

Abstract
Background and purpose:
(−)-Stepholidine has high affinity for dopamine D1 and D2 receptors. The aims of the present study were to examine the oral bioavailability and brain penetration of (−)-stepholidine and to gain understanding of mechanisms governing its transport across the enterohepatic barrier and the blood–brain barrier.
Experimental approach:
The pharmacokinetics of (−)-stepholidine was studied in rats and microdialysis was used to measure delivery to the brain. These studies were supported by biological measurement of unbound (−)-stepholidine. Membrane permeability was assessed using Caco-2 cell monolayers. Metabolite profiling of (−)-stepholidine in rat bile and plasma was performed. Finally, in vitro metabolic stability and metabolite profile of (−)-stepholidine were examined to compare species similarities and differences between rats and humans.
Key results:
Orally administered (−)-stepholidine was rapidly absorbed from the gastrointestinal tract; two plasma concentration peaks were seen, and the second peak might result from enterohepatic circulation. Due to extensive pre-systemic metabolism, the oral bioavailability of (−)-stepholidine was poor (<2%). However, the compound was extensively transported across the blood–brain barrier, demonstrating an AUC (area under concentration–time curve) ratio of brain : plasma of ∼0.7. (−)-Stepholidine showed good membrane permeability that was unaffected by P-glycoprotein and multidrug resistance-associated protein 2. In vitro (−)-stepholidine was metabolized predominantly by glucuronidation and sulphation in rats and humans, but oxidation of this substrate was very low.
Conclusions and implications:
Although (−)-stepholidine exhibits good brain penetration, future development efforts should aim at improving its oral bioavailability by protecting against pre-systemic glucuronidation or sulphation. In this regard, prodrug approaches may be useful.
Keywords: (−)-stepholidine, CNS drug, enterohepatic barrier, blood–brain barrier, oral bioavailability, brain penetration, metabolic profiling, prodrug
Introduction
Schizophrenia is a chronic psychiatric disease with positive and negative symptoms, such as auditory hallucinations, delusions and social withdrawal, as well as cognitive dysfunction (Freedman, 2003). Approximately 1% of the world's population suffers from this lifetime disease. The pathophysiology of schizophrenia involves the up-regulation of dopamine D2-like receptors (receptor nomenclature follows Alexander et al., 2008) in the subcortex, limbic cortex and ventral tegmental area, resulting in positive symptoms, and dysfunction of D1-like receptors in the prefrontal cortex leading to negative symptoms and cognitive deficits (Yang and Chen, 2005). Other neurotransmitters and their receptors, such as glutamate-NMDA, 5-HT and GABA, are additionally involved in schizophrenia. Most traditional antipsychotic agents, such as perphenazine, haloperidol and chlorpromazine act on the positive symptoms by blocking dopamine D2 receptor hyperfunction. Chronic treatment with these drugs can decrease D1 receptor levels in the prefrontal cortex, leading to serious side effects such as tardive dyskinesia. A number of recently developed atypical antipsychotic drugs (e.g. clozapine, risperidone, olanzapine and aripiprazole) use dopamine D2 receptor antagonism to attenuate positive symptoms and are also effective in treating negative symptoms via interactions with other receptors, such as 5-HT without causing extrapyramidal side effects. However, clinically significant weight gain and diabetes mellitus have been increasingly reported in patients treated with these drugs (Elias and Hofflich, 2008).
(−)-Stepholidine [(−)-SPD], a tetrahydroprotoberberine alkaloid isolated from the Chinese herb Stephania intermedia LO, has been reported to exhibit D1 receptor agonistic activity while acting as a D2 receptor antagonist (Jin et al., 2002; Mo et al., 2007; Natesan et al., 2008). (−)-SPD was found to be 18 times more potent than haloperidol with respect to binding to D1 receptors, but 14 times weaker for D2 receptors (Xu et al., 1989). In addition, (−)-SPD also exerts agonist effects at 5-HT1A receptors and inhibits lipid peroxidation and oxidative stress (playing a role in neurodegeneration leading to dyskinesia) (Jin et al., 2000). This unique pharmacological profile makes (−)-SPD a potentially useful agent in the treatment of schizophrenia, because it could show superior efficacy against both negative and positive symptoms along with cognitive deficits and reduced side effects. A recent double-blind clinical trial in 61 inpatients was performed by Wu et al. (2003) to determine the potential use of (−)-SPD as an antipsychotic drug. The subjects were recruited after diagnosis according to the Chinese Classification of Mental Disorders, II-R version, published by the Chinese Society of Psychiatry (Beijing, China). The clinical effects of (−)-SPD (31 cases, 225–625 mg·day−1 given p.o. for 8 weeks) were compared with those of perphenazine (30 cases, 16–44 mg·day−1 given p.o. for 8 weeks). As a result, (−)-SPD had affirmative effect on both the positive and negative symptoms of schizophrenia, but did not induce any obvious extrapyramidal symptoms, suggesting that (−)-SPD was superior to perphenazine in both efficacy and safety. The Chinese State Food and Drug Administration has approved oral (−)-SPD for clinical use (http://www.sfda.gov.cn).
Information on the oral bioavailability (F) and brain penetration of (−)-SPD will give us a better understanding of the pharmacological actions of p.o. administered (−)-SPD. However, such pharmacokinetic data are scarce. Although a preliminary rat pharmacokinetic study (Zhang et al., 1990) was carried out using radiolabelled (−)-SPD, the biological assay failed to differentiate the unchanged parent compound from its metabolites in biological fluids, leading to misinterpretation of some of the pharmacokinetic properties. Moreover, information about the distribution of (−)-SPD to the brain was not available from the study. Recently, Odontiadis et al. (2007) developed a method based on liquid chromatography/fluorescence for determining the concentrations of (−)-SPD in rat plasma and brain tissue homogenate samples.
Here, we have sought to examine the F of (−)-SPD and its distribution to the brain and to gain understanding of relevant mechanisms governing its transport across the enterohepatic barrier (EHB) and the blood–brain barrier (BBB). Because (−)-SPD showed antipsychotic effects in rats (Ellenbroek et al., 2006), we performed brain microdialysis study in rats, together with other rat pharmacokinetic studies. In an effort to extrapolate the pharmacokinetic data from rats to humans, we further examined comparative in vitro metabolic stability of (−)-SPD, as well as the associated metabolite profiles, between rats and humans.
Methods
Experimental animals
Animal care and experimental procedures were carried out according to protocols approved by the Review Committee of Animal Care and Use at the Shanghai Institute of Materia Medica (Shanghai, China). Male and female Sprague-Dawley rats (280–320 g) were obtained from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China). Rats were housed in rat cages (48 × 29 × 18 cm, three rats per cage) in a unidirectional airflow room under controlled temperature (20–24°C), humidity (40–70%) and a 12 h cycle of light and dark. Filtered tap water was made available ad libitum. The rodents were given commercial rat chow (Suzhou, China) ad libitum except for the overnight period prior to dosing, when food was withheld. Prior to the experiment, rats were acclimatized to the facilities and environment for 7 days. After study use, the rats were killed with CO2 gas.
Rat studies
Rats were randomly assigned to four treatment groups (three rats per group) and received a single p.o. dose of (−)-SPD at 20, 50 or 100 mg·kg−1 (via gavage; 10 mL·kg−1) or an i.v. bolus dose at 2 mg·kg−1 (from the tail vein). Serial blood samples (∼0.25 mL; 0, 5, 15 and 30 min and 1, 2, 4, 6, 8, 10, 12 and 24 h) were collected from the tail vein into heparinized polypropylene tubes. The blood samples were centrifuged at 1300×g for 10 min to obtain plasma samples, which were frozen at −70°C for later analysis.
