

Abstract
Background and purpose:
Potentiating neurosteroids are some of the most efficacious modulators of the mammalian GABAA receptor. One of the crucial interactions may be between the C20 ketone group (D-ring substituent at C17) of the neurosteroid, and the N407 and Y410 residues in the M4 domain of the receptor. In this study, we examined the contribution of hydrogen bonding between 17β-substituents on the steroid D-ring and the GABAA receptor to potentiation by neurosteroids.
Experimental approach:
Whole-cell and single-channel recordings were made from HEK 293 cells transiently expressing wild-type and mutant α1β2γ2L GABAA receptors.
Key results:
A steroid with a 17β-carbonitrile group (3α5α18nor17βCN) was a potent and efficacious potentiator of the GABAA receptor. Potentiation was also shown by a cyclosteroid in which C21 and the C18 methyl group of (3α,5α)-3-hydroxypregnan-20-one are connected within a six-membered ring containing a double bond as a hydrogen bond acceptor (3α5αCDNC12), a steroid containing a 17β-ethyl group on the D-ring (3α5α17βEt) and a steroid lacking a 17β-substituent on the D-ring (3α5α17H). Single-channel kinetic analysis indicates that the kinetic mechanism of action is the same for the neurosteroid 3α5αP, 3α5α18nor17βCN, 3α5αCDNC12, 3α5α17βEt and 3α5α17H. Interestingly, 3α5α17βEt, at up to 3 µM, was incapable of potentiating the α1N407A/Y410F double mutant receptor.
Conclusions and implications:
Hydrogen bonding between the steroid 17β-substituent and the GABAA receptor is not a critical requirement for channel potentiation. The α1N407/Y410 residues are important for neurosteroid potentiation for reasons other than hydrogen bonding between steroid and receptor.
Keywords: GABAA receptor, channel, modulation, neurosteroid
Introduction
Some of the most effective modulators of the mammalian GABAA receptor are potentiating neuroactive steroids, having possible applications as anxiolytics, anticonvulsants, sedatives and anaesthetics (Herd et al., 2007; Meldrum and Rogawski, 2007). Recent work has provided significant new insights into the mechanisms of action of potentiating steroids. The interaction of these drugs with the α1β2γ2L GABAA receptor leads to an increase in the channel open probability due to specific changes in the channel open and closed times (Akk et al., 2001; 2005;). In whole-cell recordings, the peak current is enhanced when the steroid is coapplied with GABA.
The essential features of GABAergic steroids with high potency and efficacy have been long considered a steroid skeleton with a 3α-hydroxyl group which functions as a hydrogen bond donor, and a 17β-substituent which is capable of being a hydrogen bond acceptor (Phillipps, 1974; Harrison et al., 1987). A more recent study examining GABAA receptor modulation by the neurosteroid (3α,5α)-3-hydroxypregnan-20-one (3α5αP) proposed that the 3α-hydroxyl and 20-ketone group of this steroid interact via hydrogen bonds with the α1Q241 and α1N407/Y410 residues, respectively (Hosie et al., 2006). However, there is evidence that the steroid binding site is accessible to a number of structurally distinct compounds. Steroid analogues with ring systems other than the steroid ring system are potent modulators of the GABAA receptor, and the presently available evidence suggests that these classes of compounds act via the classic steroid binding site (Li et al., 2006; Scaglione et al., 2008). In addition, the marine cembranoid eupalmerin acetate potentiates the α1β2γ2L GABAA receptor through a kinetic mechanism similar to that of the neurosteroid 3α5αP, while mutations to the putative steroid binding site residues (α1Q241, α1N407/Y410) disrupt potentiation (Li et al., 2008). These findings have raised the question of whether the interactions described for the neurosteroid 3α5αP are required for channel modulation by other steroid analogues.
Recently, we reported that the steroid 5α-pregnan-20-one (3-deoxy5αP) was able to potentiate wild-type α1β2γ2L GABAA receptors (Akk et al., 2008). Additional results obtained with steroid analogues and receptors with mutations to residues α1S240 and α1Q241 led us to conclude that the importance of residue α1Q241 for receptor modulation by steroids extends beyond its ability to act as a hydrogen bond acceptor for a steroid 3α-hydroxyl group.
In this study, we examined the role of steroid receptor hydrogen bond interactions involving the steroid 17β-substituent. Whole-cell and single-channel recordings were used to characterize GABAA receptor potentiation by steroids differing in substitutions at the 17β-position on the D-ring of the steroid. We examined a steroid with a carbonitrile group, (3α,5α,17β)-3-hydroxy-18-nor-androstane-17-carbonitrile (3α5α18nor17βCN), a cyclosteroid in which C21 and the C18 methyl group are incorporated into a six-membered ring containing a double bond (3α,5α)-13,24-cyclo-18,21-dinorchol-20(22)-en-3-ol (3α5αCDNC12), a steroid containing an ethyl group attached to C17, (3α,5α)-pregnan-3-ol (3α5α17βEt) and a steroid lacking substitution at position C17, (3α,5α)-androstan-3-ol (3α5α17H), all shown in Figure 1. The data indicated that the chemical nature of the substitution had a relatively small effect on the ability of the steroid to potentiate channel activity. Steroids with substitutions that are incapable of forming a hydrogen bond could still potentiate the receptor and demonstrated kinetic mechanisms that were indistinguishable from those observed in the presence of 3α5αP. We infer from the data that the hydrogen bond between the steroid D-ring and the binding site is not a critical requirement for potentiation of the α1β2γ2L GABAA receptor.
Figure 1.
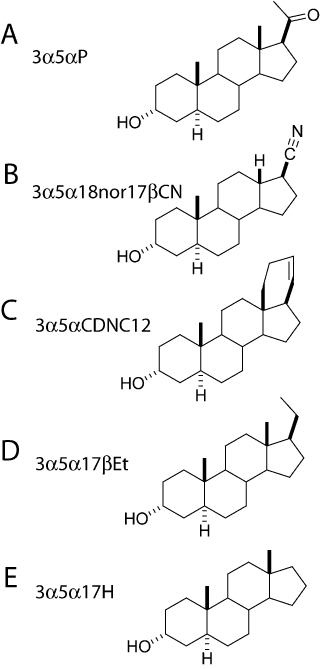
Structures of the steroid analogues. Molecular structures of the neurosteroid 3α5αP (A), and the steroid analogues 3α5α18nor17βCN (B), 3α5αCDNC12 (C), 3α5α17βEt (D) and 3α5α17H (E).
