

Abstract
Background and purpose:
Pulmonary arterial hypertension (PAH) is associated with increased contraction and proliferation of pulmonary vascular smooth muscle cells. The anti-diabetic drug metformin has been shown to have relaxant and anti-proliferation properties. We thus examined the effect of metformin in PAH.
Experimental approach:
Metformin effects were analysed in hypoxia- and monocrotaline-induced PAH in rats. Ex vivo and in vitro analyses were performed in lungs, pulmonary artery rings and cells.
Key results:
In hypoxia- and monocrotaline-induced PAH, the changes in mean pulmonary arterial pressure and right heart hypertrophy were nearly normalized by metformin treatment (100 mg·kg−1·day−1). Pulmonary arterial remodelling occurring in both experimental models of PAH was also inhibited by metformin treatment. In rats with monocrotaline-induced PAH, treatment with metformin significantly increased survival. Metformin increased endothelial nitric oxide synthase phosphorylation and decreased Rho kinase activity in pulmonary artery from rats with PAH. These effects are associated with an improvement of carbachol-induced relaxation and reduction of phenylephrine-induced contraction of pulmonary artery. In addition, metformin inhibited mitogen-activated protein kinase activation and strongly reduced pulmonary arterial cell proliferation during PAH. In vitro, metformin directly inhibited pulmonary artery smooth muscle cell growth.
Conclusions and implications:
Metformin protected against PAH, regardless of the initiating stimulus. This protective effect may be related to its anti-remodelling property involving improvement of endothelial function, vasodilatory and anti-proliferative actions. As metformin is currently prescribed to treat diabetic patients, assessment of its use as a therapy against PAH in humans should be easier.
Keywords: pulmonary artery, smooth muscle, endothelium, contraction, proliferation, biguanide, AMP kinase
Introduction
Pulmonary arterial hypertension (PAH) is a rare disease characterized by a progressive increase of pulmonary vascular resistance and pulmonary arterial pressure (PAP), leading to right ventricle (RV) hypertrophy and death (Rubin, 2006; Humbert, 2008). Although major advances in the understanding of disease development and treatment have been achieved over the last 15 years, the pathogenesis of PAH remains not clearly understood. Idiopathic, familial and connective tissue disease-associated forms of PAH display similar pathological changes and are considered to share common pathogenic mechanisms, which involve endothelial dysfunction, endothelial and smooth muscle cell proliferation and increased vasoconstriction (Humbert, 2008). Accordingly, current therapies are based on the use of drugs that improve endothelial function. Pharmacological agents targeting the endothelin-1 (receptor antagonists such as bosentan or sitaxsentan), the nitric oxide (NO) (sildenafil, type 5 phosphodiesterase inhibitor) or the prostacyclin (epoprostenol, iloprost) pathways have shown benefits for patients with PAH. However, these treatments failed to improve the long-term survival and their use is hampered by either side effects or inconvenient drug administration routes (Humbert et al., 2004).
The biguanide drug metformin has been widely used for more than 40 years for the treatment of hyperglycemia in patients with type 2 diabetes mellitus but has been recently described as a pleiotropic molecule. Besides reducing hyperglycemia by improving peripheral sensitivity to insulin, reducing gastrointestinal glucose absorption and decreasing hepatic glucose production (Caspary and Creutzfeldt, 1971; Hundal et al. 2000; Borst and Snellen, 2001), metformin has recently been shown to lower other cardiovascular risk factors (McAlister et al., 2008). Moreover, metformin seems to improve endothelial function. Indeed, metformin restores the microvascular reactivity to histamine, bradykinin or acetylcholine, of arterioles and venules from diabetic animal models (Sartoretto et al., 2005). More recently, it has been shown that it improves the flow-mediated dilatation of normoinsulinemic subjects (Romualdi et al., 2008). In addition, metformin has been shown to have antiproliferative actions, blocking the cell cycle progression in G(0)/G(1) phase (Ben Sahra et al., 2008; Zhuang and Miskimins, 2008).
On the basis of these observations, we hypothesized that metformin, already used clinically for diabetes, would decrease PAH, facilitating its translation to use in patients with PAH. To assess this hypothesis, we investigated the effects of metformin on haemodynamics and pulmonary vasculature remodelling in established models of experimental PAH.
Methods
Animals
All animal care and experimental procedures were conducted in accordance with international guidelines for the care and use of laboratory animals. Male Wistar rats (250 g) were used. The normoxic rats were housed in room air at normal atmospheric pressure. Chronic hypoxic PAH was obtained by housing animals in a hypobaric chamber at 380 mmHg (Vacucell 111L, Medcenter, Munich, Germany) for 21 days. Metformin (Calbiochem) was administered by daily intraperitoneal (IP) injections at a dose of 100 mg·kg−1·day−1, starting either at the first or at the 14th day of the 21 days of hypoxia. Four groups of rats were used: normoxic rats (control group), normoxic rats receiving metformin, hypoxic rats receiving saline and hypoxic rats treated by metformin.
For monocrotaline (MCT)-induced pulmonary hypertension, rats received a subcutaneous injection of saline (controls) or MCT (Sigma, Saint Quentin Fallavier, France; 60 mg·kg−1·rat−1) to induce PAH. Experimental groups were further divided in untreated rats (receiving saline), and treated rats that received daily IP injections of metformin (100 mg·kg−1·day−1), starting at the day of MCT injection (day 0). Rats were examined at day 30. Survival curves were established from day 0 to day 55.
Cardiac ultrasonography
Cardiac ultrasonography (SONOS 5500 echocardiograph, Philips, 12-MHz sector scare transducer) was performed by the same operator on anesthetized rats, placed in the left lateral decubitus position after thorax epilation. The wall thickness of the RV was measured. The peak tricuspid regurgitation velocity was measured by continuous wave. Pulmonary artery flow was recorded by pulsed-wave Doppler echocardiography and the acceleration time of pulmonary artery blood flow was measured from the onset of ejection to the time of peak velocity.
Haemodynamic measures
Haemodynamic measures were made as previously described (Guilluy et al., 2005). Rats were anaesthesized by IP injection of ketamine and xylazine. Haemodynamic parameters were measured using a venous catheter inserted in the right jugular vein. The catheter was introduced in the right atrium, the RV and then in the pulmonary artery. Systolic RV pressure, systolic, diastolic and mean PAP were then measured (Hewlett-Packard, M1106B).
Measurement of right ventricular hypertrophy
After death, the heart was removed and the blood washed out. Right ventricular hypertrophy was measured using the ratio of RV weight to left ventricle (LV) plus interventricular septum weight [RV/(LV + S)].