A bile duct cannulation study was used to investigate the metabolism and enterohepatic recirculation of (−)-SPD. Three rats were anaesthetized with ether for the surgical procedure. The bile ducts were cannulated using polyethylene tubing, and serial bile samples were collected at 0, 15 ± 5 and 30 ± 10 min and 1 ± 0.17, 2 ± 0.17, 4 ± 0.17, 6 ± 0.17, 8 ± 0.17 and 12 ± 0.17 h after a p.o. dose of (−)-SPD at 50 mg·kg−1. During bile collection, sodium taurocholate solution (pH 7.4) was infused into the duodenal cannula (∼1 mL·h−1). The collected samples were stored at −70°C pending analysis.
Pharmacokinetic data analysis
Plasma pharmacokinetic parameters were estimated by a non-compartmental method using the Kinetica 2000 software package (version 3.0; InnaPhase Corp., Philadelphia, PA, USA). The maximal concentration (Cmax) and time taken to achieve peak concentration (Tpeak) were observed values with no interpolation. The area under the concentration–time curve up to 12 h (AUC0→12 h) was calculated by trapezoidal rule. The t1/2 was calculated using the relationship 0.693/k, where k was the elimination rate constant. The total plasma clearance (CL) for i.v. dosing was estimated by dividing the administered dose by the AUC0→12 h. The distribution volume at steady status (Vss) for i.v. dosing was estimated by multiplying the CL by the mean residence time (MRT). Dose proportionality analyses were carried out using the method described by Smith et al. (2000).
Rat brain microdialysis
Microdialysis probes were surgically implanted in three rats. After anaesthetization with i.p. urethane at 1.5 g·kg−1, each rodent was placed in a stereotaxic frame with incisor bar. The bar was adjusted to achieve a horizontal skull, as indicated by ventral coordinates for bregma and lambda. A CMA/11 microdialysis probe (a 4 mm cuprophane membrane with a 6000 Da molecular weight cut-off and 14 mm stainless steel shaft; CMA/Microdialysis, Stockholm, Sweden) was implanted and aimed at the medial prefrontal cortex, using coordinates relative to the bregma and dural surface (AP +3.2, ML +0.8, DV −6.0 mm) according to the atlas of rat brain (Paxinos and Watson, 1998).
The microdialysis probe was perfused with an artificial cerebrospinal fluid (ACSF; 125 mM NaCl, 0.5 mM NaH2PO4, 2.5 mM Na2HPO4, 1.2 mM CaCl2, 2.5 mM KCl and 1.0 mM MgCl2, pH 7.4) at 1.0 µL·min−1. The rat was allowed to equilibrate for 1 h prior to initiation of sample collection. After blank control dialysate samples were collected, the animal received a bolus i.v. dose of (−)-SPD at 2 mg·kg−1 (from the tail vein) followed by a 60 min dialysate sampling. The dialysates were collected at 5 min intervals using a CMA/142 microfraction collector (CMA/Microdialysis, Solna, Sweden). For all samples, a 5 min delay was incorporated into the sampling procedure to compensate for the dead volume between the active membrane and the sample collection outlet.
The concentration of (−)-SPD in brain extracellular fluid (bECF) was calculated from that in the corresponding dialysate sample according to the following equation:
| (1) |
where CbECF was the concentration of (−)-SPD in the bECF; Cd was the measured concentration of (−)-SPD in the dialysate sample; Rin vivo was the recovery of (−)-SPD when the microdialysis probe was inserted into the rat brain; Rin vitro was the recovery of the probe immersed in ACSF containing 100 ng·mL−1 (−)-SPD and perfused with (−)-SPD-free ACSF; Din vitro was the delivery of (−)-SPD calculated by measuring the loss in the compound concentration between the perfusate and the dialysate when the probe was immersed in (−)-SPD-free aCSF and perfused with aCSF solution containing 100 ng·mL−1 (−)-SPD; Din vivo was the delivery of (−)-SPD determined in the same way as Din vitro except that the probe was inserted into the rat brain. In the current study, the Rin vitro, Din vivo and Din vitro of (−)-SPD were 20.4%, 18.1% and 20.5% respectively, yielding an Rin vivo of 18.0%.
An additional three rats of the same batch were given an i.v. bolus dose of (−)-SPD at 2 mg·kg−1, and serial blood samples (0, 5, 15, 25, 35 and 55 min) were collected for measurement of unbound (−)-SPD. To assess the extent of brain penetration, the ratio of unbound (−)-SPD in brain to that in plasma (the partition coefficient, Kp) was calculated as AUCbECF/AUCplasma.
Caco-2 cell culture and transport study
Caco-2 cells (American Type Culture Collection, Manassas, VA, USA) were cultured as described in our earlier publication (Dai et al., 2008). Bidirectional transport experiments were performed in triplicate at initial (−)-SPD concentrations of 50, 20, 8, 3.2, 1.3 and 0.51 µM in Hank's balanced salt solution in the presence or absence of the selective P-glycoprotein (P-gp) inhibitor, verapamil, or the multidrug resistance-associated protein 2 (MRP2) inhibitor, indomethacin. After incubation for 20 min at 37°C, samples were collected from both the receiver and donor compartments, and the recovery of (−)-SPD was determined. Propranolol, atenolol, rhodamine123 and sulphasalazine were used as control compounds for high permeability, low permeability, P-gp and MRP2 substrates respectively. The Papp expressed in cm·s−1 was calculated according to the following equation:
| (2) |
where ΔQ/Δt was the linear appearance rate of (−)-SPD on the receiver side; A was the surface area of the cell monolayer; C0 was the initial (−)-SPD concentration on the donor side. The apical efflux ratio was defined as Papp(basolateral→apical)/Papp(apical→basolateral). An efflux ratio greater than three was considered a positive result, suggesting that the tested compound was an in vitro substrate for the efflux transporter(s). Sampling from the donor side was also carried out at 20 min to determine the recovery of (−)-SPD.
Metabolic incubations
In vitro metabolic stability and metabolite profile of (−)-SPD were examined with tissue extracts of rat liver microsomes (RLM), rat liver cytosol (RLC), rat small intestine microsomes (RIM), rat small intestine cytosol (RIC), pooled human liver microsomes (HLM), pooled human liver cytosol (HLC), human small intestine cytosol (HIC) and human small intestine microsomes (HIM). RIC and HIC were prepared from the corresponding intestine S9 by centrifugation for 1 h at 100 000×g and 4°C. Metabolism was studied at a standard substrate concentration of 1 µM and a standard total protein concentration of 0.5 mg·mL−1. Incubations were performed in duplicate at 37°C for 0, 5, 15, 30, 60 or 120 min in 96-well plates. For cytochrome P450 (CYP)-mediated oxidation, incubation mixtures in a total assay volume of 50 µL consisted of liver or intestine microsomes, (−)-SPD, 100 mM potassium phosphate buffer (pH 7.4) and a NADPH-generating system comprising 3.3 mM magnesium chloride, 3.3 mM glucose 6-phosphate, 0.5 U·mL−1 glucose-6-phosphate dehydrogenase and 1.3 mM nicotinamide adenine dinucleotide phosphate (NADP). Before commencement of the reaction by adding the NADPH-generating system, the incubation mixture was preincubated for 5 min at 37°C. In addition, microsomal incubation mixtures (50 µL) for UDP-glucuronosyltransferase (UGT)-mediated glucuronidation contained the microsomes (as above), 10 mM magnesium chloride, 50 mM Tris-HCl buffer (pH 7.5) and 2 mM uridine 5′-diphosphoglucuronic acid (UDPGA). Cytosolic incubation mixtures (50 µL) for sulphotransferase (SULT)-mediated sulphation contained liver or intestine cytosol, 8 mM dithiothreitol, 10 mM magnesium chloride, 50 mM Tris-HCl buffer (pH 7.4) and 0.1 mM 3′-phosphoadenosine-5′-phosphosulphate (PAPS). After incubation, the reactions were terminated by adding 100 µL of ice-cold methanol. The resulting samples were centrifuged at 3000×g for 10 min, and the supernatants were applied to bioanalytical assay. Negative controls were incubated in the absence of glucose-6-phosphate dehydrogenase, UDPGA or PAPS. For positive controls, midazolam or dextromethorphan, substituting for (−)-SPD as the substrate, were used as to test the CYP-catalytic activity of the microsomes. In addition, quercetin was used to assess UGT activity of the microsomes and SULT activity of the cytosol respectively. In the determination of the in vitro t1/2, the (−)-SPD concentrations at different incubation times were converted to percentage of remaining, using the initial concentration as 100%. The slope of the linear regression of ln (% remaining) with incubation time (−k) was used to calculate the t1/2 value according to the following equation:
| (3) |
Quantification of (−)-SPD in various biological samples
Validated liquid chromatography/tandem mass spectrometry-based (LC/MS/MS-based) methods were applied, and a Thermo Fisher TSQ Quantum mass spectrometer (San Jose, CA, USA) was interfaced via an ESI probe with an Agilent 1100 liquid chromatograph (Waldbronn, Germany). The LC method was similar to our earlier report (Zhao et al., 2007), except for the gradient programme, that is, 0–0.5 min from 20% (v/v) to 100% methanol, 0.5–3.0 min at 100% methanol, 3.1 min back to 20% methanol and 3.1–7 min at 20% methanol. For measurement of unbound (−)-SPD in plasma, a high-speed, rapid ultrafiltration method (Guo et al., 2006) was used. The brain dialysate and the transport medium samples were analysed following centrifugation at 16 060×g for 3 min. For examination of conjugated (−)-SPD, the rat plasma samples were treated with or without hydrochloric acid (Zhao et al., 2007). The MS/MS measurement was performed in the positive ion ESI mode, and the precursor-to-product ion pair for selected reaction monitoring of (−)-SPD was m/z 328→178 under the collision energy 22 V. The linearity dynamic ranges for measuring (−)-SPD were 0.192–600 ng·mL−1 for all of the tested biological samples.