Methods
Cell preparation
The experiments were carried out on HEK 293 cells expressing rat wild-type and mutant α1β2γ2L GABAA receptors. The receptors were transiently expressed using a modified technique based on calcium phosphate precipitation (Akk, 2002). The α1N407A/Y410F mutations were generated using QuikChange (Stratagene, San Diego, CA, USA). The effects of the mutations on channel activation have been described previously (Li et al., 2006; 2007;). The α1 subunit is epitope (FLAG) tagged in the amino terminal of the subunit (Ueno et al., 1996). Cells expressing high levels of receptors were determined using a bead-binding technique where the presence of the FLAG peptide was detected with a mouse monoclonal antibody to the FLAG epitope (M2, Sigma-Aldrich, St Louis, MO, USA), which had been adsorbed to beads with a covalently attached goat anti-mouse IgG antibody (Dynal, Great Neck, NY, USA).
Electrophysiological assays
The experiments were carried out using standard single-channel patch clamp and whole-cell voltage clamp methods. The bath solution contained (in mM): 140, NaCl; 5, KCl; 1, MgCl2; 2, CaCl2; 10, glucose; and 10 HEPES (pH 7.4). In single-channel recordings, the pipette solution contained (in mM): 120, NaCl; 5, KCl; 10, MgCl2; 0.1, CaCl2; 20, tetraethylammonium chloride; 5, 4-aminopyridine; 10, glucose; 10, HEPES (pH 7.4). In whole-cell recordings, the pipette solution contained (in mM): 140, CsCl; 4, NaCl; 4, MgCl2; 0.5, CaCl2; 5, EGTA; 10, HEPES (pH 7.4).
The agonist (GABA) and steroid modulators were added to the pipette solution in single-channel recordings, or applied through the bath using an SF-77B fast perfusion stepper system (Warner Instruments, Hamden, CT, USA) in whole-cell experiments. The steroids were initially dissolved in dimethyl sulphoxide (DMSO) at 5–10 mM concentration, and diluted immediately before the experiment. The maximal DMSO concentration in diluted steroid solutions was 0.1%. We have previously found that channel activation by GABA is not affected by the presence of up to 0.3% DMSO (Li et al., 2007). All experiments were carried out at room temperature (19–22°C).
The recording and analysis of single-channel currents have been described in detail previously (Akk et al., 2001; 2004; Steinbach and Akk, 2001). The pipette potential was held at +60 to +80 mV, which translates to an approximately −100 to −120 mV potential difference across the patch membrane. The channel activity was recorded using an Axopatch 200B amplifier (Molecular Devices, Union City, CA, USA), low-pass filtered at 10 kHz and acquired with a Digidata 1320 series interface at 50 kHz using pClamp software (Molecular Devices). Prior to kinetic analysis, the currents were low-pass filtered at 2–3 kHz, and the data were idealized using the segmented-k-means algorithm (Qin et al., 1996). The open and closed times were estimated from the idealized currents using a maximum likelihood method, which incorporates a correction for missed events (QuB Suite; http://www.qub.buffalo.edu). The kinetic analysis was limited to clusters, that is, episodes of intense activity originating from the activation of a single ion channel, or fragments of clusters containing no overlapping currents.
The recording and analysis of whole-cell currents were carried out as described previously (Li et al., 2006). The cells were clamped at −60 mV. The cells were exposed to GABA (at an approximate EC25 concentration) and steroids for 4 s with 30 s wash-outs separating successive applications. The current traces were low-pass filtered at 2 kHz and digitized at 10 kHz. The analysis of whole-cell currents was carried out using the pClamp 9.0 software package, and was aimed at determining the peak amplitude. Each cell was, prior to testing the effects of steroids, examined using two GABA concentrations (5 µM and 1 mM) to determine the approximate GABA EC50 for the cell in order to verify the expression of γ subunit in the receptor complexes (Boileau et al., 2003; Li et al., 2006).
Data analysis
Statistical analyses were carried out using paired Student's t-test (Excel, Microsoft, Richmond, WA, USA) or analysis of variance with Dunnett's correction (Systat 7.0; Systat Software, Inc., Point Richmond, CA, USA).
Materials
The 3α5α18nor17βCN was prepared from a 17-keto-18-norsteroid precursor using standard methods for the conversion of a steroid 17-ketone group into a 17β-carbonitrile group (Han et al., 1996). The compound had spectroscopic properties consistent with the assigned structure, was chromatographically pure and gave the correct elemental analysis. The 3α5αCDNC12 was prepared as described previously (Jiang et al., 2003).
The 3α5α17βEt was prepared as follows. To a stirred solution of diethyl azodicarboxylate (0.17 mL, 0.39 mM, 40% in toluene) and (3β,5α)-pregnan-3-ol (purchased from Steraloids, Newport, RI, USA, 95 mg, 0.31 mM) dissolved in anhydrous tetrahydrofuran (0.6 mL), trifluoroacetic acid (0.03 mL, 0.38 mM) was added at room temperature, followed by the addition of solid triphenylphosphine (100 mg, 0.38 mM). After stirring the reaction for 10 min, sodium benzoate (55 mg, 0.38 mM) was added and the reaction was stirred overnight. Because a large amount of starting steroid was still detected by thin layer chromatography, additional reagents (diethyl azodicarboxylate, 0.08 mL, 0.18 mM; triphenyl phosphine, 50 mg, 0.19 mM; sodium benzoate, 27 mg, 0.19 mM) were added, and the reaction was stirred for another 20 h. Volatiles were removed under reduced pressure. The resultant crude 3α-benzoate ester was then hydrolysed by refluxing the crude product overnight with NaHCO3 (90 mg, 1.07 mM) in methanol (10 mL). The solvents were evaporated, and the crude product was extracted with methylene chloride. The combined organic extracts were washed with water and brine, and dried over anhydrous Na2SO4. The crude 3α5α17βEt product was further purified by column chromatography on silica gel using 10% ethyl acetate in hexane as eluent. This procedure yielded pure 3α5α17βEt (68%, 65 mg) which had: melting point 186–88°C; reported melting point 186°C (Pancrazi and Khuong-Huu, 1975).
