers. As a positive control for the detection of NF-κB activation, cells were stimulated by PMA or lipopolysaccharide (LPS), as both agents were previously demonstrated to induce NF-κB nuclear translocation in human monocytes (Lavagno et al., 2004). At this point, we decided to evaluate the effects of PPARγ ligands on PMA-stimulated monocytes from smoker and non-smoker CHD patients. As shown in Figure 5B, rosiglitazone, used at the near maximal 5 µM concentration, decreased the PMA-induced NF-κB translocation in both cell populations. GW9662, used at 1 µM, almost completely reversed this effect in monocytes from non-smoker CHD patients, but was less effective in smokers. Similar results were obtained in MDMs from smoker and non-smoker CHD patients (Figure 5C).
Figure 5.
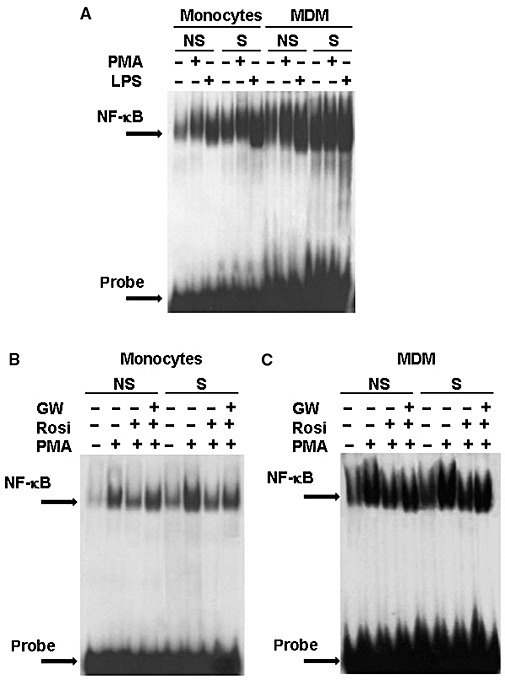
Electrophoretic mobility shift assay for NF-κB in cells from coronary heart disease (CHD) patients. In (A), monocytes and monocyte-derived macrophages (MDMs) from smoker and non-smoker CHD patients stimulated by phorbol-12-myristate 13-acetate (PMA) (10−6 M) or lipopolysaccharide (500 ng·mL−1) for 1 h. In (B), effects of rosiglitazone (rosi; 5 × 10−6 M) and GW9662 (GW; 10−6 M) on monocytes from smoker and non-smoker CHD patients stimulated by PMA. In (C), effects of rosiglitazone (rosi; 5 × 10−6 M) and GW9662 (GW; 10−6 M) on MDM from smoker and non-smoker CHD patients stimulated by PMA. Each gel is representative of two others.
Discussion and conclusions
Tobacco smoke is recognized as the leading cause of morbidity and avoidable mortality (Benowitz, 2008; Chiolero et al., 2008), and markedly affects monocyte/macrophage function (Bardelli et al., 2005; Gunella et al., 2006; Karimi et al., 2006; Benowitz, 2008; Caito et al., 2008). Indeed, the ability of tobacco smoke to accelerate atherosclerosis and cardiovascular diseases is widely recognized: smoking worsens heart failure, increases insulin resistance and, in diabetic patients, markedly accelerates the progression of cardiovascular events (Benowitz, 2008; Chiolero et al., 2008).
Although concerns about the cardiovascular safety of rosiglitazone in diabetic patients have recently been expressed (Nissen and Wolski, 2007), PPARγ is generally considered as an anti-inflammatory and anti-atherogenic factor. However, it is not known whether tobacco smoke affects PPARγ expression in cells from CHD patients, and there was no definite information on the level of expression. Therefore, we planned this observational pilot study, in which 85 CHD patients of both sexes were consecutively enrolled and stratified according to their smoking habit as smokers, non-smokers and ex-smokers. Some non-homogeneous parameters (e.g. gender, family history of CHD, diabetes prevalence, drug therapy) observed among the three CHD subgroups can be attributed to the comprehensive inclusion criteria used, as our main aims were to: (i) get a snapshot of the constitutive expression of PPARγ protein in circulating monocytes and MDMs isolated from CHD patients in real life; (ii) evaluate whether or not a varied PPARγ expression could affect the spontaneous release of inflammatory cytokines; (iii) ascertain the role (if any) of cigarette smoking on PPARγ protein expression; and (iv) get new insight on PPARγ function in cells from CHD patients.