UPLC/MS- and UPLC/MS/MS-based metabolite profiling
The metabolite profiling of (−)-SPD in rat plasma and bile samples, as well as in incubation samples, was initially conducted using full scan UPLC/MS (m/z 50–1000) in both the positive- and negative-ion ESI modes, according to the molecular mass gains or losses predicted for the possible metabolites compared with those of the parent compound. The metabolite prediction was carried out according to literature list of common biotransformations of drugs (Kostiainen et al., 2003; Anari et al., 2004) and was also facilitated by the PALLAS MetabolExpert 3.6 software (CompuDrug International, Sedona, AZ, USA). The metabolite identity was confirmed according to the pharmacokinetic behaviours. The metabolites were characterized by UPLC/MS/MS at the collision energy 20 V in the full scan ranging from m/z 50 to 600. For sample clean-up, a methanol-based protein precipitation method was used at the precipitant-to-sample volume ratio 2:1. UPLC separations were achieved using a Waters 1.7 µm BEH C18 column (100 × 2.1 mm i.d.; Dublin, Ireland) under a biliary gradient programme, that is, 0–10 min from 0% to 10% methanol, 10–20 min from 10% to 30% methanol, 20–22 min from 30% to 70% methanol, 22–25 min at 70% methanol and 25–30 min at 0% methanol.
Data analysis
Results are shown as means ± SEM.
Materials
Purified (−)-SPD (free base, 99.2%) was provided by the Phytochemical Department of Shanghai Institute of Materia Medica (Shanghai, China). For p.o. administration, (−)-SPD was prepared by triturating a suspension in 0.5% (w/v) sodium carboxymethyl cellulose (CMC-Na) to concentrations of 2, 5 and 10 mg·mL−1. For i.v. administration, the compound (0.5 mg·mL−1) was dissolved in 0.5% (w/v) N,N-dimethylacetamide, 9.5% (w/v) cremophor EL and 90% (v/v) saline.
Verapamil hydrochloride, cremophor EL, β-NADP, glucose-6-phosphate monosodium salt, glucose-6-phosphate dehydrogenase, UDPGA and PAPS were purchased from Sigma-Aldrich (St. Louis, MO, USA). Indomethacin, midazolam, dextromethorphan and quercetin were obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Urethane, CMC-Na and other chemical reagents of analytical reagent grade were obtained from Sinopharm Chemical Reagent Co. (Shanghai, China). HPLC-grade methanol was purchased from Merck (Darmstadt, Germany). Ethyl acetate was obtained from Tedia (Fairfield, OH, USA). HPLC-grade water was prepared with a Direct-Q 3 UV water purifying system (Millipore, Bedford, MA, USA).
Sprague-Dawley RLM, RLC, RIM, rat small intestine S9, pooled HLM, pooled HLC, human small intestine S9 and HIM were obtained from XenoTech, LLC (Lenexa, KS, USA).
Results
Oral bioavailability and plasma pharmacokinetics of (−)-SPD in rats
The mean plasma concentration–time profiles of unbound (−)-SPD, after the rats received a bolus i.v. dose at 2 mg·kg−1 or a single p.o. dose at 20, 50 or 100 mg·kg−1, are shown in Figure 1. The pharmacokinetic parameters are summarized in Table 1. The plasma level of (−)-SPD declined rapidly during the first 6 h after the i.v. administration, as indicated by a short mean t1/2 of around 0.8 h. The highest plasma concentration of unbound (−)-SPD, measured at the first blood sampling (5 min post dosing), had a mean value of 352 ng·mL−1. Thereafter, a minor peak concentration (averaging 4.7 ng·mL−1) was observed between 6 and 12 h post dosing. The mean CL of unbound (−)-SPD (17.8 L·h−1·kg−1) was found to be about fivefold greater than the average rat hepatic blood flow (3.4 L·h−1·kg−1) (Davies and Morris, 1993), suggesting that first-pass hepatic metabolism might limit the exposure of the tested compound. The mean Vss of unbound (−)-SPD (30.6 L·kg−1) was greater than the approximate amount of rat total body water (0.67 L·kg−1), suggesting that the tested compound could be extensively distributed to the tissues. Significant sexual dimorphism was observed in the AUC0→12 h and CL values; female rats exhibited greater AUC0→12 h but a slower CL, compared with male rats (Table 1).
Figure 1.
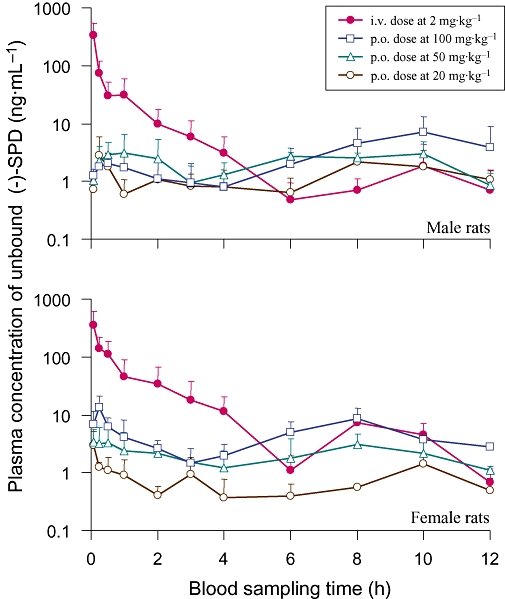
Mean unbound plasma level-time profiles of (−)-stepholidine [(−)-SPD] after a single p.o. dose (20, 50 or 100 mg·kg−1) or an i.v. bolus dose (2 mg·kg−1) to male (upper panel) and female (lower panel) rats.
Table 1.