The structures of the steroid compounds are given in Figure 1. Other chemicals including GABA and salts were purchased from Sigma-Aldrich.
Results
A steroid with the 17β-carbonitrile group potentiates the α1β2γ2L GABAA receptor
Our previous data have indicated that some steroids containing a C17 carbonitrile group are potent potentiators of the GABAA receptor (Akk et al., 2004). Here, we examined the potentiation of the α1β2γ2L GABAA receptor by (3α,5α,17β)-3-hydroxy-18-nor-androstane-17-carbonitrile (3α5α18nor17βCN; Figure 1B). In whole-cell recordings from 13 cells, co-application of 1 µM 3α5α18nor17βCN with 5 µM GABA enhanced the peak response to 320 ± 46% of control (mean ± SEM; P < 0.001, paired t-test). Sample current traces are shown in Figure 2A.
Figure 2.
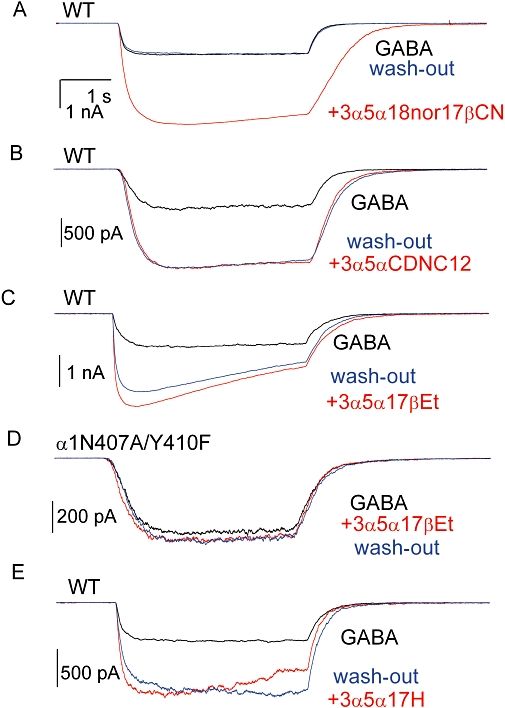
Channel modulation by the neuroactive steroids. The wild-type α1β2γ2L receptors were activated by 5 µM GABA (∼EC25) in the absence and presence of 1 µM 3α5α18nor17βCN (A), 1 µM 3α5αCDNC12 (B), 3 µM 3α5α17βEt (C) or 3 µM 3α5α17H (E). The α1N407A/Y410F double mutant receptor was activated by 10 µM GABA (∼EC25) in the absence and presence of 3 µM 3α5α17βEt (D). All steroid analogues potentiated the wild-type receptor. Exposure to 3α5α17βEt did not modulate the current response from receptors containing the α1N407A/Y410F double mutation. Note the lack of wash-out following exposure to 3α5αCDNC12, 3α5α17βEt or 3α5α17H.
We next determined the concentration–effect relationship for 3α5α18nor17βCN. The experiments were conducted on nine cells exposed to GABA in the presence of 30–3000 nM steroid. Each steroid application was followed by one or more applications of GABA alone, to assure full wash-out of the steroid and prevent a slowly accumulating potentiating effect. The data show that the concentration eliciting half-maximal effect is 148 ± 16 nM, and the maximal effect is 365 ± 9% of control. The summary of the data is shown in Figure 3A.
Figure 3.
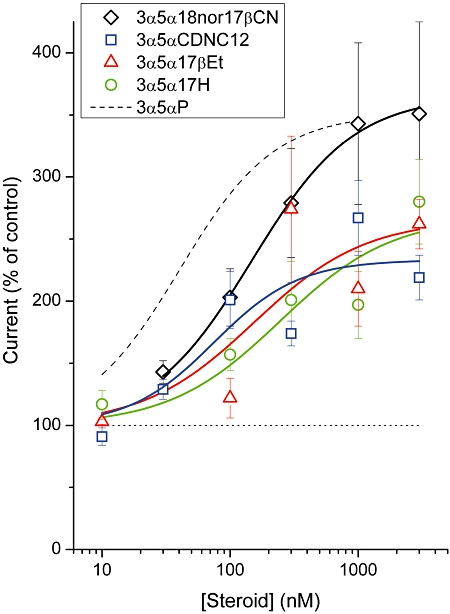
Summary of the steroid analogue concentration–effect relationships. The wild-type α1β2γ2L receptors were activated by 5 µM GABA (∼EC25) in the absence and presence of the steroids 3α5α18nor17βCN, 3α5αCDNC12, 3α5α17βEt and 3α5α17H. The data points show mean ± SEM from four to eleven cells. The effect by 3α5α18nor17βCN was determined by exposing a cell to various concentrations of steroid for 4 s, separated by 30 s wash-out periods and control exposures to GABA alone. The effects by 3α5αCDNC12, 3α5α17βEt and 3α5α17H were determined by exposing a cell to GABA followed by an application of GABA + a single concentration of the steroid. No wash-out of the effect was observed following an up to 5 min exposure to bath solution. The curves were fitted to the Hill equation with an offset fixed at 100%. For 3α5α18nor17βCN, the best-fit parameters are: maximal potentiation = 365 ± 9%, EC50= 148 ± 16 nM and nH= 1.1 ± 0.1. For 3α5αCDNC12, the best-fit parameters are: maximal potentiation = 233 ± 31%, EC50= 75 ± 62 nM and nH= 1.3 ± 1.2. For 3α5α17βEt, the best-fit parameters are: maximal potentiation = 266 ± 48%, EC50= 154 ± 188 nM and nH= 1 (fixed). For 3α5α17H, the best-fit parameters are: maximal potentiation = 268 ± 32%, EC50= 246 ± 173 nM and nH= 1 (fixed). The dashed line corresponds to data in the presence of the neurosteroid 3α5αP. The best-fit parameters for 3α5αP are: maximal potentiation = 351%, EC50= 41 nM and nH= 1.2 (Akk et al., 2008).