Histological analysis
Histological examination of pulmonary vascular remodelling was performed as previously described (Guilluy et al., 2005). The left lung was removed. Left bronchus was slowly injected with 4% paraformaldehyde in PBS to distend air-cells and the entire piece was put in the same fixation solution to be processed for paraffin-embedded sections (10 µm). Three transversal sections per lung were stained with haematoxylin-eosin-saffron and Weigert's staining. A total of 15–30 arterial vessels (20 to 60 µm in diameter) were examined in each section. The wall thickness was quantified by measuring the area using Metaview software (Universal Imaging Co., West Chester, PA, USA). The ratio area of the lumen (LA) to the area of the entire vessel (arterial wall plus lumen; VA) ×100 was calculated. The medial area (MA, in %) was calculated using the formula MA = 100 – LA/VA × 100. Muscularity of distal pulmonary arteries was assessed as previously described (Chassagne et al., 2000) and expressed as a percentage of the total number of arteries examined (30–60 in each condition). Proliferation was assessed using immunohistochemistry on lung sections stained with anti-proliferating cell nuclear antigen (PCNA) antibody (Dako, Glostrup, Denmark). All these analyses were performed by one observer unaware of the group from which the sections were taken.
Western blot analysis
Pulmonary arteries (extralobar, main and first branches) were harvested and put in NETF buffer (100 mM NaCl, 2 mM EGTA, 50 mM Tris-Cl, pH 7.4 and 50 mM NaF) containing 1% NP-40, 2 mM orthovanadate, protease and phosphatase inhibitor cocktails (Sigma). Lysates were obtained using Polytron material and then sonicated. Protein concentration of each sample was determined and equal amounts of protein were loaded in each lane of polyacrylamide/SDS gels. After electrophoresis and transfer to nitrocellulose membrane, samples were analysed by Western blot with antibodies against acetyl CoA carboxylase (ACC) and phospho-ACC (Cell Signaling, Cell Signaling Technology, Inc., Danvers, MA, USA), Endothelial NO synthase (eNOS) and phospho-eNOS (Cell Signaling), RhoA (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-ERK (Santa Cruz), phospho-JNK (Cell Signaling), phospho-p38 (Cell Signaling) and PCNA. Rho kinase activity was quantified by Western blot analysis for phosphorylated myosin phosphatase target subunit (MYPT) using a rabbit polyclonal anti-phospho-MYPT (Thr696) (Santa Cruz). Equal loading was confirmed by reprobing the membrane with anti-β-actin antibody (Sigma, Saint Quentin Fallavier, France). Immunoreactive bands were visualized using horseradish peroxidase-conjugated secondary antibody and subsequent ECL kit detection (Amersham Pharmacia Biotech, Orsay, France). Quantification was made by densitometric analysis with QuantyOne (Biorad, Hercules, CA, USA).
Proliferation
Smooth muscle cells from rat pulmonary artery explants were cultured in DMEM with 10% foetal calf serum, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin. Secondary cultures were obtained by serial passages after the cells were harvested with 0.5 g·L−1 trypsin and 0.2 g·L−1 EDTA and reseeded in fresh medium. After dissociation, cells at passage 2 were seeded on a six-well plate, washed and maintained in serum-free medium for 24 h, and then stimulated by platelet-derived growth factor (PDGF) (Peprotech France, Levallois Perret) during 24 h, with or without 4 mM metformin. Proliferation was assessed using both Western blot on cell lysates with antibody against PCNA and bromodeoxyuridine (BrdU) proliferation assay.
Contraction experiments
Pulmonary arteries were collected in physiological saline solution (in mM; 130 NaCl, 5.6 KCl, 1 MgCl2, 2 CaCl2, 11 glucose, 10 Tris, pH 7.4 with HCl) and cut in rings. Rings of pulmonary arteries were suspended under isometric conditions, connected to a force transducer (Pioden controls Ltd, Canterbury, UK) and set up at a tension of 300–500 mg in Krebs-Henseleit solution at 37°C bubbled with 95% O2-5% CO2. After equilibration, the response to KCl 60 mM was determined. Endothelial-dependent relaxation was tested by adding increasing concentrations of carbachol to rings pre-contracted by phenylephrine (1 µM). Concentration-response curves to phenylephrine were obtained by measuring the amplitude of the contractile responses to increasing phenylephrine concentrations. For the analysis of the ex vivo effect of metformin, pulmonary arterial rings from normoxic rats were treated by metformin (4 mM, 2.5 h) and contraction measurements were performed in the continuous presence of 4 mM metformin. Amplitude of the phenylephrine-induced contraction was expressed in mg per mg of tissue (mg·mg−1).
Statistical analysis
Values are expressed as mean ± SEM. In experiments with comparison of two conditions, a non-paired Student's t-test was used. Differences among multiple groups were tested with anova (one-way anova, Fisher's test, Holm-Sidak method). P < 0.05 was considered significant.
Materials
Ketamine and xylazine were from Merial (Lyon, France). All other products were from Sigma.
Results
Beneficial effect of metformin on PAH
Rats maintained in a hypobaric chamber for 21 days displayed an increased hematocrit (66.0 ± 1.4% vs. 45.4 ± 1.9 in controls, n= 10, P < 0.001), attesting to the hypoxic condition. The rats exposed to chronic hypoxia developed PAH characterized by an increase in mean PAP (P < 0.001), thickening of the RV wall (P < 0.001) and decrease of the pulmonary flow acceleration time (P < 0.001) (Figure 1A–C). Right ventricular remodelling in hypoxic rats was also demonstrated by the marked increase in the ratio of RV weight to LV plus septum [RV/(LV + S)] (P < 0.001) (Figure 1D). Metformin treatment (100 mg·kg−1·day−1) applied daily for the entire duration of hypoxia exposure almost completely prevented PAH. Mean PAP, RV wall thickness and the RV/(LV + S) ratio remained all to near normal levels (Figure 1A, B and D), and the pulmonary flow acceleration time was partially normalized (Figure 1C). The protective action of metformin in hypoxic rats depended of the dose used as shown by the gradual increase in the effect of metformin concentrations ranging from 0.1 to 100 mg·kg−1·day−1 on mean PAP and RV/(LV + S) (Figure 1E and F).
Figure 1.