Plasma PK of unbound (−)-SPD in rats
| PK parameter | i.v. 2 mg·kg−1 |
p.o. |
||
|---|---|---|---|---|
| 20 mg·kg−1 | 50 mg·kg−1 | 100 mg·kg−1 | ||
| Male rats | ||||
| AUC0→12 h (h·ng·mL−1) | 120 ± 45 | 19.2 ± 4.9 | 27.6 ± 7.6 | 49.2 ± 24.9 |
| MRT (h) | 1.40 ± 0.35 | 5.05 ± 1.75 | 3.22 ± 0.52 | 4.85 ± 1.57 |
| T1max (h) | – | 0.22 ± 0.10 | 0.75 ± 0.25 | 0.58 ± 0.22 |
| C1max (ng·mL−1) | 341 ± 114a | 3.12 ± 1.68 | 4.35 ± 1.33 | 2.29 ± 0.44 |
| T2max (h) | 8.67 ± 1.15 | 8.67 ± 0.67 | 7.33 ± 1.33 | 10.0 ± 0.0 |
| C2max (ng·mL−1) | 2.11 ± 1.33 | 2.62 ± 1.30 | 3.70 ± 0.20 | 7.17 ± 3.55 |
| t11/2 (h) (i.v., 0–1 h) | 0.15 ± 0.03 | – | – | – |
| t21/2 (h) (i.v., 1–6 h; p.o. 1–4 h) | 0.75 ± 0.14 | 1.22 ± 0.25 | 1.07 ± 0.16 | 1.73 ± 0.99 |
| CL (L·h−1·kg−1) | 24.1 ± 10.7 | – | – | – |
| Vss (L·kg−1) | 25.3 ± 3.9 | – | – | – |
| F (%) | – | 1.60 ± 0.41 | 0.92 ± 0.25 | 0.82 ± 0.42 |
| Female rats | ||||
| AUC0→12 h (h·ng·mL−1) | 244 ± 161 | 9.78 ± 2.09 | 28.2 ± 10.1 | 66.2 ± 8.9 |
| MRT (h) | 2.51 ± 0.17 | 4.01 ± 0.78 | 4.22 ± 1.02 | 4.31 ± 0.05 |
| T1max (h) | – | 0.14 ± 0.06 | 0.14 ± 0.06 | 0.25 ± 0.00 |
| C1max (ng·mL−1) | 362 ± 146a | 3.25 ± 1.04 | 3.59 ± 1.45 | 13.8 ± 4.6 |
| T2max (h) | 8.00 ± 0.00 | 10.0 ± 0.00 | 8.00 ± 1.16 | 8.67 ± 0.67 |
| C2max (ng·mL−1) | 7.26 ± 5.76 | 1.44 ± 0.38 | 3.11 ± 0.89 | 6.50 ± 1.88 |
| t11/2 (h) (i.v., 0–1 h) | 0.33 ± 0.06 | – | – | – |
| t21/2 (h) (i.v., 1–6 h; p.o. 1–4 h) | 0.92 ± 0.09 | 1.07 ± 0.23 | 1.74 ± 0.26 | 1.05 ± 0.17 |
| CL (L·h−1·kg−1) | 11.5 ± 4.7 | – | – | – |
| Vss (L·kg−1) | 35.8 ± 23.7 | – | – | – |
| F (%) | – | 0.40 ± 0.09 | 0.46 ± 0.17 | 0.54 ± 0.07 |
All values are given as mean ± SEM (n= 3).
(−)-SPD, (−)-stepholidine; AUC, area under concentration–time curve; CL, total plasma clearance; MRT, mean residence time; PK, pharmacokinetics; Vss, distribution volume at steady status.
The highest measured concentration at 5 min after i.v. administration.
The double-peak phenomena in the plasma concentration–time curves of unbound (−)-SPD after p.o. administration were observed in rats, regardless of dose levels; the tested compound reached its first (C1max) and second (C2max) peak concentrations within 1 h and at 6–10 h post dosing respectively. Log-transformed plots of the AUC0→12 h, C1max and C2max versus the p.o. dose were analysed using power regression. The results revealed that these exposure parameters did not increase linearly over the tested dose range (20–100 mg·kg−1), but instead were directly related to dose. The systemic exposure of unbound (−)-SPD after p.o. administration was found to be markedly lower than that after i.v. dosing when the AUC0→12 h was corrected for dose; the F of (−)-SPD was very low, with a mean value of <2%. Due to issues with solubility, the i.v. dose level of (−)-SPD (2 mg·kg−1) was much lower than the lowest tested p.o. dose (20 mg·kg−1), potentially affecting the accuracy of the estimated F.
Brain penetration of (−)-SPD
Orally administered (−)-SPD is targeted towards CNS delivery. Figure 2 shows the time-dependent changes in levels in bECF and unbound levels in plasma of (−)-SPD (2 mg·kg−1), given i.v. to rats. We found that the rat brain distribution of the tested compound was rapid, with both the highest unbound bECF level (520 ± 39 ng·mL−1) and the highest unbound plasma level (547 ± 74 ng·mL−1) of (−)-SPD being measured at 5 min after administration of an i.v. bolus dose at 2 mg·kg−1. In addition, (−)-SPD extensively penetrated the BBB after dosing, demonstrating a mean Kp of 0.71, which was calculated from the AUC0→55 min values of unbound compound in bECF (104 ± 17 h·ng·mL−1) and in plasma (146 ± 41 h·ng·mL−1). The bECF level decreased rapidly at a half-life of 0.11 h, which was comparable with the plasma values (0.13 h).
Figure 2.
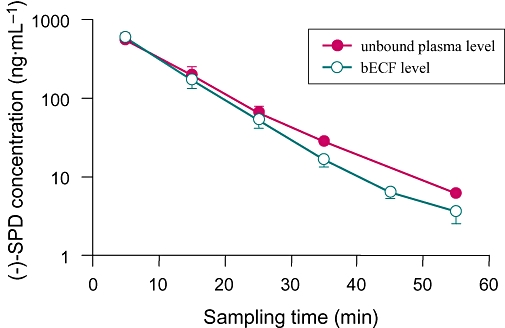
Time courses of mean brain extracellular fluid (bECF) levels and unbound plasma levels of (−)-stepholidine [(−)-SPD] after an i.v. bolus dose (2 mg·kg−1) to male rats.
Membrane permeability of (−)-SPD measured in Caco-2 monolayers
Based on our in-house sigmoid correlation between the percentage of dose absorbed after p.o. administration in humans and the corresponding log of the Caco-2 Papp values of a series of reference compounds (Lu et al., 2008), a compound might be absorbed poorly, moderately or well with a Papp value of < 1 × 10−6, 1 to 10 × 10−6 or 10 × 10−6 cm·s−1 respectively. The membrane permeability of (−)-SPD was good, with a mean Papp(apical→basolateral) of (40.8 ± 8.7) × 10−6 cm·s−1. The Caco-2 cells were found to show high levels of P-gp and MRP2 expression, demonstrating mean apical efflux ratios of 9.3 and 4.3 for rhodamine123 and sulphasalazine respectively. The efflux ratios for rhodamine123 and sulphasalazine could be reduced to 1.5 and 1.0 by treatment of the P-gp inhibitor verapamil and the MRP2 inhibitor indomethacin respectively. The bidirectional Papp values were comparable and were not affected by the initial concentration on the donor side (data not shown), suggesting the presence of a passive diffusion mechanism for (−)-SPD across the Caco-2 monolayer. The mean efflux ratio was 1.0 for (−)-SPD, which was not affected by verapamil and indomethacin. The transport recovery of (−)-SPD was 98%.