We have previously shown using single-channel patch clamp that in the presence of 50 µM GABA, the intracluster open and closed time histograms are adequately described using sums of three exponentials, and that co-application of steroids with GABA has specific effects on the open and closed time distributions. For example, co-application of 3α5αP with GABA increases the mean duration and prevalence of OT3 (the longest-lived open time component), and decreases the prevalence of CT3 (the longest intracluster closed time component) (Akk et al., 2005; see also Tables 1 and 2). Some steroid analogues may selectively affect a subset of these kinetic components (Akk et al., 2004; Li et al., 2007).
Table 1.
Summary of single-channel kinetic analysis of the open time distributions from the α1β2γ2L receptor under control conditions, and in the presence of steroids
| Agonist, modulator | OT1 (ms) | Fraction OT1 | OT2 (ms) | Fraction OT2 | OT3 (ms) | Fraction OT3 | n |
|---|---|---|---|---|---|---|---|
| 50 µM GABA | 0.30 ± 0.06 | 0.20 ± 0.04 | 3.0 ± 0.6 | 0.67 ± 0.06 | 6.8 ± 2.9 | 0.13 ± 0.07 | 5 |
| +1 µM 3α5αP | 0.41 ± 0.04* | 0.39 ± 0.07** | 2.4 ± 0.9ns | 0.23 ± 0.03*** | 14.1 ± 2.1* | 0.38 ± 0.04* | 4 |
| +1 µM 3α5α18nor17βCN | 0.24 ± 0.12ns | 0.25 ± 0.14ns | 2.6 ± 1.8ns | 0.30 ± 0.09*** | 16.3 ± 4.4** | 0.46 ± 0.10*** | 4 |
| +3 µM 3α5αCDNC12 | 0.39 ± 0.09ns | 0.35 ± 0.07ns | 4.6 ± 3.6ns | 0.25 ± 0.12*** | 16.5 ± 1.7** | 0.40 ± 0.11*** | 4 |
| +10 µM 3α5α17βEt | 0.45 ± 0.11* | 0.39 ± 0.03* | 2.6 ± 1.2ns | 0.22 ± 0.03*** | 17.0 ± 2.2** | 0.39 ± 0.01*** | 4 |
| +10 µM 3α5α17H | 0.38 ± 0.03ns | 0.39 ± 0.12* | 1.5 ± 0.2ns | 0.23 ± 0.12*** | 14.8 ± 4.3** | 0.38 ± 0.05*** | 6 |
The intracluster open time histograms were fitted to a sum of three exponentials. The table gives the mean durations ± SD (OT1-3) and average relative contributions ± SD (fraction OT1-3) for the three open time components, and the number of patches under each condition (n). Statistical analysis was carried out using analysis of variance with Dunnett's correction (Systat 7.0, Systat Software, Inc., Point Richmond, CA, USA). The significance levels apply to comparison to control condition (50 µM GABA). The data for 3α5αP are from Akk et al. (2008).
P < 0.05;
P < 0.01;
P < 0.001; ns, not significant.
Summary of single-channel kinetic analysis of the closed time distributions from the α1β2γ2L receptor under control conditions, and in the presence of steroids
| Agonist, modulator | CT1 (ms) | Fraction CT1 | CT2 (ms) | Fraction CT2 | CT3 (ms) | Fraction CT3 | n |
|---|---|---|---|---|---|---|---|
| 50 µM GABA | 0.15 ± 0.01 | 0.59 ± 0.09 | 1.4 ± 0.3 | 0.13 ± 0.04 | 13.6 ± 4.0 | 0.28 ± 0.06 | 5 |
| +1 µM 3α5αP | 0.22 ± 0.04* | 0.64 ± 0.12ns | 1.4 ± 0.2ns | 0.30 ± 0.10* | 14.3 ± 1.2ns | 0.05 ± 0.01*** | 4 |
| +1 µM 3α5α18nor17βCN | 0.16 ± 0.05ns | 0.65 ± 0.09ns | 1.1 ± 0.5ns | 0.22 ± 0.06** | 11.3 ± 2.8ns | 0.13 ± 0.04*** | 4 |
| +3 µM 3α5αCDNC12 | 0.16 ± 0.03ns | 0.60 ± 0.03ns | 1.1 ± 0.2ns | 0.30 ± 0.02*** | 11.1 ± 2.4ns | 0.11 ± 0.02*** | 4 |
| +10 µM 3α5α17βEt | 0.23 ± 0.04ns | 0.67 ± 0.06ns | 1.3 ± 0.2ns | 0.21 ± 0.03* | 14.8 ± 5.0ns | 0.12 ± 0.05*** | 4 |
| +10 µM 3α5α17H | 0.26 ± 0.06** | 0.63 ± 0.04ns | 1.3 ± 0.1ns | 0.32 ± 0.03*** | 11.0 ± 2.3ns | 0.05 ± 0.01*** | 6 |
The intracluster closed time histograms were fitted to a sum of three exponentials. The table gives the mean durations ± SD (CT1-3) and average relative contributions ± SD (fraction CT1-3) for the three closed time components, and the number of patches under each condition (n). Statistical analysis was carried out using analysis of variance with Dunnett's correction (Systat 7.0, Systat Software, Inc., Point Richmond, CA, USA). The significance levels apply to comparison to control condition (50 µM GABA). The data for 3α5αP are from Akk et al. (2008).
P < 0.05;
P < 0.01;
P < 0.001; ns, not significant.