The results obtained here indicate that, at variance from our previous data in healthy donors (Amoruso et al., 2007), smoking had no major effect on the expression of PPARγ protein in monocytes from CHD patients, while it markedly affected constitutive PPARγ expression in MDMs and partially differentiated macrophages. Indeed, PPARγ protein expression in MDMs significantly varied between CHD smokers (who presented the lowest amount) and ex-smokers (who presented the highest amount); non-smokers showing intermediate levels. We have no clear explanation for this fact, but we can rule out a variation in response to selective agonists among the three subgroups, as both the endogenous ligand 15d-PGJ2 (used at 10 µM) and the synthetic ligand rosiglitazone (used at 5 µM) increased PPARγ expression about 2.5-fold (15d-PGJ2) and 3.5-fold (rosiglitazone) in MDMs from the three CHD subgroups, independently of smoker status (data not shown). It is also interesting to note that the highest PPARγ expression was observed in the ex-smoker CHD patients who had quit cigarette smoking 1–2 years before, and then declined in the following years, reaching the mean value of non-smokers in about 5–10 years. It is well known that cardiovascular risk is significantly reduced 1 year after giving up smoking and equals the non-smokers' risk in 15 years (Benowitz, 2008); therefore, it is tempting to speculate that the enhanced PPARγ expression in ex-smoker CHD patients could represent a protective mechanism. In this study, 21 out of 85 (about 25%) consecutively enrolled patients were current smokers, in good agreement with the estimated prevalence of smoking in CHD patients (Brown and Mensah, 2007). In fact, despite medical advice, smoking remains widespread in CHD patients, its deleterious effects further contributing to the severity of the disease.
In different in vitro cell systems, PPARγ agonists exert anti-inflammatory activity, which has been largely related to their ability to down-regulate pro-inflammatory cytokine production (Jiang et al., 1998; Lee et al., 2006; Li and Palinski, 2006; Amoruso et al., 2007). In this study, we found an inverse relation between PPARγ expression and basal release of inflammatory cytokines. Thus, MDMs from smoker CHD patients presented the lowest expression of PPARγ and the highest spontaneous release of TNF-α and IL-6. On the contrary, PPARγ expression in MDMs from the ex-smoker CHD group was significantly higher than in non-smokers and smokers, and this enhanced PPARγ protein expression was accompanied by a significantly decreased spontaneous release of TNF-α and IL-6, with no major variation in the release of the anti-inflammatory IL-10.
Numerous studies have highlighted the role of TNF-α and IL-6 as relevant markers of cardiovascular risk; increased plasma concentrations of both cytokines have been repeatedly found in CHD and levels of pro-inflammatory cytokines correlated with the severity of coronary artery occlusion (Ridker et al., 2000; Blake and Ridker, 2002; Skoog et al., 2002). The role of PPARγ in the regulation of cytokine secretion was further confirmed by our results in monocytes: in this cell type, PPARγ expression was similar among the three CHD groups, no major changes occurring in basal cytokine release. Moreover, in both monocytes and MDMs from selected CHD patients, rosiglitazone dose dependently inhibited PMA-evoked TNF-α release, with IC50 values in the nM range. The selective PPARγ antagonist GW9662 reversed this effect, demonstrating that the effects of rosiglitazone were PPARγ mediated and corroborating the hypothesis that cytokine release is under PPARγ control. However, GW9662 was less effective in cells (especially, MDMs) from CHD smokers. We have no definite explanation for this fact, but we can suggest some possible reasons. First, cytokine release is controlled by different signal pathways and mediators, including NF-κB which, as demonstrated here, is constitutively enhanced in cells from CHD smokers. Second, the lower PPARγ expression in MDMs from CHD smokers could be associated with a lower PPARγ activity. Moreover, when we evaluated NF-κB regulation by PPARγ ligands, we also observed a reduced effect of GW9662 in cells from CHD smokers, as compared to non-smokers.
Although the mechanisms for PPARγ-mediated down-regulation of pro-inflammatory cytokine production are not fully elucidated, experimental evidence supports a transrepressive mechanism in which transcription factors such as NF-κB, AP-1 and NF-AT are inhibited by binding of activated PPARγ. In keeping with previous results (Ricote et al., 1998; Lee et al., 2006; Caito et al., 2008), we showed here that rosiglitazone inhibited the nuclear translocation of NF-κB in PMA-stimulated monocytes and MDMs from both CHD smokers and non-smokers, and that GW9662 antagonized this effect, despite a lesser effect in cells from CHD smokers.