Metabolism of (−)-SPD
Ingested (−)-SPD was extensively metabolized in rats. Metabolite profiling assays revealed the occurrence of 17 (−)-SPD metabolites (M1–M17) in bile and 11 metabolites (M2–M11 and M13) in plasma following p.o. administration of (−)-SPD to rats. The detection and CID data on these metabolites are summarized in Table 2. No metabolites were detected in bile or plasma samples from untreated rats. Following p.o. administration of (−)-SPD, the concentrations of the various metabolites increased and then decreased in bile or plasma over time. The metabolite peaks appeared in the chromatograms with the retention times of 11.1–17.2 min, while (−)-SPD had a retention time of 17.9 min. The molecular masses of M1 and M2 were consistent with those of the glucuronide of SPD (503 Da), M3 and M4 with the sulphate of SPD (407 Da), M5 with the disulphate of SPD (487 Da), M6 and M7 with the glucuronide of sulphated SPD (583 Da), M8 and M9 with the glucuronide of methylated SPD (517 Da), M10 and M11 with the glucuronide of N-oxidized SPD (519 Da), M12–M14 with the glucuronide of N-oxidized demethyl SPD (505 Da) and M15–M17 with the sulphate of N-oxidized demethyl SPD (409 Da). Further MS/MS analyses provided structural evidence for the assignment of the metabolite peaks. Based on this, we were able to propose a metabolic pathway of (−)-SPD in rats, as depicted in Figure 3.
Table 2.
PI, FI and LC retention times (tR) of (−)-SPD metabolites, as well as the exposure data
| Metabolite | PIa (m/z) | FIb (m/z) | tR (min) |
Detected in |
AUCM/AUCSPDc | AUCb/AUCpd | |
|---|---|---|---|---|---|---|---|
| Bile | Plasma | ||||||
| SPD | 328 | 119, 135, 151, 163, 178 | 17.9 | − | + | – | – |
| M1 (SPD-O-glucuronide) | 504 | 151, 163, 178, 328; [176] | 12.8 | + | − | – | – |
| M2 (SPD-O-glucuronide) | 504 | 151, 163, 178, 328; [176] | 13.9 | + | + | 134 | 79 |
| M3 (SPD-O-sulphate) | 408 | 135, 151, 163, 178, 328; [80] | 16.0 | + | + | 0.12 | 1906 |
| M4 (SPD-O-sulphate) | 408 | 135, 151, 163, 178, 328; [80] | 17.2 | + | + | 0.15 | 239 |
| M5 (SPD-O-disulphate) | 488 | 151, 163, 178, 328, 408; [80, 160] | 14.5 | + | + | 0.69 | 62 |
| M6 (sulphated SPD-O-glucuronide) | 584 | 151, 163, 178, 328, 408, 504; [80, 176] | 11.1 | + | + | 0.01 | 1022 |
| M7 (sulphated SPD-O-glucuronide) | 584 | 151, 163, 178, 328, 408, 504; [80, 176] | 12.8 | + | + | 0.02 | 13876 |
| M8 (methylated-SPD-O-glucuronide) | 518 | 165, 192, 342; [176] | 16.6 | + | + | 0.02 | 2174 |
| M9 (methylated-SPD-O-glucuronide) | 518 | 151, 163, 178, 342; [176] | 17.4 | + | + | 0.11 | 550 |
| M10 (N-oxidized SPD-O-glucuronide) | 520 | 151, 163, 178 344; [176] | 14.0 | + | + | 0.03 | 67 |
| M11 (N-oxidized SPD-O-glucuronide) | 520 | 151, 163, 178 344; [176] | 15.2 | + | + | 0.11 | 456 |
| M12 (N-oxidized demethyl-SPD-O-glucuronide) | 506 | 151, 165, 178, 330; [176] | 12.8 | + | − | – | – |
| M13 (N-oxidized demethyl-SPD-O-glucuronide) | 506 | 151, 165, 178, 330; [176] | 13.9 | + | + | 2.59 | 93 |
| M14 (N-oxidized demethyl-SPD-O-glucuronide) | 506 | 151, 165, 178, 330; [176] | 14.4 | + | − | – | – |
| M15 (N-oxidized demethyl-SPD-O-sulphate) | 410 | 151, 163, 178, 330; [80] | 14.4 | + | − | – | – |
| M16 (N-oxidized demethyl-SPD-O-sulphate) | 410 | 151, 163, 178, 330; [80] | 16.0 | + | − | – | – |
| M17 (N-oxidized demethyl-SPD-O-sulphate) | 410 | 151, 163, 178, 330; [80] | 17.2 | + | − | – | – |
(−)-SPD, (−)-stepholidine; AUC, area under concentration–time curve; FI, fragment ions; PI, parent ions.
[M+H]+ generated in the positive ion ESI mode.
Produced from the [M+H]+ by CID. The number in bold is for the base peak, and the FI of relative abundance <5% are not shown. The number in square brackets is for neutral loss. The collision energy was set from 5 to 50 V.
AUCM/AUCSPD: plasma metabolite-to-(−)-SPD ratio in AUC0→12 h, which were calculated from the peak areas because of the reference standards of metabolites not available. When a plasma metabolite showed an AUCM/AUCD greater than 0.1, it was considered a major metabolite of (−)-SPD.
AUCb/AUCp: bile-to-plasma distribution ratios in AUC0→12 h, which were calculated from the peak areas.
Figure 3.
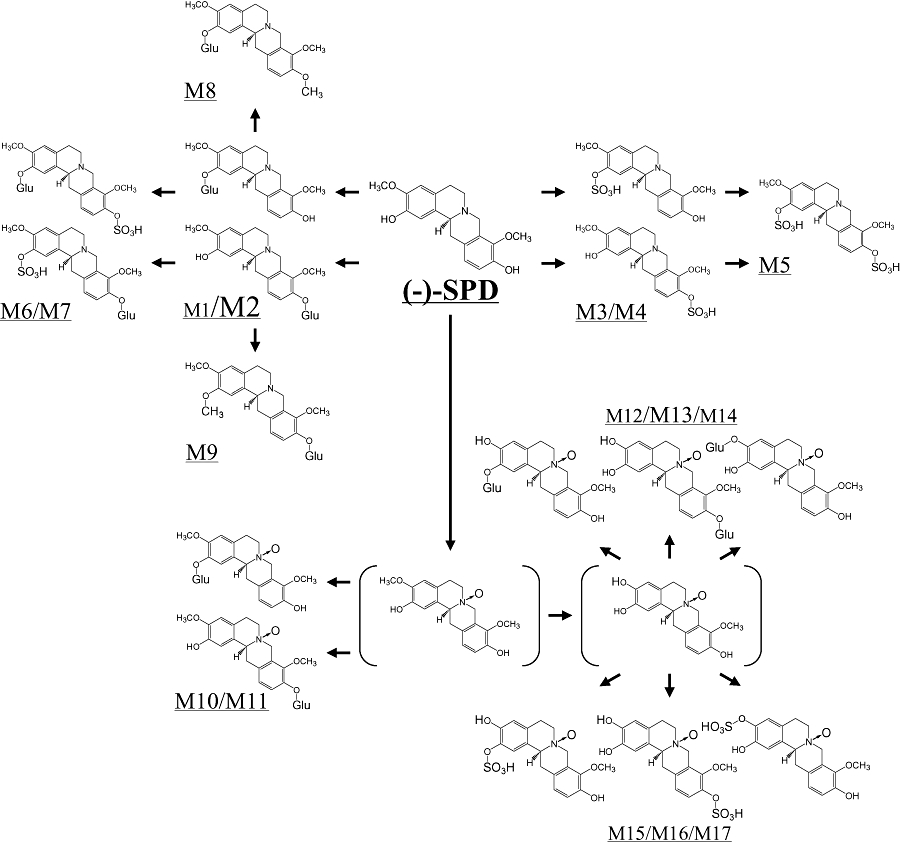
Proposed metabolic pathways of (−)-stepholidine [(−)-SPD] in rats.