We examined the kinetic mechanism of potentiation by 3α5α18nor17βCN using cell-attached single-channel patch clamp. The receptors were exposed to 50 µM GABA + 1 µM steroid. The data indicate that the application of steroid affected the channel open and closed time distributions. Compared to the control data (GABA alone), the currents in the presence of steroid exhibited a prolongation of the mean OT3, an increase in the prevalence of OT3 and a decrease in the prevalence of CT3. Sample current traces are shown in Figure 4A,B, and the data are summarized in Tables 1 and 2. Overall, the data indicate that 3α5α18nor17βCN acts mechanistically similarly to 3α5αP, and that neither the substitution of the 17β-acetyl group with a 17β-carbonitrile group nor the removal of the C18 methyl group affects steroid interactions with the α1β2γ2L GABAA receptor. We note that the carbonitrile group can act as a hydrogen bond acceptor in lieu of the C20 ketone group.
Figure 4.
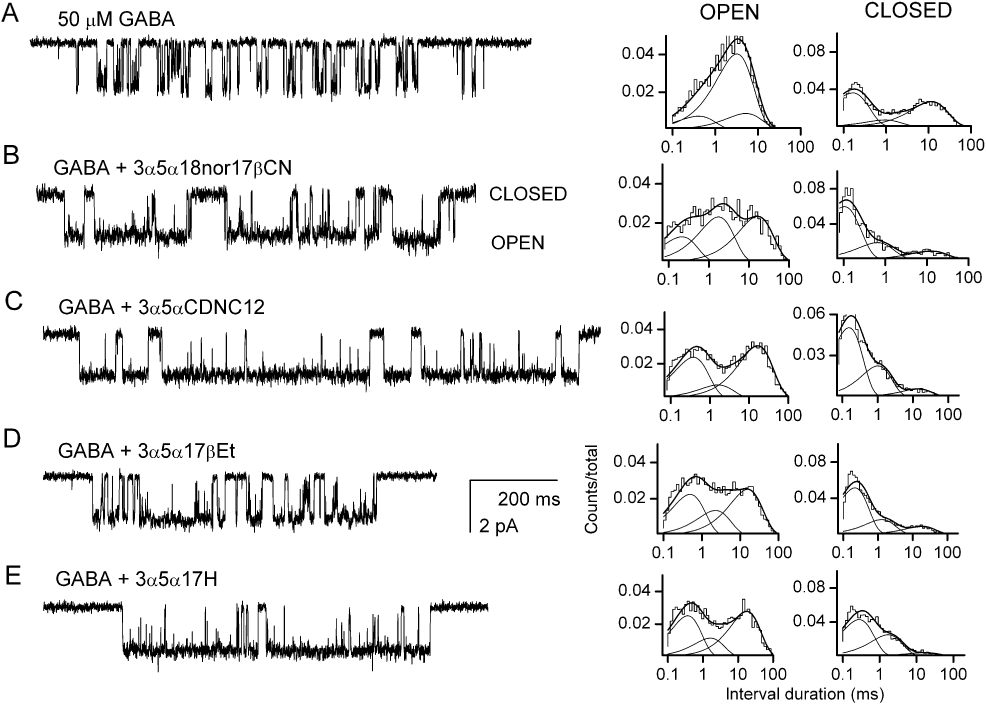
Single-channel currents in the presence of GABA and steroid analogues. (A) A sample single-channel cluster elicited by 50 µM GABA, and the open and closed time histograms. Channel openings are shown as downward deflections. The open times were 0.37 ms (13%), 2.9 ms (72%) and 4.8 ms (15%). The closed times were 0.14 ms (54%), 0.9 ms (10%) and 10.6 ms (36%). (B) A sample single-channel cluster elicited by 50 µM GABA in the presence of 1 µM 3α5α18nor17βCN, and the open and closed time histograms. The open times were 0.20 ms (23%), 1.6 ms (39%) and 15.2 ms (38%). The closed times were 0.10 ms (68%), 0.6 ms (21%) and 11.6 ms (10%). (C) A sample single-channel cluster elicited by 50 µM GABA in the presence of 3 µM 3α,5αCDNC12, and the open and closed time histograms. The open times were 0.34 ms (40%), 1.5 ms (12%) and 15.2 ms (48%). The closed times were 0.14 ms (64%), 0.9 ms (28%) and 13.0 ms (7%). (D) A sample single-channel cluster elicited by 50 µM GABA in the presence of 10 µM 3α5α17βEt, and the open and closed time histograms. The open times were 0.45 ms (38%), 2.1 ms (22%) and 14.3 ms (40%). The closed times were 0.20 ms (68%), 1.2 ms (21%) and 16.7 ms (11%). (E) A sample single-channel cluster elicited by 50 µM GABA in the presence of 10 µM 3α5α17H, and the open and closed time histograms. The open times were 0.33 ms (41%), 1.5 ms (18%) and 16.1 ms (42%). The closed times were 0.26 ms (60%), 1.6 ms (35%) and 14.9 ms (6%).
We also tested direct activation by the steroid. A cell was successively exposed to 3 µM 3α5α18nor17βCN or 5 µM GABA (∼EC25), and the extent of direct activation was estimated by comparing the respective peak macroscopic responses. In five cells, the response to steroid was 10 ± 3% (mean ± SEM) of that to GABA.
Potentiation of the GABAA receptor by a cyclosteroid
We next tested the ability of the cyclosteroid (3α,5α)-13,24-cyclo-18,21-dinorchol-20(22)-en-3-ol (3α5αCDNC12; Figure 1C) to potentiate the GABAA receptor. This cyclosteroid contains a double bond in the newly formed ring that can potentially act as a hydrogen bond acceptor in its interactions with the GABAA receptor. In seven cells, co-application of 1 µM 3α5αCDNC12 enhanced the peak response to 267 ± 30% of control (mean ± SEM; P < 0.01). A sample current trace is shown in Figure 2B.