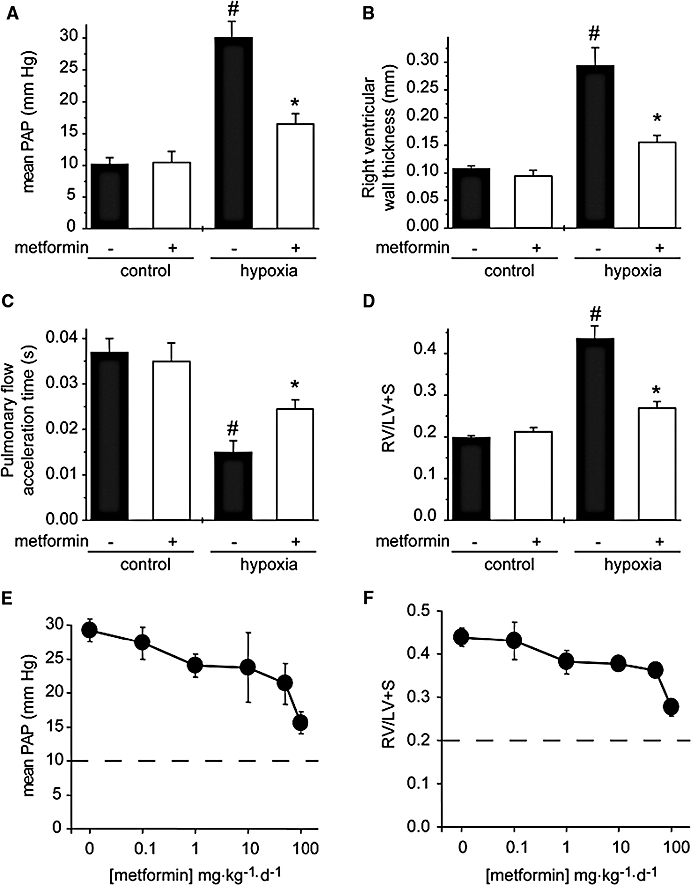
Metformin prevents chronic hypoxia-induced PAH. (A) Mean PAP, (B) right ventricular wall thickness, (C) pulmonary artery flow acceleration and (D) [RV/(LV + S)] ratio determined in control rats (normoxia), rats chronically treated for 21 days with metformin (100 mg·kg−1·day−1), rats exposed to hypoxia for 21 days, and metformin-treated rats exposed to hypoxia. (E) Mean PAP and (F) [RV/(LV + S)] ratio determined in rats exposed to hypoxia for 21 days non-treated (0) or treated with metformin doses ranging from 0.1 to 100 mg·kg−1·day−1. Dotted lines indicated the control values in normoxic rats (#P < 0.001 vs. control, *P < 0.001 vs. untreated, n= 5–16). LV, left ventricle; PAH, pulmonary arterial hypertension; PAP, pulmonary arterial pressure; RV, right ventricle.
To further define the effect of metformin in the progression of PAH in hypoxic rats, we also assessed the effect of metformin treatment only during the last 7 days of the 21 days of hypoxia. Mean PAP, RV wall thickness, the RV/(LV + S) ratio and the pulmonary flow acceleration time were all significantly reduced by metformin administration (Figure 2).
Figure 2.
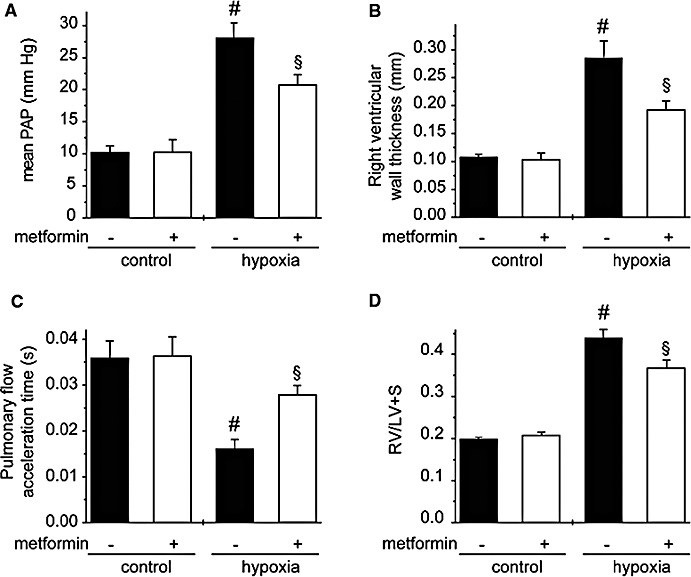
Metformin limits progression of PAH in hypoxic rats. (A) Mean PAP, (B) right ventricular wall thickness, (C) pulmonary artery flow acceleration and (D) [RV/(LV + S)] ratio determined in control rats (normoxia), rats treated for 7 days with metformin (100 mg·kg−1·day−1), rats exposed to hypoxia for 21 days (hypoxia) non-treated and treated with metformin for the last 7 days of hypoxia (#P < 0.001 vs. control, §P < 0.05 vs. untreated, n= 5–9). LV, left ventricle; PAH, pulmonary arterial hypertension; PAP, pulmonary arterial pressure; RV, right ventricle.
The rats challenged with MCT developed severe PAH characterized by an elevated mean PAP (Figure 3A) and an increase in the RV/(LV + S) ratio (Figure 3B). MCT-induced PAH is associated with a low survival rate (30% at day 30, Figure 3C). Metformin treatment improved all the parameters of MCT-induced PAH with a significant decrease of mean PAP at day 30 (Figure 3A), decrease of the RV/(LV + S) ratio (Figure 3B) and a strong improvement of the survival rate to 61% at day 30 (Figure 3C).
Figure 3.
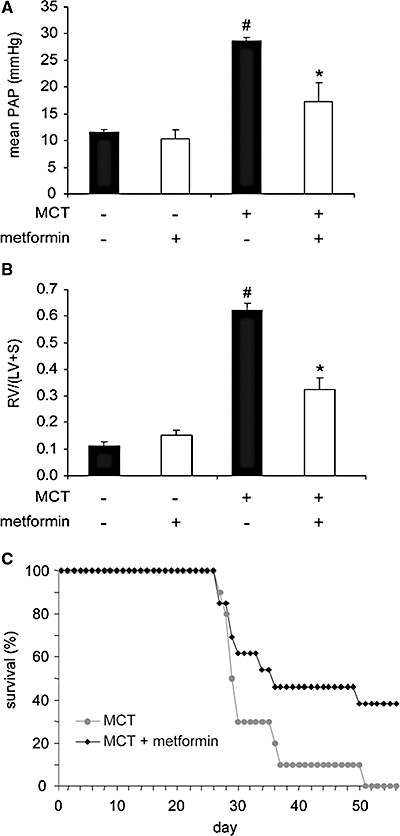
Metformin prevents MCT-induced PAH. (A) Mean PAP and, (B) (RV/LV + S) ratio determined in control rats, rats chronically treated for 30 days with metformin (100 mg·kg−1·day−1), MCT-injected rats at day 30 post MCT injection, and MCT-injected rats at day 30 post MCT injection treated for 30 days by metformin (#P < 0.001 vs. control, *P < 0.001 vs. untreated MCT-injected rats, n= 5–10). (C) Survival rates of metformin-treated MCT-injected rats (grey) versus untreated MCT-injected rats. LV, left ventricle; MCT, monocrotaline; PAH, pulmonary arterial hypertension; RV, right ventricle.