According to the plasma AUC0–12 h calculated from the LC/MS/MS peak abundance in comparison with that of the parent (−)-SPD, M2 was the predominant metabolite in plasma, with a plasma metabolite-to-parent compound ratio in AUC0→12 h (AUCM/AUCSPD) of 134, whereas M13 exhibited an AUCM/AUCSPD value of ∼3. M1, M3–M11 were demonstrated to be minor plasma metabolites, with AUCM/AUCSPD ratios of only 0.01 and 0.69 respectively. These plasma metabolites were also measured in the rat bile samples collected after p.o. administration of (−)-SPD. In addition, M1, M12 and M14–M17 were found only in bile, but not in plasma. Unlike the metabolites, parent (−)-SPD was not detected in bile. The bile-to-plasma distribution ratios in AUC0→12 h (AUCb/AUCp) of M2 was 79, suggesting that this metabolite was extensively excreted through active biliary transportation. We also found that SPD sulphates tended to be more extensively excreted into bile than the glucuronides (Table 2). Further quantitative information on the metabolism of (−)-SPD by conjugation was obtained by comparison of the AUC values for (−)-SPD in acidic hydrolysed plasma versus unhydrolysed plasma. After p.o. administration (50 mg·kg−1), the mean AUC0→12 h of SPD derived from hydrolysis treatment (33 315 h·ng·mL−1) was much greater than that obtained in the absence of hydrolysis treatment (101 h·ng·mL−1), resulting in an AUChydrolysis/AUCunhydrolysis ratio of 330. The mean AUC0→12 h values of SPD in the presence and absence of hydrolysis treatment were 4218 and 382 h·ng·mL−1 respectively, following i.v. administration (2 mg·kg−1) and the resulting AUChydrolysis/AUCunhydrolysis ratio was 11. The results also demonstrated that extensive first-pass conjugations occurred for p.o. administered (−)-SPD.
Comparative rat and human metabolic behaviours towards (−)-SPD in vitro
In order to gain information on species similarities and differences in (−)-SPD metabolism between rats and humans, a range of tissue extracts (RLM, RLC, RIM, RIC, HLM, HLC, HIM and HIC) were used to examine the metabolic stability and metabolite profile of the compound. Using NADPH as cofactor, RLM, RIM, HLM and HIM showed in vitro t1/2 values for midazolam, that is, 0.06, 1.46, 0.05 and 0.13 h respectively, or 0.10, 8.20, 1.15 and 5.73 h respectively, for dextromethorphan. These microsomal preparations also exhibited in vitro t1/2 values with the cofactor UDPGA, that is, 0.12, 0.05, 0.12 and 0.13 h respectively, for the substrate quercetin. Meanwhile, with PAPS as cofactor, RLC, RIC, HLC and HIC showed in vitro t1/2 values of quercetin, that is, 0.09, 2.47, 0.17 and 0.21 h respectively. These data indicated that both the rat and human subcellular enzymic preparations had appropriate catalytic activities, supporting their use for our study purpose.
As shown in Figure 4, (−)-SPD was very labile in UDPGA-RLM, UDPGA-RIM and PAPS-HLC, demonstrating quite short in vitro t1/2 values 0.08, 0.06 and 0.10 h respectively. Meanwhile, the compound exhibited moderate lability in PAPS-RLC, UDPGA-HLM, UDPGA-HIM and PAPS-HIC, indicative of an in vitro t1/2 value ranging from 0.18 to 0.41 h. High metabolic stability of (−)-SPD was observed when the compound was in RLM, RIM, HLM and HIM with NADPH as cofactor. Due to this metabolic lability of (−)-SPD, in vitro metabolite profiling was performed with the incubation samples of UDPGA-RLM, PAPS-RLC, UDPGA-RIM, PAPS-RLC, UDPGA-HLM, PAPS-HLC, UDPGA-HIM and PAPS-HIC, and the results are summarized in Table 3. These in vitro data suggested that UGT- or SULT-mediated conjugations represented the major metabolic pathways of (−)-SPD in rats and humans. However, significant CYP-mediated oxidation of the compound occurred neither in rats nor in humans. The major species difference in (−)-SPD metabolism observed in vitro was that the glucuronidation was predominant for rats, with significantly less sulphation, but that, in extracts of human tissues, sulphation and glucuronidation appeared to be equally major metabolic pathways. This in vitro metabolite profile of (−)-SPD in rats was quite consistent with that observed in vivo.
Figure 4.
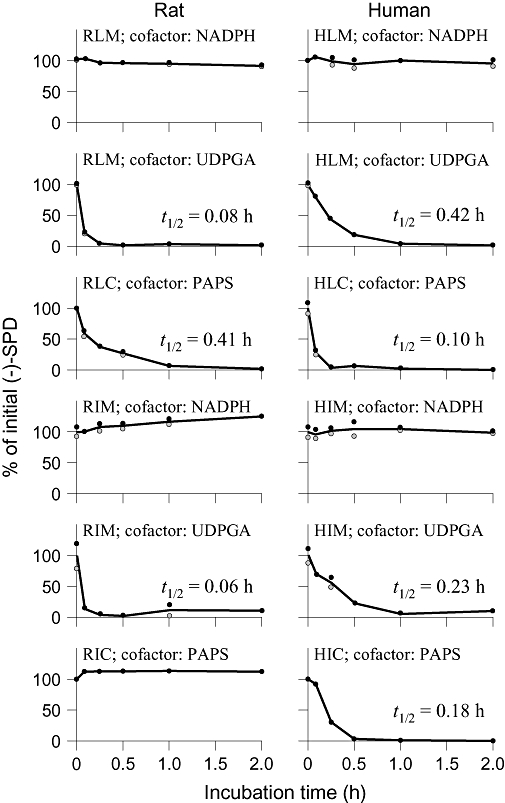
Metabolic stability profiles of (−)-SPD in RLM, RLC, RIM, RIC, HLM, HLC, HIM and HIC. NADPH, UDPGA and PAPS were used as cofactors for CYP450-, UGT- and SULT-mediated depletions of (−)-SPD respectively. The initial (−)-SPD concentration for the incubation (0–2 h) was 1 µM, and each incubation was performed in duplicate. (−)-SPD, (−)-stepholidine; CYP, cytochrome P450; HIC, human small intestine cytosol; HIM, human small intestine microsomes; HLC, human liver cytosol; HLM, human liver microsomes; PAPS, 3′-phosphoadenosine-5′-phosphosulphate; RIC, rat small intestine cytosol; RIM, rat small intestine microsomes; RLC, rat liver cytosol; RLM, rat liver microsomes; SULT, sulphotransferase; UDPGA, uridine 5′-diphosphoglucuronic acid; UGT, UDP-glucuronosyltransferases.
Table 3.
Metabolite profiles of (−)-SPD after incubation with enzyme preparations from rat or human tissues
| Enzyme | Metabolite |
Metabolite abundancea(×106) |
|
|---|---|---|---|
| Rat | Human | ||
| Liver microsomes | M2 (SPD-O-glucuronide) | 1.01 | 0.30 |
| Liver cytosol | M3 (SPD-O-sulphate) | 0.03 | 0.01 |
| M4 (SPD-O-sulphate) | 0.02 | 0.71 | |
| Small intestine microsomes | M2 (SPD-O-glucuronide) | 0.97 | 0.08 |
| Small intestine cytosol | M4 (SPD-O-sulphate) | 0.01 | 0.54 |
All values are given as mean (n= 2). The incubation time was 5 min, and the protein concentration was 0.5 mg·mL−1.
(−)-SPD, (−)-stepholidine; LC/MS/MS, liquid chromatography/tandem mass spectrometry.
The LC/MS/MS response in peak area of the metabolite of (−)-SPD from the incubation sample.