The steroid 3α5αCDNC12 is very hydrophobic (logP = 6.92, calculated using Advanced Chemistry Development software, version 8.19), which is likely to account for the inability to wash out the potentiating effect (Figure 2B). In order to gain insight into the concentration–effect relationship, we measured the potentiating effect of 10, 30, 100 and 300 nM, and 3 µM 3α5αCDNC12 on individual cells. In these experiments, each cell was only once exposed to the steroid, so that a single data point was obtained from a cell. The data show that the presence of 10 nM 3α5αCDNC12 was without effect on the currents elicited by 5 µM GABA (91 ± 7%, n= 4 cells, P > 0.30). In contrast, the co-application of 100 nM steroid significantly enhances the peak response (201 ± 23%, n= 4 cells, P < 0.05). The concentration–effect relationship is summarized in Figure 3. We estimate that the EC50 for the 3α5αCDNC12 potentiation curve was 75 ± 62 nM. Direct activation by 3 µM 3α5αCDNC12 resulted in a response that was 3 ± 1% of the peak current from receptors activated by 5 µM GABA (n= 5 cells).
We conducted single-channel patch clamp experiments to determine the kinetic mechanism of action of 3α5αCDNC12. In these experiments, 3 µM 3α5αCDNC12 was co-applied with 50 µM GABA. The data indicate that the steroid acts by enhancing the mean duration and prevalence of OT3, and reducing the prevalence of CT3 (Figure 4C). Therefore, the kinetic mechanism of action of 3α5αCDNC12 is similar to that of 3α5αP and 3α5α18nor17βCN. These data are summarized in Tables 1 and 2.
The steroid (3α,5α)-pregnan-3-ol potentiates the wild-type GABAA receptor, but not a receptor containing the α1N407A/Y410F double mutation
We next tested the ability of the steroid (3α,5α)-pregnan-3-ol (3α5α17βEt; Figure 1D) to potentiate the GABAA receptor. This steroid contains a 17β-ethyl group that is unable to form a hydrogen bond with the GABAA receptor. To our surprise, we found that 3α5α17βEt potentiated the activity from the α1β2γ2L receptor. The application of 3 µM 3α5α17βEt enhanced the macroscopic peak current elicited by 5 µM GABA to 262 ± 20% of control (mean ± SEM; n= 11 cells; P < 0.001), indicating that a group capable of forming a hydrogen bond is not required as a 17β-substituent on the D-ring. A sample whole-cell current trace is shown in Figure 2C. Similar to 3α5αCDNC12, the effect of 3α5α17βEt was not reversed following wash-outs with bath up to 5 min. Accordingly, in order to determine the concentration dependency of this steroid, we measured the effect of 10, 100 and 300 nM, and 1 µM 3α5α17βEt using a new cell for each data point. The findings demonstrate that the presence of 10 nM (103 ± 2%; n= 4 cells; P > 0.31) or 100 nM 3α5α17βEt (122 ± 16%; n= 4 cells, P > 0.26) was without effect on receptors activated by 5 µM GABA. When 300 nM steroid was co-applied with GABA, the peak response was enhanced to 274 ± 59% of control (n= 5 cells, P < 0.05). Our estimate for the concentration producing a half-maximal effect is 154 ± 188 nM. These results are summarized in Figure 3. The application of 3 µM 3α5α17βEt alone yielded a peak response that was 7 ± 2% of the peak current from receptors activated by 5 µM GABA (n= 5 cells).
Sample single-channel currents elicited by 50 µM GABA and 10 µM 3α5α17βEt are shown in Figure 4D. The data demonstrate that the steroid acts by enhancing the mean duration and fraction of OT3, and reducing the prevalence of CT3. We note that the same kinetic parameters were modified when the receptor is exposed to 3α5αP (Akk et al., 2005). The findings are summarized in Tables 1 and 2.
A previous work has shown reduced sensitivity to potentiating neurosteroids in a receptor containing the α1N407A/Y410F double mutation, and suggested that the mutations act by disrupting a hydrogen bond with the steroid 17β-acetyl group (Hosie et al., 2006). We examined the effect of the α1N407A/Y410F double mutation on channel potentiation by 3α5α17βEt. The mutant receptors were activated by 10 µM GABA (EC25; Li et al., 2006) in the absence and presence of 3 µM 3α5α17βEt. The presence of steroid was without effect on the peak current (113 ± 41%; n= 17 cells; P > 0.2). A sample current trace is shown in Figure 2D.
The steroid 3α5α17H potentiates the α1β2γ2L GABAA receptor
We also tested whether a steroid that has no substituent at C17 (3α5α17H; Figure 1E) can potentiate the GABAA receptor. In seven cells, the co-application of 3 µM 3α5α17H with 5 µM GABA enhanced the peak current to 280 ± 34% of control (P < 0.01). Sample current responses are shown in Figure 2E.
The concentration–effect relationship for this steroid was measured over a range of 10, 100 and 300 nM, and 1 µM 3α5α17H, using a new cell for each data point. The presence of 10 nM (117 ± 11%, n= 4 cells, P > 0.23) or 100 nM 3α5α17H (157 ± 13%, n= 3 cells, P > 0.05) was without effect on receptors activated by 5 µM GABA. When 300 nM steroid was co-applied with GABA, the peak response was enhanced to 201 ± 31% of control (n= 4 cells, P < 0.05). The concentration producing a half-maximal effect was 246 ± 173 nM (Figure 3). The steroid 3α5α17H was capable of directly activating GABAA receptors. Exposure of the receptors to 3 µM steroid resulted in a macroscopic peak response that was 4 ± 1% of that observed in the presence of 5 µM GABA (n= 4 cells).
The kinetic mechanism of potentiation of 3α5α17H was examined using single-channel patch clamp by co-applying 10 µM steroid with 50 µM GABA. A sample single-channel cluster is shown in Figure 4E. The data show that 3α5α17H potentiated the receptor by enhancing the mean duration and prevalence of OT3, and reducing the prevalence of CT3 (Tables 1 and 2). This indicates that the mode of action of steroid is unchanged when the C17 acetyl group is replaced with a hydrogen atom.
Discussion and conclusions
We have examined the effect of 17β-substituents with different hydrogen-bonding capabilities on steroid potentiation of α1β2γ2L GABAA receptors. The steroids used in the study were 3α5α18nor17βCN, 3α5αCDNC12, 3α5α17βEt and 3α5α17H (Figure 1). The carbonitrile and double bond found in 3α5α18nor17βCN and 3α5αCDNC12, respectively, are both hydrogen bond acceptor groups. By contrast, 3α5α17βEt and 3α5α17H do not contain hydrogen bond acceptor groups. Overall, the results indicate that the C17 substitution has a relatively small effect on the ability of the steroid to potentiate receptor function.