Anti-remodelling effect of metformin in the pulmonary vasculature
Histological examination of the pulmonary vasculature showed that medial wall thickness of small pulmonary arteries (20–60 µm in diameter) and distal muscularization were markedly increased in hypoxic rats at day 21 (Figure 4). Treatment with metformin resulted in a normalization of pulmonary arterial wall thickness and muscularization (Figure 4).
Figure 4.
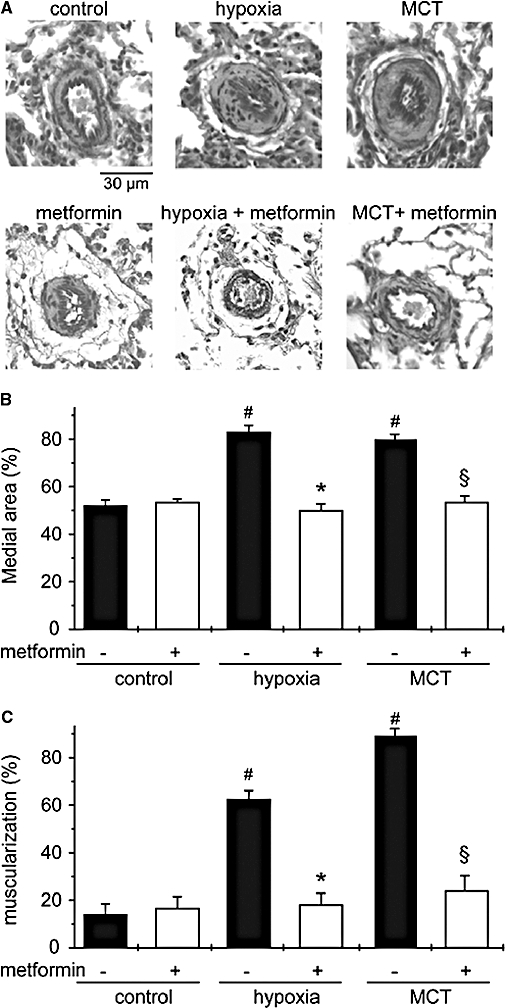
Metformin prevents PAH-associated pulmonary arterial wall remodelling. Representative sections of lung tissue (A), quantification of the relative thickness of small pulmonary artery (20–60 µm) wall (B) and percentage of distal muscularization of normally non-muscular arteries (C) in samples from control, hypoxic (21 days) and MCT-injected rats (day 30), untreated and treated by metformin (#P < 0.001 vs. control, *P < 0.001 vs. untreated hypoxic rats, §P < 0.001 vs. untreated MCT-injected rats). MCT, monocrotaline; PAH, pulmonary arterial hypertension.
Similarly, lung specimens from MCT-treated rats (30 days) displayed severe thickening and muscularization of small artery wall and metformin treatment also significantly reduced pulmonary arterial remodelling in MCT-treated rats (Figure 4). The progressive arterial wall remodelling occurring in PAH resulted from both pulmonary arterial cell proliferation and excessive vasoconstriction. We thus assessed the effect of metformin on these two different processes.
Metformin reduces pulmonary artery contraction and improves endothelial function
To analyse potential effect of metformin on contractile properties of pulmonary artery, we analysed by Western blot, expression and activity of markers of endothelial function and arterial contraction in lysates of pulmonary artery from control and hypoxic rats, treated or not by metformin. As metformin has been shown to stimulate AMP kinase (AMPK) activity (Zhou et al., 2001), the efficiency of metformin treatment was first checked by monitoring the level of phosphorylation of ACC, a representative downstream target of AMPK. The increased ACC phosphorylation observed in pulmonary arteries from hypoxic rats treated with metformin showed the efficiency of the treatment (Figure 5A).
Figure 5.
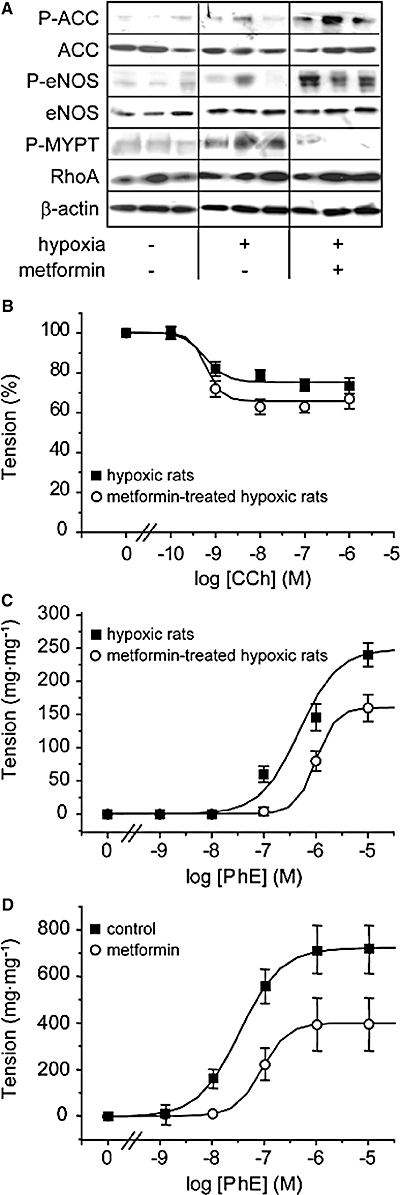
Metformin improves endothelial function and reduces pulmonary artery contractility. (A) ACC phosphorylation and expression, eNOS phosphorylation and expression, Rho kinase activity (assessed by the extent of phosphorylation of MYPT) and RhoA expression have been analysed by Western blot in pulmonary artery from control rats and rats exposed to hypoxia for 21 days, untreated and treated with metformin (three different samples representative of each condition). Equal protein loading was checked by examination of β-actin expression (B) Cumulative concentration-response curves for carbachol (CCh)-induced relaxation of phenylephrine (PhE; 1 µM)-contracted pulmonary artery rings from hypoxic (21 days) and metformin-treated hypoxic rats. Tension is expressed as percentage of the amplitude of the phenylephrine-induced contraction. (C) Cumulative concentration-response curves for the contraction induced by phenylephrine in pulmonary artery rings from hypoxic (21 days) and metformin-treated hypoxic rats. (D) Cumulative concentration-response curves for the contraction induced by phenylephrine in pulmonary artery rings from normoxic rats under control condition and in the presence of metformin (4 mM). ACC, acetyl CoA carboxylase; CCh, carbachol; eNOS, endothelial nitric oxide synthase; MYPT, myosin phosphatase target subunit.