Discussion and conclusions
Many drugs that act upon the CNS are given orally. Good F and brain penetration are two highly desirable pharmacokinetic properties for drugs that are targeted towards oral delivery and CNS action. Obtaining information about the transport characteristics of potential drug candidates across the EHB versus the BBB has become an important part of drug discovery and development. In the current study, we evaluated the F of (−)-SPD in rats and its delivery to the CNS, as both are important components of rational strategy for lead optimization. Because of the functional complexity of the EHB and BBB, we first used in vivo methods that provided the most reliable measurements of (−)-SPD transport across these biological barriers in rats. To study delivery of the drug to the CNS, we used microdialysis because it enabled us to measure the unbound drug concentration at the site of action in the brain; in theory, this should reflect the dynamic effects of the drug (Upton, 2007). (−)-SPD binds moderately to rat plasma proteins (77–82%) or to human plasma proteins (85%). In the current study, we developed a sensitive LC/MS/MS method and used it to measure unbound (−)-SPD in the dialysate, applying a high sampling frequency (5 min per collection) that was sufficient to characterize the tested compound given by i.v. bolus injection. Although Caco-2 cell monolayers are often used as an in vitro model for the intestinal epithelium, this epithelial model can also be used as a surrogate BBB model. In the current study, we used Caco-2 cell monolayers to qualitatively assess the membrane permeability of (−)-SPD and obtained some valuable mechanistic insights to support the findings of our rat-based assays. In addition, UPLC/MS- and UPLC/MS/MS-based metabolite profiling was performed with the rat bile and plasma samples, providing additional mechanistic information about first-pass effects on the F of (−)-SPD. Finally, comparative in vitro metabolic stability and metabolite profile of (−)-SPD between rats and humans were studied to support extrapolation of the in vivo pharmacokinetic data from rats to humans. The strategy illustrated may be adopted for supporting optimization of other CNS leads that are targeted towards oral delivery.
Structurally, (−)-SPD is characterized as a four-ring tetrahydroprotoberberine with substituent methoxyl and hydroxyl groups at each benzene ring (Figure 3). Our results from the rat-based assays indicated that orally administered (−)-SPD was poorly available to systemic circulation, but the compound exhibited good entry into the CNS, suggesting that the drug was able to pass easily through the BBB. Consistent with this finding, the Caco-2 data indicated that (−)-SPD possessed good membrane permeability that was not affected by the efflux transporters, P-gp or MRP2. Chemoinformatic assessment of (−)-SPD was performed as described in our earlier publications (Dai et al., 2008; Lu et al., 2008). The results suggested that the compound had favourable physiochemical properties for supporting good membrane permeability, including lipophilicity (LogP 2.3; within the favourable range of 0–5), molecular mass (327 Da; <500 Da), hydrogen-bonding capacity (HBA + HBD: 5 + 2; <12) and molecular flexibility (NROTB: 2; <10). Furthermore, (−)-SPD was defined as moderately soluble, with a calculated aqueous solubility value of 0.86 mM. The in vitro and in silico data revealed that the membrane permeability would not account for the inconsistency in the ability of (−)-SPD to cross the BBB, but not the EHB. Furthermore, our results suggest that P-gp- and/or MRP2-mediated efflux transport of (−)-SPD across the biological barriers is likely to be limited.
Our metabolite profiling suggested that (−)-SPD absorbed from the gastrointestinal tract was rapidly and predominantly eliminated by glucuronidation of a phenolic hydroxyl group (the plasma AUCM2/AUCSPD ratio being 134 according to peak area calculation), and less so by sulphation, methylation, demethylation and/or N-oxidation. In addition, acidic hydrolysis of plasma samples after p.o. administration resulted in a >1000-fold increase in the AUC0→12 h of (−)-SPD compared with that in plasma samples that were not exposed to hydrolytic treatment. The result demonstrated that the poor F of (−)-SPD observed in rats was mainly due to extensive pre-systemic metabolism. We also found that the major metabolite M2 was extensively reduced by biliary excretion; the bile-to-plasma distribution ratio was 79, suggesting the involvement of active transportation.
Different metabolite profiles of (−)-SPD were observed in bile and in plasma (Table 2). There might be two reasons for these differences. First, the biliary metabolites, which were not measured in plasma, were minor metabolites compared with M2. Second, the rapid and extensive biliary excretion of these metabolites involved active transport mechanisms. Enterohepatic circulation of a drug occurs via biliary excretion and intestinal reabsorption (Roberts et al., 2002; Shou et al., 2005). Our results suggest that (−)-SPD appeared to undergo such enterohepatic circulation, as indicated by the double peaks observed in the plasma concentration–time curves after p.o. and i.v. administration. According to our experimental observations, the process of enterohepatic circulation might occur via secretion of (−)-SPD into the bile predominantly as glucuronides, which may then be deposited back into the intestinal lumen. After deconjugation of the glucuronides by β-D-glucuronidase from the colonic bacteria, a fraction of the released (−)-SPD could then be reabsorbed by the enterocytes and recirculated through the liver.
The brain, unlike most other organs of the body, is completely separated from the blood by various barriers, especially the BBB (Abbott, 2004). We found that (−)-SPD could rapidly and extensively distributed into the rat brain and the resulting exposure level could be high. Similar to the situations in the liver and the intestine, CYP-mediated metabolism of (−)-SPD in brain may also be quite poor. In addition, UGTs have been reported to be of low expression in the human brain (Ohno and Nakajin, 2009). We expect that brain CYP- or UGT-mediated metabolism is not a limiting factor for (−)-SPD crossing the BBB. Finally, our in vitro data of the species similarities and differences in metabolism of (−)-SPD suggested that the compound might also be subject to extensive pre-systemic elimination in humans. Different from the metabolic situation in rats, the sulphates, together with the glucuronides, might also be major metabolites of (−)-SPD in humans. Several SULT1A isoenzymes, such as SULT1A1 and 1A3, have been identified in the human brain (Gamage et al., 2006); whether these SULT enzymes could limit the exposure of human brain to (−)-SPD remains to be examined.
An early structure–activity relationship study suggested that attachment of a hydroxyl group to each benzene ring was very important for the affinity of (−)-SPD towards dopamine receptors, which gives the compound more potent intrinsic activities than other tetrahydroprotoberberines (Xu et al., 1989). The coexistence of two hydroxyl groups at the same benzene ring or substitution of the hydroxyl groups by methyl or other functional groups has been shown to decrease the affinity towards the dopamine receptors. However, our current pharmacokinetic study indicated that these phenolic hydroxyl groups of (−)-SPD were also molecular targets for the unwanted pre-systemic metabolism.
Prodrug approaches to drug design have led to the development of compounds with better drug-like properties (Rautio et al., 2008). The term ‘prodrug’ was first proposed by Albert (1958) and refers to chemically modified versions of the pharmacologically active agent that must undergo enzymatic or chemical transformation in vivo in order for the active drug to be released. To circumvent the undesirable pre-systemic metabolism identified herein, a prodrug approach involving modification of the hydroxyl group(s) may represent a promising strategy for overcoming the barriers limiting the efficacy of oral delivery of (−)-SPD.
In summary, although (−)-SPD possesses favourable properties for delivery to the CNS, rapid and extensive pre-systemic metabolism restricts its F. Prodrug development may be a tool for improving the pharmacokinetic properties of (−)-SPD, thereby allowing the drug to overcome the obstacles barring its oral delivery. In the current study, we assessed some key pharmacokinetic properties of (−)-SPD and identified the barrier blocking its F, which are essential steps in guiding future strategies for prodrug development.
Acknowledgments
This work was supported by Grants 2004CB720305 and 2005CB523403 from the Chinese Ministry of Science and Technology, Grants 30213204 and 30873120 from the National Natural Science Foundation of China, Grant 04DZ19215 from the Science & Technology Commission of Shanghai Municipality and Grant 07G603J049 from SIMM-CAS.