The lack of wash-out with some of the steroids used (3α5α17H, 3α5α17βEt and 3α5αCDNC12) prevented us from completing full concentration–effect measurements on the same cell. We had to resort to measuring a single data point per cell (i.e. each cell was exposed to a single steroid application). The data from several cells were averaged to construct the concentration–effect relationships shown in Figure 3. We note that there is some inherent variability in such approach. Our estimates for the concentrations producing a half-maximal effect are 75 nM for 3α5αCDNC12, 154 nM for 3α5α17βEt and 246 nM for 3α5α17H. For comparison, the best-fit estimate for EC50 for potentiation is 148 nM for 3α5α18nor17βCN (Figure 3) and 41 nM for the neurosteroid 3α5αP (dashed line in Figure 3; Akk et al., 2008). The finding that the concentrations producing half-maximal potentiation of the α1β2γ2L GABAA receptor was relatively similar for all test compounds, irrespective of the hydrogen-bonding capability of the 17β-substituent, was unexpected.
Single-channel kinetic measurements indicate that the mechanisms of action are strikingly similar for 3α5α17H, 3α5α17βEt, 3α5αCDNC12 and 3α5α18nor17βCN. The presence of any of these steroid analogues results in increases in the mean duration and prevalence of OT3, and a decrease in the prevalence of CT3. Exposure to the neurosteroid 3α5αP leads to modification of the same kinetic variables. We note that supramaximal concentrations of the steroid analogues were used in single-channel studies and that the extent of kinetic modifications was similar for each drug. We infer that a hydrogen bond between the 17β-substituent on the steroid D-ring and the GABAA receptor is not required to observe the archetypal steroid effect (Akk et al., 2004; 2005;).
We were surprised that our data indicated that steroids which lack a hydrogen bond acceptor at the C17 position could be potent and efficacious potentiators in our assays. Some previous studies had indicated that such steroids could act on GABAA receptors. An analogue of the anaesthetic steroid alfaxalone which contains a 17β-ethyl substituent can produce anaesthesia in rodents, albeit at a higher dose than alphaxalone (Phillipps, 1974). Similarly, 3α5α17H can inhibit the binding of t-butylbicyclophosphorothionate to rat brain membrane preparations, although, again, only at relatively high concentrations (Bolger et al., 1996). These reports support the conclusion that the hydrogen-bonding substituent at C17 is not required for potentiation, although they do suggest that potency or efficacy is affected. We note that 3α5α17βEt does not appear to potentiate the responses of α1β2γ2L GABAA receptors when they are expressed in Xenopus oocytes (S. Mennerick, pers. comm.). The reason for this difference in effect between the two expression systems is not known, but the difference indicates that steroid potentiation may be affected by additional factors in the experimental systems employed.
Interestingly, the steroid analogue 3α5α17βEt did not potentiate the α1N407A/Y410F double mutant receptor. A previous study (Hosie et al., 2006) had suggested that these mutations prevent the hydrogen bond interaction between the C20 ketone of 3α5αP and the receptor. Our experiments show that a hydrogen bonding group on the D-ring is not required for GABAA receptor potentiation. Furthermore, the experiments on the α1N407A/Y410F mutant receptor suggest that the mutations do not act by interrupting hydrogen bonding between the steroid D-ring and the receptor. At present, we do not understand the exact mechanism for the effect of the α1N407A/Y410F double mutation.
Previous studies on the human oestrogen receptor have indicated that hormone binding is stabilized by hydrogen bonds formed via the C3-OH and C17-OH groups (Tanenbaum et al., 1998). In contrast, a study of the human progesterone receptor proposed that the D-ring (C20) oxygen is not involved in making hydrogen bond contacts with the receptor (Williams and Sigler, 1998), and it has been shown that synthetic E-17-halomethylene steroids, incapable of forming hydrogen bonds with the receptor in the D-ring region, bind to the progesterone receptor with higher affinity than progesterone itself (Hillisch et al., 2003). Our previous work on the steroid interactions with the GABAA receptor showed that the C3-OH group is not critical to the steroid's ability to potentiate the receptor (Akk et al., 2008). The present work indicates that a hydrogen bond between the D-ring and the GABAA receptor is not required for channel potentiation. Unfortunately, a steroid with neither a C3 hydroxyl nor a C17 hydrogen bond acceptor is too insoluble for study. Hence, it is possible that a single interaction with the receptor, at either end of the steroid molecule, might be required for channel modulation.
In summary, we have shown that the nature of the 17β-substituent on the steroid D-ring has a relatively small effect on the ability of the steroid analogue to potentiate the α1β2γ2L GABAA receptor, and that steroids incapable of forming the hydrogen bond in the D-ring region are potent modulators of the receptor. The single-channel studies indicate that these steroids potentiate the GABAA receptor through kinetic mechanisms indistinguishable from the ones previously described for the neurosteroid 3α5αP. We infer that formation of the hydrogen bond between the 17β-substituent and the α1β2γ2L GABAA receptor is not required for channel potentiation.
Acknowledgments
We thank John Bracamontes for molecular biological work. This work was supported by a National Institutes of Health grant GM47969 (to D.F.C. and J.H.S.). J.H.S. is the Russell and Mary Shelden Professor of Anesthesiology.
Glossary
Abbreviations:
- 3-deoxy5αP
5α-pregnan-20-one
- 3α5α17βEt
(3α,5α)-pregnan-3-ol
- 3α5α17H
(3α,5α)-androstan-3-ol
- 3α5α18nor17βCN
(3α,5α,17β)-3-hydroxy-18-nor-androstane-17-carbonitrile
- 3α5αCDNC12
(3α,5α)-13,24-cyclo-18,21-dinorchol-20(22)-en-3-ol
- 3α5αP
(3α,5α)-3-hydroxypregnan-20-one
Conflicts of interest
None.