Phosphorylation of eNOS was used as a marker of endothelial function and phosphorylation of MYPT as a marker of RhoA/Rho kinase activation and arterial smooth muscle cell contraction (Figure 5A). Metformin treatment strongly increased the level of eNOS phosphorylation in hypoxic rat (6.3 ± 1.2-fold over hypoxic rats, n= 8, P < 0.001) (Figure 5A). This effect was associated with a marked decrease in hypoxia-induced MYPT phosphorylation (4.8 ± 0.9-fold over control in hypoxic rats vs. 0.8 ± 0.7-fold over control in metformin treated rats, n= 8, P < 0.001), suggesting that metformin decreased RhoA/Rho kinase activity, without change of RhoA expression (Figure 5A). These data therefore suggested that metformin treatment might limit pulmonary artery vasoconstriction in hypoxic rats. To test this suggestion, we then assessed the contractile properties of pulmonary artery rings from untreated and metformin-treated hypoxic rats (Figure 5B). Endothelial NO releasing capacity of the endothelium was assessed by measuring carbachol-induced relaxation of pulmonary artery rings contracted by phenylephrine (1 µM). As shown in Figure 5B, carbachol-induced relaxation was increased in metformin-treated hypoxic rat, indicating that metformin limited hypoxia-induced endothelial dysfunction. This observation is in agreement with the increased phosphorylation of eNOS observed in pulmonary arteries form metformin-treated hypoxic rats (Figure 5A). Cumulative contraction-response curve to phenylephrine further showed that contractile responses were significantly reduced in pulmonary artery rings from metformin-treated hypoxic rats (Figure 5C), in agreement with the reduced RhoA/Rho kinase activation observed in pulmonary arteries from metformin-treated hypoxic rats (Figure 5A). This inhibitory effect of metformin on pulmonary artery contraction was also observed in control pulmonary arteries, treated ex vivo by metformin (Figure 5D). The maximal amplitude of phenylephrine-induced contraction was reduced by 50% in pulmonary arterial rings treated by metformin (4 mM, 2.5 h). Taken together, these results provide evidence that metformin-induced inhibition of pulmonary vasoconstriction and improvement of endothelial function could participate in its beneficial effect on PAH.
Metformin inhibits pulmonary artery cell proliferation
In lung sections from hypoxic rats, PCNA labelling showed cell proliferation in pulmonary arteries, compared with control (Figure 6A) and manifested as a strong increase in the number of PCNA positive pulmonary artery sections (Figure 6A). In parallel to normalization of vessel morphology, the number of PCNA positive vessels was restored to basal levels in hypoxic rats treated with metformin (Figure 6A). To get further insights into the antiproliferative properties of metformin, we next examined, by Western blot, the activation of the main members of the three major subgroups of the mitogen-activated protein kinase family, namely extracellular regulated kinase (ERK), p38 and C-Jun NH2-terminal kinase (JNK), in pulmonary artery lysates. As previously described (Welsh et al., 2001), hypoxia induced activation of ERK (3.3 ± 0.8-fold over control, n= 8), a strong increase in p38 activity (5.6 ± 1.1-fold over control, n= 8) and no significant change in JNK activity (Figure 6B). Hypoxia-induced activation of ERK and p38 was strongly reduced in metformin-treated rats (1.1 ± 0.5 and 3.3 ± 0.7-fold over control, respectively, n= 8, P < 0.001) (Figure 6B). To analyse whether metformin directly affected pulmonary artery smooth muscle cell proliferation, we next analysed in vitro rat pulmonary artery smooth muscle cell proliferation induced by PDGF. PDGF (20 ng·mL−1) induced pulmonary artery smooth muscle cell proliferation, as shown by Western blot, by a threefold increase in PCNA expression (Figure 7A) and the increased number of BrDU positive cells (Figure 7A). Both PCNA expression and the number of BrdU positive cells were strongly reduced by metformin (4 mM), indicating that metformin directly inhibited pulmonary artery smooth muscle cell proliferation (Figure 7A and B). This anti-proliferative action of metformin could thus partly account for its beneficial effect on pulmonary artery remodelling and PAH.
Figure 6.
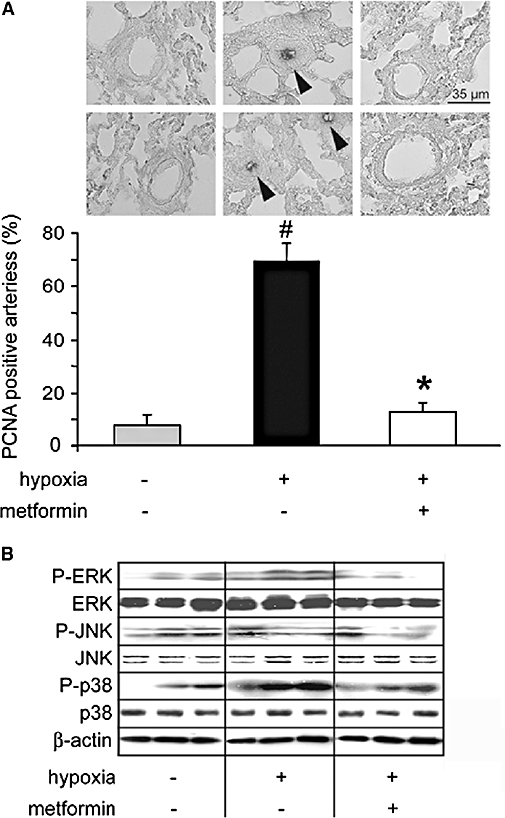
Metformin decreased pulmonary arterial cell proliferation associated with hypoxic PAH. (A) Representative PCNA staining and quantification of the percentage of PCNA-positive pulmonary arteries in lungs from normoxic rats (normoxia), hypoxic (21 days) and metformin-treated hypoxic rats (#P < 0.001 vs. control, *P < 0.001 vs. untreated). (B) ERK, p38 and JNK phosphorylation and expression analysed by Western blot in pulmonary arteries from control rats, rats exposed to hypoxia for 21 days untreated and treated with metformin (three different samples representative of each condition). β-actin amounts were also assessed in each sample. JNK, C-Jun NH2-terminal kinase; PAH, pulmonary arterial hypertension; PCNA, proliferating cell nuclear antigen.