Glossary
Abbreviations:
- (−)-SPD
(−)-stepholidine
- ACSF
artificial cerebrospinal fluid
- AUC
area under concentration–time curve
- BBB
blood–brain barrier
- bECF
brain extracellular fluid
- CL
total plasma clearance
- Cmax
maximal concentration
- CYP
cytochrome P450
- EHB
enterohepatic barrier
- F
oral bioavailability
- Kp
partition coefficient defined as the ratio of unbound drug in brain to that in plasma
- HIC
human small intestine cytosol
- HIM
human small intestine microsomes
- HLC
human liver cytosol
- HLM
human liver microsomes
- LC/MS/MS
liquid chromatography/tandem mass spectrometry
- MRP2
multidrug resistance-associated protein 2
- MRT
mean residence time
- PAPS
3′-phosphoadenosine-5′-phosphosulphate
- P-gp
P-glycoprotein
- RIC
rat small intestine cytosol
- RIM
rat small intestine microsomes
- RLC
rat liver cytosol
- RLM
rat liver microsomes
- SULT
sulphotransferase
- Tpeak
time taken to achieve peak concentration
- UDPGA
uridine 5′-diphosphoglucuronic acid
- UGT
UDP-glucuronosyltransferases
- Vss
distribution volume at steady status
Conflict of interest
The authors state no conflict of interest.
References
- Abbott NJ. Prediction of blood-brain barrier permeation in drug discovery from in vivo, in vitro and in silico models. Drug Discov Today: Technologies. 2004;1:407–416. doi: 10.1016/j.ddtec.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Albert A. Chemical aspects of selective toxicity. Nature. 1958;182:421–422. doi: 10.1038/182421a0. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathine A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153:s1–s209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anari MR, Sanchez RI, Bakhtiar R, Franklin RB, Baillie TA. Integration of knowledge-based metabolic predictions with liquid chromatography data-dependent tandem mass spectrometry for drug metabolism studies: application to studies on the biotransformation of indinavir. Anal Chem. 2004;76:823–832. doi: 10.1021/ac034980s. [DOI] [PubMed] [Google Scholar]
- Dai J-Y, Yang J-L, Li C. Transport and metabolism of flavonoids from Chinese herbal remedy Xiaochaihu-tang across human intestinal Caco-2 cell monolayers. Acta Pharmacol Sin. 2008;29:1086–1093. doi: 10.1111/j.1745-7254.2008.00850.x. [DOI] [PubMed] [Google Scholar]
- Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- Elias AN, Hofflich H. Abnormalities in glucose metabolism in patients with schizophrenia treated with atypical antipsychotic medications. Am J Med. 2008;121:98–104. doi: 10.1016/j.amjmed.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Zhang X-X, Jin G-Z. Effects of (−)-stepholidine in animal models for schizophrenia. Acta Pharmcol Sin. 2006;27:1111–1118. doi: 10.1111/j.1745-7254.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- Freedman R. Schizophrenia. N Engl J Med. 2003;349:1738–1749. doi: 10.1056/NEJMra035458. [DOI] [PubMed] [Google Scholar]
- Gamage N, Barnett A, Hempel N, Duggleby RG, Windmill KF, Martin JL, et al. Human sulfotransferases and their role in chemical metabolism. Toxicol Sci. 2006;90:5–22. doi: 10.1093/toxsci/kfj061. [DOI] [PubMed] [Google Scholar]
- Guo B, Li C, Wang G-J, Chen L-S. Rapid and direct measurement of free concentrations of highly protein-bound fluoxetine and its metabolite norfluoxetine in plasma. Rapid Commun Mass Spectrom. 2006;20:39–47. doi: 10.1002/rcm.2265. [DOI] [PubMed] [Google Scholar]
- Jin G-Z, Zhu Z-T, Fu Y. Stepholidine: a potential novel antipsychotic drug with dual D1 receptor agonist and D2 receptor antagonist actions. Trends Pharmacol Sci. 2002;23:4–7. doi: 10.1016/s0165-6147(00)01929-5. [DOI] [PubMed] [Google Scholar]
- Jin X-L, Shao Y, Wang M-J, Chen L-J, Jin G-Z. Tetrahydroprotoberberines inhibit lipid peroxidation and scavenge hydroxyl free radicals. Acta Pharmacol Sin. 2000;21:477–480. [PubMed] [Google Scholar]
- Kostiainen R, Kotiaho T, Kuuranne T, Auriola S. Liquid chromatography/atmospheric pressure ionization-mass spectrometry in drug metabolism studies. J Mass Spectrom. 2003;38:357–372. doi: 10.1002/jms.481. [DOI] [PubMed] [Google Scholar]
- Lu T, Yang J-L, Gao X-M, Chen P, Du F-F, Sun Y, et al. Plasma and urinary tanshinol from Salvia miltiorrhiza (Danshen), can be used as pharmacokinetic markers for cardiotonic pills, a cardiovascular herbal medicine. Drug Metab Dispos. 2008;36:1578–1586. doi: 10.1124/dmd.108.021592. [DOI] [PubMed] [Google Scholar]
- Mo J, Guo Y, Yang Y-S, Shen J-S, Jin G-Z, Zhen X-C. Recent developments in studies of l-Stepholidine and its analogs: chemistry, pharmacology and clinical implications. Curr Med Chem. 2007;14:2996–3002. doi: 10.2174/092986707782794050. [DOI] [PubMed] [Google Scholar]
- Natesan S, Reckless GE, Barlow KBL, Odontiadis J, Nobrega JN, Baker GB, et al. The antipsychotic potential of l-stepholidine-a naturally occurring dopamine receptor D1 agonist and D2 antagonist. Psychopharmacology. 2008;199:275–289. doi: 10.1007/s00213-008-1172-1. [DOI] [PubMed] [Google Scholar]
- Odontiadis J, MacKenzie EM, Natesan S, Mamo D, Kapur S, Baker GB. Quantification of l-stepholidine in rat brain and plasma by high performance liquid chromatography combined with fluorescence detection. J Chromatogr B. 2007;850:544–547. doi: 10.1016/j.jchromb.2006.11.039. [DOI] [PubMed] [Google Scholar]
- Ohno S, Nakajin S. Determination of mRNA expression of human UDP-glucurosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab Dispos. 2009;37:32–40. doi: 10.1124/dmd.108.023598. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 1998. [Google Scholar]
- Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, et al. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. 2002;41:751–790. doi: 10.2165/00003088-200241100-00005. [DOI] [PubMed] [Google Scholar]
- Shou M, Lu W, Kari PH, Xiang C, Liang Y-X, Rodrigues AD, et al. Population pharmacokinetic modeling for enterohepatic recirculation in Rhesus monkey. Eur J Pharm Sci. 2005;26:151–161. doi: 10.1016/j.ejps.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Forgue ST, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17:1278–1283. doi: 10.1023/a:1026451721686. [DOI] [PubMed] [Google Scholar]
- Upton RN. Cerebral uptake of drugs in humans. Clin Exp Pharmacol Physiol. 2007;34:695–701. doi: 10.1111/j.1440-1681.2007.04649.x. [DOI] [PubMed] [Google Scholar]
- Wu D-C, Xing X-Z, Wang W-A, Xie H, Wang Y-H, Jin G-Z, et al. A double blind comparison trial of l-stepholidine and perphenazine in treatment of schizophrenia. Chin J New Drugs Clin Rem. 2003;22:155–160. [Google Scholar]
- Xu S-X, Yu L-P, Han Y-R, Chen Y, Jin G-Z. Effects of tetrahydroprotoberberines on dopamine receptor subtypes in brain. Acta Pharmacol Sin. 1989;10:104–110. [PubMed] [Google Scholar]
- Yang CR, Chen L. Targeting prefrontal cortical dopamine D1 and N-methyl-D-aspartate receptor interactions in schizophrenia treatment. Neuroscientist. 2005;11:452–470. doi: 10.1177/1073858405279692. [DOI] [PubMed] [Google Scholar]
- Zhang Z-D, Zhou C-M, Jin G-Z, Zhang X, Yang L. Pharmacokinetics and autoradiography of [3H] or [14C] stepholidine. Acta Pharmacol Sin. 1990;11:289–292. [PubMed] [Google Scholar]
- Zhao Y, Wang L, Bao Y-W, Li C. A sensitive method for the detection and quantification of ginkgo flavonols from plasma. Rapid Commun Mass Spectrom. 2007;21:971–981. doi: 10.1002/rcm.2907. [DOI] [PubMed] [Google Scholar]