Tobacco smoke by itself profoundly affects NF-κB activation, as previously shown by us (Bardelli et al., 2005; Gunella et al., 2006) and others (Karimi et al., 2006; Caito et al., 2008). In this paper, we found a constitutively enhanced NF-κB translocation in cells from CHD smokers (as compared to non-smokers), which was further stimulated by LPS or PMA. This is in agreement with recent findings by Caito et al. (2008), who demonstrated that cigarette smoke extract, in MonoMac6 cells (a human monocyte/macrophage cell line), activated NF-κB, decreased the level of nuclear PPARγ protein and disrupted the association between PPARγ and p65 subunit of NF-κB. Moreover, PPARγ activation by natural or synthetic ligands resulted in differential inhibitory effects on cigarette smoke-mediated cytokine release (Caito et al., 2008).
Different research groups using PPARγ ligands have provided a range of results, depending on the PPARγ agonist used, the different concentrations used, the single cell type and/or the nature of the cell stimulus. In the majority of in vitro data, the agonist concentration represents a relevant issue and needs to be carefully considered. Concentrations of rosiglitazone >1 µM are not systemically achieved after daily treatment with 8 mg of this TZD (Cox et al., 2000), and the plasma concentration of rosiglitazone falls below 20 nM, 12 h after 2 mg rosiglitazone (Thompson et al., 2007). In our hands, rosiglitazone inhibited PMA-induced TNF-α release in monocytes and MDMs from smoker and non-smoker CHD patients, with IC50 values (between 9 and 33 nM) well within its therapeutic plasma concentrations, and with a major effect in non-smoker patients.
Therefore, our results further confirmed PPARγ as an important regulator of cytokine secretion (Jiang et al., 1998; Ricote et al., 1998; Amoruso et al., 2007) and provided ex vivo evidence that the level of expression of PPARγ regulated the spontaneous release of pro-inflammatory cytokines.
Considering the role of inflammatory mediators in CHD (Ridker et al., 2000; Blake and Ridker, 2002; Skoog et al., 2002), and that the expression of a given receptor is regulated via a complex network of different mediators, our results demonstrated that: (i) PPARγ protein expression in MDMs from CHD patients was strongly affected by tobacco smoking, smoker and ex-smoker patients presenting the lowest and the highest protein content, respectively; (ii) PPARγ was a potent inhibitory regulator of pro-inflammatory cytokine secretion.
Therefore, in keeping with the current understanding of the pathophysiology of atherosclerosis, and the well-recognized anti-inflammatory and anti-atherogenic properties of PPARγ (Li and Palinski, 2006; Straus and Glass, 2007; Rigamonti et al., 2008; Szanto and Roszer, 2008), we suggest that the reduced PPARγ expression in MDMs from smoker CHD patients, together with the enhanced cytokine release, could participate in the deleterious effects of tobacco smoke. Although the clinical relevance of these findings needs to be more extensively investigated, this paper demonstrates a smoking-related effect on PPARγ expression in MDMs from CHD patients that must be considered when treating patients with PPARγ agonists.
Acknowledgments
This study was supported by Regione Piemonte, Ricerca Sanitaria Finalizzata 2006 (N. 1211), and PRIN, Progetti di Ricerca di interesse nazionale (N. 2004065227) grants. We dedicate this paper to Professor Enrico Genazzani to remember our last friendly conversation.
Glossary
Abbreviations:
- CHD
coronary heart disease
- EMSA
electrophoretic mobility shift assay
- GW 9662
2-chloro-5-nitro-N-phenylbenzamide
- IL-6
interleukin-6
- IL-10
interleukin-10
- LPS
lipopolysaccharide
- M4d
partially differentiated macrophage
- MDM
monocyte-derived macrophage
- NF-κB
nuclear factor-kappaB
- PMA
phorbol-12-myristate 13-acetate
- PPARγ
peroxisome proliferator-activated receptor-γ
- TNF-α
tumour necrosis factor-α
- TZD
thiazolidinedione
Conflict of interest
The authors state no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoruso A, Bardelli C, Gunella G, Fresu LG, Ferrero V, Brunelleschi S. Quantification of PPARgamma protein in monocyte/macrophages from healthy smokers and non-smokers: a possible direct effect of nicotine. Life Sci. 2007;81:906–915. doi: 10.1016/j.lfs.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Amoruso A, Bardelli C, Gunella G, Ribichini F, Brunelleschi S. A novel activity for substance P: stimulation of peroxisome proliferator-activated receptor-γ protein expression in human monocytes and macrophages. Br J Pharmacol. 2008;154:144–152. doi: 10.1038/bjp.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardelli C, Gunella G, Varsaldi F, Balbo P, Del Boca E, Bernardone IS, et al. Expression of functional NK1 receptors in human alveolar macrophages: superoxide anion production, cytokine release and involvement of NF-κB pathway. Br J Pharmacol. 2005;145:385–396. doi: 10.1038/sj.bjp.0706198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL. Clinical pharmacology of nicotine: implications for understanding, preventing, and treating tobacco addiction. Clin Pharmacol Ther. 2008;83:531–541. doi: 10.1038/clpt.2008.3. [DOI] [PubMed] [Google Scholar]