Figure 7.
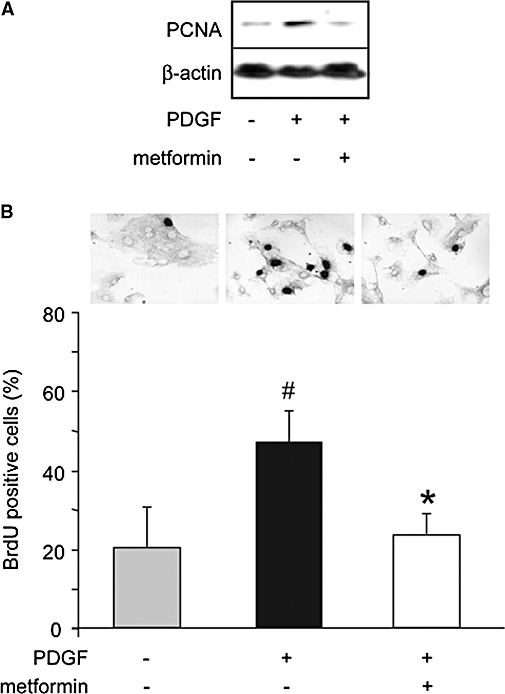
Metformin inhibits PDGF-induced pulmonary artery smooth muscle cell proliferation. (A) Analysis by Western blot of the expression of PCNA in rat pulmonary artery smooth muscle cells, cultured under basal conditions or stimulated by PDGF (20 ng·mL−1) for 24h, without or with metformin (4 mM). (B) Typical BrdU labelling of rat pulmonary artery smooth muscle cells under basal condition and stimulated by PDGF (20 ng·mL−1) for 24h, without or with metformin (4 mM). Quantification of proliferation was expressed as the percentage of BrdU positive pulmonary arterial smooth muscle cells (#P < 0.001 vs. control, *P < 0.001 vs. PDGF alone). BrdU, bromodeoxyuridine; PCNA, proliferating cell nuclear antigen; PDGF, platelet derived growth factor.
Discussion
The novel finding of the present study was the identification of the anti-diabetic drug metformin as an effective therapeutic agent in two well-established models of severe PAH. Metformin improved ultrasonographic parameters used for evaluation of the severity of PAH (free RV wall thickness, pulmonary flow acceleration time) and normalized haemodynamic parameters and RV hypertrophy. We further showed that metformin had an efficient anti-remodelling effect on pulmonary vasculature, improved endothelial function, decreased pulmonary artery contractility and inhibited pulmonary artery cell proliferation.
Several lines of evidence support the protective properties of metformin in the systemic vasculature. In humans, metformin, at therapeutic concentration, is shown to improve vascular endothelial reactivity in type 2 diabetic patients, independently of its anti-hyperglycemic effects (de Aguiar et al., 2006). Furthermore, metformin is described to have anti-hypertensive properties in hypertensive obese women and in patients with type 2 diabetes (Giugliano et al., 1993). These observations therefore suggest that metformin may affect the risk of cardiovascular diseases through mechanisms other than lowering glycemia, possibly by direct vascular effects. In agreement with this hypothesis, metformin acts in vitro as an inhibitor of pro-inflammatory responses in human vascular smooth muscle cells, macrophages and endothelial cells (Hattori et al., 2006; Isoda et al., 2006). In addition, in endothelial cells, metformin possesses antioxidant properties that could participate in its cardiovascular protective effects (Ouslimani et al., 2005). Metformin is also shown to decrease arterial contraction, probably through an intracellular calcium-lowering effect in arterial smooth muscle cells (Dominguez et al., 1996). Metformin reduced arterial pressure in spontaneously hypertensive rats (Bhalla et al., 1996), and rapidly relaxed contractions to adrenoceptor agonists in aortic rings from spontaneously hypertensive rats (Lee and Peuler, 2001). Metformin corrected the altered endothelium-dependent response of resistance arteries to vasodilator agents in diabetic rats (Sartoretto et al., 2005), probably by increasing NOS activity and NO production (Morrow et al., 2003; Davis et al., 2006). Our data demonstrate for the first time the effects of metformin on pulmonary artery contraction and endothelial function, supporting its protective effect in experimental models of PAH. Metformin improved endothelial-dependent vasodilation of pulmonary arteries from hypoxic rats. This effect might result from the increased eNOS phosphorylation and activity, in agreement with previous observations in systemic arteries (Morrow et al., 2003; Davis et al., 2006). In addition, metformin directly reduced pulmonary artery contraction, in association with a reduction of the activation of the RhoA/Rho kinase pathway. This result is therefore in agreement with recent data from pharmacological studies showing that inhibition of Rho kinase reduced the PAP and pulmonary arterial remodelling in rat and mouse models of PAH (Abe et al., 2004; Fagan et al., 2004; Jernigan et al., 2004; Nagaoka et al., 2004; Guilluy et al., 2005; Nagaoka et al., 2005). As found for sildenafil, an inhibitory action of metformin on the RhoA/Rho kinase signalling could thus participate in the beneficial effect of metformin on PAH.
In addition to its action on pulmonary artery vasoreactivity, we also identified anti-proliferative effect of metformin on pulmonary arterial cells. Metformin has been shown to block cell cycle progression in G(0)/G(1) phase (Ben Sahra et al., 2008; Zhuang and Miskimins, 2008) and a potential reduction of cancer risk has been attributed to metformin through a possible anti-proliferative effect (Phoenix et al., 2009). In the cardiovascular system, metformin inhibited leptin-induced human arterial smooth muscle cell proliferation, possibly through the inhibition of reactive oxygen species production (Li et al., 2005). Structural changes observed in animal models of PAH resemble some characteristics of human pulmonary hypertension, particularly the marked medial wall thickening (Jeffery and Wanstall, 2001). In agreement with previous reports, we found a marked increase of arterial cell proliferation in pulmonary arteries of hypoxic rats. Metformin treatment resulted in near normal vessel morphology and reduced the proliferation of pulmonary arterial smooth muscle cells. Inhibition of pulmonary arterial smooth muscle cell proliferation and vascular remodelling is likely a direct effect of metformin because metformin also effectively inhibited proliferation of cultured pulmonary arterial smooth muscle cells. Although not studied in detail here, this anti-proliferative action could result from the observed inhibition of ERK and p38 activation.