References
- Table 2.Akk G. Contributions of the non-α subunit residues (loop D) to agonist binding and channel gating in the muscle nicotinic acetylcholine receptor. J Physiol. 2002;544:695–705. doi: 10.1113/jphysiol.2002.029413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Bracamontes J, Steinbach JH. Pregnenolone sulfate block of GABAA receptors: mechanism and involvement of a residue in the M2 region of the α subunit. J Physiol. 2001;532:673–684. doi: 10.1111/j.1469-7793.2001.0673e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Bracamontes JR, Covey DF, Evers A, Dao T, Steinbach JH. Neuroactive steroids have multiple actions to potentiate GABAA receptors. J Physiol. 2004;558:59–74. doi: 10.1113/jphysiol.2004.066571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, et al. Neurosteroid access to the GABAA receptor. J Neurosci. 2005;25:11605–11613. doi: 10.1523/JNEUROSCI.4173-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Li P, Bracamontes J, Reichert DE, Covey DF, Steinbach JH. Mutations of the GABAA receptor α1 subunit M1 domain reveal unexpected complexity for modulation by neuroactive steroids. Mol Pharmacol. 2008;74:614–627. doi: 10.1124/mol.108.048520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Li T, Benkwitz C, Czajkowski C, Pearce RA. Effects of the γ2S subunit incorporation on GABAA receptor macroscopic kinetics. Neuropharmacology. 2003;44:1003–1012. doi: 10.1016/s0028-3908(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Bolger MB, Wieland S, Hawkinson JE, Xia H, Upasani R, Lan RC. In vitro and in vivo activity of 16,17-dehydro-epipregnanolones: 17,20-bond torsional energy analysis and D-ring conformation. Pharm Res. 1996;13:1488–1494. doi: 10.1023/a:1016019327120. [DOI] [PubMed] [Google Scholar]
- Han M, Zorumski CF, Covey DF. Neurosteroid analogues. 4. The effect of methyl substitution at the C-5 and C-10 positions of neurosteroid on electrophysiological activity at GABAA receptors. J Med Chem. 1996;39:4218–4232. doi: 10.1021/jm960304p. [DOI] [PubMed] [Google Scholar]
- Harrison NL, Majewska MD, Harrington JW, Barker JL. Structure–activity relationships for steroid interaction with the γ-aminobutyric acidA receptor complex. J Pharmacol Exp Ther. 1987;241:346–353. [PubMed] [Google Scholar]
- Herd MB, Belelli D, Lambert JJ. Neurosteroid modulation of synaptic and extrasynaptic GABAA receptors. Pharmacol Ther. 2007;116:20–34. doi: 10.1016/j.pharmthera.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Hillisch A, von Langen J, Menzenbach B, Droescher P, Kaufmann G, Schneider B, et al. The signficance of the 20-carbonyl group of progesterone in steroid receptor binding: a molecular dynamics and structure-based ligand design study. Steroids. 2003;68:869–878. doi: 10.1016/j.steroids.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HMA, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Jiang X, Manion BD, Benz A, Rath NP, Evers AS, Zorumski CF, et al. Neurosteroid analogues. 9. Conformationally constrained pregnanes: structure–activity studies of 13,24-cyclo-18,21-dinorcholane analogues of the GABA modulatory and anesthetic steroids (3α,5α)- and (3α,5β)-3-hydroxypregnan-20-one. J Med Chem. 2003;46:5334–5348. doi: 10.1021/jm030302m. [DOI] [PubMed] [Google Scholar]
- Li P, Covey DF, Steinbach JH, Akk G. Dual potentiating and inhibitory actions of a benz[e]indene neurosteroid analog on recombinant α1β2γ2 GABAA receptors. Mol Pharmacol. 2006;69:2015–2026. doi: 10.1124/mol.106.022590. [DOI] [PubMed] [Google Scholar]
- Li P, Bracamontes J, Katona BW, Covey DF, Steinbach JH, Akk G. Natural and enantiomeric etiocholanolone interact with distinct sites on the rat α1β2γ2L GABAA receptor. Mol Pharmacol. 2007;71:1582–1590. doi: 10.1124/mol.106.033407. [DOI] [PubMed] [Google Scholar]
- Li P, Reichert DE, Rodriguez AD, Manion BD, Evers AS, Eterovic VA, et al. Mechanisms of potentiation of the mammalian GABAA receptor by the marine cembranoid eupalmerin acetate. Br J Pharmacol. 2008;153:598–608. doi: 10.1038/sj.bjp.0707597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancrazi A, Khuong-Huu Q. Steroidal alkaloids. CLXXII. Photochemistry of azido steroids. Tetrahedron. 1975;31:2041–2048. [Google Scholar]
- Phillipps GH. Structure–activity relationships in steroidal anaesthetics. In: Halsey MJ, Millar RA, Sutton JA, editors. Molecular Mechanisms in General Anaesthesia. A Glaxo Symposium. Edinburgh/London/New York: Churchill Livingstone; 1974. pp. 32–47. [Google Scholar]
- Qin F, Auerbach A, Sachs F. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys J. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione JB, Jastrzebska I, Krishnan K, Li P, Akk G, Manion BD, et al. Neurosteroid analogues. 14. Alternative ring system scaffolds: GABA modulatory and anesthetic actions of cyclopenta[b]phenanthrenes and cyclopenta[b]anthracenes. J Med Chem. 2008;51:1309–1318. doi: 10.1021/jm701128r. [DOI] [PubMed] [Google Scholar]
- Steinbach JH, Akk G. Modulation of GABAA receptor gating by pentobarbital. J Physiol. 2001;537:715–733. doi: 10.1111/j.1469-7793.2001.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domains. Proc Natl Acad Sci USA. 1998;95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Zorumski C, Bracamontes J, Steinbach JH. Endogenous subunits can cause ambiguities in the pharmacology of exogenous γ-aminobutyric acidA receptors expressed in human embryonic kidney 293 cells. Mol Pharmacol. 1996;50:931–938. [PubMed] [Google Scholar]
- Williams SP, Sigler PB. Atomic structure of progesterone complexed with its receptor. Nature. 1998;393:392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]