- Blake GJ, Ridker PM. Inflammatory biomarkers and cardiovascular risk prediction. J Intern Med. 2002;252:283–294. doi: 10.1046/j.1365-2796.2002.01019.x. [DOI] [PubMed] [Google Scholar]
- Brown DW, Mensah GA. Smoking among adults with coronary heart disease. Prev Med. 2007;44:85–86. doi: 10.1016/j.ypmed.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Caito S, Yang SR, Kode A, Edirisinghe I, Rajendrasozhan A, Phipps RP, et al. Rosiglitazone and 15-deoxy-delta12,14-prostaglandin J2, PPARγ agonists, differentially regulate cigarette smoke-mediated pro-inflammatory cytokine release in monocyte/macrophages. Antioxid Redox Signal. 2008;10:253–260. doi: 10.1089/ars.2007.1889. [DOI] [PubMed] [Google Scholar]
- Chiolero A, Faeh D, Paccaud F, Cornuz J. Consequences of smoking for body weight, body fat distribution and insulin resistance. Am J Clin Nutr. 2008;87:801–809. doi: 10.1093/ajcn/87.4.801. [DOI] [PubMed] [Google Scholar]
- Cox PJ, Ryan DA, Hollis FJ, Harris AM, Miller AK, Vousden M, et al. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos. 2000;28:772–780. [PubMed] [Google Scholar]
- Gunella G, Bardelli C, Amoruso A, Viano I, Balbo P, Brunelleschi S. Macrophage stimulating protein (MSP) differently affects human alveolar macrophages from smoker and non-smoker patients: evaluation of respiratory burst, cytokine release and NF-κB pathway. Br J Pharmacol. 2006;148:478–489. doi: 10.1038/sj.bjp.0706751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPARγ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Karimi K, Sarir H, Mortaz E, Smit JJ, Hosseini H, De Kimpe SJ, et al. Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir Res. 2006;7:66. doi: 10.1186/1465-9921-7-66. doi: 10.1186/1465-9921-7-66 PMID: 16620395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavagno L, Gunella G, Bardelli C, Spina S, Fresu LG, Viano I, et al. Anti-inflammatory drugs and tumour necrosis factor-alpha production from monocytes: role of transcription factor NF-kappaB and implication for rheumatoid arthritis therapy. Eur J Pharmacol. 2004;501:199–208. doi: 10.1016/j.ejphar.2004.07.101. [DOI] [PubMed] [Google Scholar]
- Lee SY, Kang EJ, Hur GY, Jung KH, Jung CH, Lee SY, et al. Peroxisome proliferator-activated receptor gamma inhibits cigarette-smoke solution-induced mucin production in human airway epithelial (NCI-H292) cells. Am J Physiol. 2006;291:L84–90. doi: 10.1152/ajplung.00388.2005. [DOI] [PubMed] [Google Scholar]
- Li AC, Palinski W. Peroxisome proliferator-activated receptors: how their effects on macrophages can lead to the development of a new drug therapy against atherosclerosis. Annu Rev Pharmacol Toxicol. 2006;46:1–39. doi: 10.1146/annurev.pharmtox.46.120604.141247. [DOI] [PubMed] [Google Scholar]
- Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular risk. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Stamfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–1772. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPARα, PPARγ, and LXRs in mice and men. Arterioscler Thromb Vasc Biol. 2008;28:1050–1059. doi: 10.1161/ATVBAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- Skoog T, Dichtl W, Skoglund-Andersson C, Karpe F, Tang R, Bond MG, et al. Plasma tumour necrosis factor-α and early carotid atherosclerosis in healthy middle-aged men. Eur Heart J. 2002;23:376–383. doi: 10.1053/euhj.2001.2805. [DOI] [PubMed] [Google Scholar]
- Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Szanto A, Roszer T. Nuclear receptors in macrophages: a link between metabolism and inflammation. FEBS Lett. 2008;582:106–116. doi: 10.1016/j.febslet.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Taylor BV, Oudit GY, Kaalman PG, Liu P. Clinical and pathophysiological effects of active and passive smoking on cardiovascular system. Can J Cardiol. 1998;14:1129–1139. [PubMed] [Google Scholar]
- Thompson PW, Bayliffe AI, Warren AP, Lamb JR. Interleukin-10 is upregulated by nanomolar rosiglitazone treatment of mature dendritic cells and human CD4 + T cells. Cytokine. 2007;39:184–191. doi: 10.1016/j.cyto.2007.07.191. [DOI] [PubMed] [Google Scholar]