An interesting and surprising point is that another antidiabetic drug, rosiglitazone, an agonist at the peroxisome proliferator-activated receptor (PPAR)-γ and a thiazolidinedione compound, has also been recently described as a protective agent against pulmonary hypertension in particular mice models. The insulin-resistant apoE-deficient (apoE-/-) mice on a high-fat diet spontaneously develop PAH (Hansmann et al., 2007). Treatment of apoE-/- mice with rosiglitazone resulted in markedly higher plasma adiponectin, improved insulin sensitivity, and complete regression of PAH, right ventricular hypertrophy and abnormal pulmonary artery muscularization (Hansmann et al., 2007). The potential protective role of PPAR-γ was then further supported by the observation that mice with targeted deletion of PPAR-γ in smooth muscle cells spontaneously developed PAH (Hansmann et al., 2008). In addition, thiazolidinedione treatment was also shown to inhibit MCT- or hypoxia-induced PAH in rats (Matsuda et al., 2005; Crossno et al., 2007). It is unlikely that the effect of metformin described here involved activation of PPAR-γ as, in agreement with previous reports (Gao et al., 2008; Raso et al., 2009), we found down-regulation of PPAR-γ in metformin-treated animals (not shown). However, the most relevant hypothesis for the beneficial effect of both types of antidiabetic drugs against PAH is that both PPAR-γ agonists and metformin may act through converging target(s), and the well-known metabolic fuel gauge AMPK seems to be a good candidate. Indeed, the numerous biological effects of metformin are mediated, at least in part by AMPK, and it is known that PPAR-γ agonists improve insulin sensitivity, at least in part, by increasing the expression and release of adiponectin, an adipokine that activates AMPK. Therefore, both PPAR-γ agonists and metformin could be considered as indirect AMPK activators (Fryer et al., 2002). The increased phosphorylation of the AMPK kinase target, ACC, in pulmonary arteries from hypoxic rats treated with metformin suggests that metformin stimulated the activity of AMPK. The analysis of the role of AMPK in the protective effects of both metformin and PPAR-γ agonists against PAH thus deserves a complete study, including the use of AMPK deficient mice.
In conclusion, we show that treatment with metformin prevented the development of PAH in recognized animal models of the human disease. The demonstrated anti-proliferative and vasodilatory action of metformin on the pulmonary artery may contribute to its beneficial effects. A recent study showing that the prevalence of insulin resistance is higher in female PAH patients than in the general population, suggested that interventions that enhance insulin sensitivity in patients with PAH could be of clinical benefit (Zamanian et al., 2009). Our present study would support this hypothesis. As metformin is currently used to lower glycemia in diabetic patients, use of metformin as a new therapy for PAH might therefore be plausible. Furthermore, the effective doses of metformin used in this study (1–100 mg·kg−1·day−1) are in the range of concentrations used in diabetic patients (0.001–500 mg·kg−1·day−1). Metformin has been widely used for many years, world-wide and its safety profile is well known. The most common side effects are usually self-limiting mild and transient gastrointestinal symptoms and there is no risk of hypoglycaemia in normoglycemic subjects (Caspary and Creutzfeldt, 1971; Borst and Snellen, 2001). The most serious side effect is lactic acidosis that occurs in 0.024–0.084 cases per 1000 patients each year. This side effect has mostly been described in patients overdosed with metformin or with hepatic or renal insufficiency. Metformin is considered to be safe even in patients with heart failure and may be used in such patients, despite current theoretical contraindications (McCormack et al., 2005; Tahrani et al., 2007).
Acknowledgments
This work is supported by grants from INSERM and from the Région Pays de La Loire (Provasc project). Malvyne Rolli-Derkinderen and Eric Dumas de la Roque were supported by CNRS. We thank N. Vaillant for excellent technical assistance and the IBISA platform Cardiex for functional explorations.
Glossary
Abbreviations:
- ACC
acetyl CoA carboxylase
- AMPK
AMP kinase
- BrdU
bromodeoxyuridine
- CCh
carbachol
- JNK
C-Jun NH2-terminal kinase
- LV
left ventricle
- MCT
monocrotaline
- MYPT
myosin phosphatase target subunit
- PAH
pulmonary arterial hypertension
- PAP
pulmonary arterial pressure
- PCNA
proliferating cell nuclear antigen
- PDGF
platelet-derived growth factor
- PPAR
peroxisome proliferator-activated receptor
- RV
right ventricle
References
- Abe K, Shimokawa H, Morikawa K, Uwatoku T, Oi K, Matsumoto Y, et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res. 2004;94:385–393. doi: 10.1161/01.RES.0000111804.34509.94. [DOI] [PubMed] [Google Scholar]
- de Aguiar LG, Bahia LR, Villela N, Laflor C, Sicuro F, Wiernsperger N, et al. Metformin improves endothelial vascular reactivity in first-degree relatives of type 2 diabetic patients with metabolic syndrome and normal glucose tolerance. Diabetes Care. 2006;29:1083–1089. doi: 10.2337/diacare.2951083. [DOI] [PubMed] [Google Scholar]
- Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- Bhalla RC, Toth KF, Tan E, Bhatty RA, Mathias E, Sharma RV. Vascular effects of metformin. Possible mechanisms for its antihypertensive action in the spontaneously hypertensive rat. Am J Hypertens. 1996;9:570–576. doi: 10.1016/0895-7061(95)00356-8. [DOI] [PubMed] [Google Scholar]
- Borst SE, Snellen HG. Metformin, but not exercise training, increases insulin responsiveness in skeletal muscle of Sprague-Dawley rats. Life Sci. 2001;69:1497–1507. doi: 10.1016/s0024-3205(01)01225-5. [DOI] [PubMed] [Google Scholar]
- Caspary WF, Creutzfeldt W. Analysis of the inhibitory effect of biguanides on glucose absorption: inhibition of active sugar transport. Diabetologia. 1971;7:379–385. doi: 10.1007/BF01219474. [DOI] [PubMed] [Google Scholar]
- Chassagne C, Eddahibi S, Adamy C, Rideau D, Marotte F, Dubois-Rande JL, et al. Modulation of angiotensin II receptor expression during development and regression of hypoxic pulmonary hypertension. Am J Respir Cell Mol Biol. 2000;22:323–332. doi: 10.1165/ajrcmb.22.3.3701. [DOI] [PubMed] [Google Scholar]
- Crossno JT, Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, et al. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol. 2007;292:L885–L897. doi: 10.1152/ajplung.00258.2006. [DOI] [PubMed] [Google Scholar]
- Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. doi: 10.2337/diabetes.55.02.06.db05-1064. [DOI] [PubMed] [Google Scholar]
- Dominguez LJ, Davidoff AJ, Srinivas PR, Standley PR, Walsh MF, Sowers JR. Effects of metformin on tyrosine kinase activity, glucose transport, and intracellular calcium in rat vascular smooth muscle. Endocrinology. 1996;137:113–121. doi: 10.1210/endo.137.1.8536601. [DOI] [PubMed] [Google Scholar]
- Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, et al. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L656–L664. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- Gao Y, Xue J, Li X, Jia Y, Hu J. Metformin regulates osteoblast and adipocyte differentiation of rat mesenchymal stem cells. J Pharm Pharmacol. 2008;60:1695–1700. doi: 10.1211/jpp.60/12.0017. [DOI] [PubMed] [Google Scholar]
- Giugliano D, De Rosa N, Di Maro G, Marfella R, Acampora R, Buoninconti R, et al. Metformin improves glucose, lipid metabolism, and reduces blood pressure in hypertensive, obese women. Diabetes Care. 1993;16:1387–1390. doi: 10.2337/diacare.16.10.1387. [DOI] [PubMed] [Google Scholar]
- Guilluy C, Sauzeau V, Rolli-Derkinderen M, Guerin P, Sagan C, Pacaud P, et al. Inhibition of RhoA/Rho kinase pathway is involved in the beneficial effect of sildenafil on pulmonary hypertension. Br J Pharmacol. 2005;146:1010–1018. doi: 10.1038/sj.bjp.0706408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circ. 2007;115:1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–1188. doi: 10.1161/01.HYP.0000221429.94591.72. [DOI] [PubMed] [Google Scholar]
- Humbert M. Update in pulmonary arterial hypertension 2007. Am J Respir Crit Care Med. 2008;177:574–579. doi: 10.1164/rccm.200801-029UP. [DOI] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–1436. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26:611–617. doi: 10.1161/01.ATV.0000201938.78044.75. [DOI] [PubMed] [Google Scholar]
- Jeffery TK, Wanstall JC. Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol Ther. 2001;92:1–20. doi: 10.1016/s0163-7258(01)00157-7. [DOI] [PubMed] [Google Scholar]
- Jernigan NL, Walker BR, Resta TC. Chronic hypoxia augments protein kinase G-mediated Ca2+ desensitization in pulmonary vascular smooth muscle through inhibition of RhoA/Rho kinase signaling. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1220–L1229. doi: 10.1152/ajplung.00196.2004. [DOI] [PubMed] [Google Scholar]
- Lee JM, Peuler JD. A possible indirect sympathomimetic action of metformin in the arterial vessel wall of spontanously hypertensive rats. Life Sci. 2001;69:1085–1092. doi: 10.1016/s0024-3205(01)01202-4. [DOI] [PubMed] [Google Scholar]
- Li L, Mamputu JC, Wiernsperger N, Renier G. Signaling pathways involved in human vascular smooth muscle cell proliferation and matrix metalloproteinase-2 expression induced by leptin: inhibitory effect of metformin. Diabetes. 2005;54:2227–2234. doi: 10.2337/diabetes.54.7.2227. [DOI] [PubMed] [Google Scholar]
- McAlister FA, Eurich DT, Majumdar SR, Johnson JA. The risk of heart failure in patients with type 2 diabetes treated with oral agent monotherapy. Eur J Heart Fail. 2008;10:703–708. doi: 10.1016/j.ejheart.2008.05.013. [DOI] [PubMed] [Google Scholar]
- McCormack J, Johns K, Tildesley H. Metformin's contraindications should be contraindicated. CMAJ. 2005;173:502–504. doi: 10.1503/cmaj.045292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda Y, Hoshikawa Y, Ameshima S, Suzuki S, Okada Y, Tabata T, et al. Effects of peroxisome proliferator-activated receptor gamma ligands on monocrotaline-induced pulmonary hypertension in rats. Nihon Kokyuki Gakkai Zasshi. 2005;43:283–288. [PubMed] [Google Scholar]
- Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278:31629–31639. doi: 10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, et al. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287:L665–L672. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, et al. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:494–499. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]
- Ouslimani N, Peynet J, Bonnefont-Rousselot D, Therond P, Legrand A, Beaudeux JL. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metab. 2005;54:829–834. doi: 10.1016/j.metabol.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Phoenix KN, Vumbaca F, Claffey KP. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERalpha negative MDA-MB-435 breast cancer model. Breast Cancer Res Treat. 2009;113:101–111. doi: 10.1007/s10549-008-9916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raso GM, Esposito E, Iacono A, Pacilio M, Cuzzocrea S, Canani RB, et al. Comparative therapeutic effects of metformin and vitamin E in a model of non-alcoholic steatohepatitis in the young rat. Eur J Pharmacol. 2009;604:125–131. doi: 10.1016/j.ejphar.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Romualdi D, Costantini B, Selvaggi L, Giuliani M, Cristello F, Macri F, et al. Metformin improves endothelial function in normoinsulinemic PCOS patients: a new prospective. Hum Reprod. 2008;23:2127–2133. doi: 10.1093/humrep/den230. [DOI] [PubMed] [Google Scholar]
- Rubin LJ. Pulmonary arterial hypertension. Proc Am Thorac Soc. 2006;3:111–115. doi: 10.1513/pats.200510-112JH. [DOI] [PubMed] [Google Scholar]
- Sartoretto JL, Melo GA, Carvalho MH, Nigro D, Passaglia RT, Scavone C, et al. Metformin treatment restores the altered microvascular reactivity in neonatal streptozotocin-induced diabetic rats increasing NOS activity, but not NOS expression. Life Sci. 2005;77:2676–2689. doi: 10.1016/j.lfs.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Tahrani AA, Varughese GI, Scarpello JH, Hanna FW. Metformin, heart failure, and lactic acidosis: is metformin absolutely contraindicated? BMJ. 2007;335:508–512. doi: 10.1136/bmj.39255.669444.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DJ, Peacock AJ, MacLean M, Harnett M. Chronic hypoxia induces constitutive p38 mitogen-activated protein kinase activity that correlates with enhanced cellular proliferation in fibroblasts from rat pulmonary but not systemic arteries. Am J Respir Crit Care Med. 2001;164:282–289. doi: 10.1164/ajrccm.164.2.2008054. [DOI] [PubMed] [Google Scholar]
- Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, et al. Insulin resistance in pulmonary arterial hypertension. Eur Respir J. 2009;33:318–324. doi: 10.1183/09031936.00000508. L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Miskimins WK. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]