

Abstract
Background and purpose:
It has been previously shown that high levels of nitric oxide (NO), from NO donors, kill neurones, but the mechanisms are unclear.
Experimental approach:
The effects of NO donors on the electrical properties of rat cultured cerebellar granule cells (CGC neurones) were investigated using the whole-cell patch-clamp technique.
Key results:
The NO donor (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA-NONOate or NOC-18) caused a rapid, persistent, but fully reversible inward current that was associated with an increase in baseline noise and was concentration dependent (100 µM–10 mM). The response to 3 mM DETA-NONOate was completely inhibited by 1 mM gadolinium, but not by NO scavengers (1 mM haemoglobin or 1 mM PTIO) or glutamate receptor antagonists (10 µM MK-801 or 60 µM CNQX). Application of decomposed 3 mM DETA-NONOate or 3 mM nitrite had no effect. In contrast, the NO donor S-nitrosoglutathione (GSNO) caused a rapid, persistent, but fully reversible outward current that was also concentration dependent (1–10 mM). The 3 mM GSNO response was unaltered by NO scavengers, glutamate antagonists or gadolinium, but was mimicked by decomposed 3 mM GSNO and 3 mM oxidized glutathione.
Conclusions and implications:
These results suggest that DETA-NONOate directly activates cation-selective channels, causing an inward current in CGCs. In contrast, GSNO causes an outward current in these cells. Some of the effects of these NO donors are independent of NO, and thus caution is required in interpreting results when using high concentrations of these compounds.
Keywords: nitric oxide, cell death, GSNO, cation, NO donor, S-nitrosoglutathione, excitotoxicity, cerebellar granule cell, cultured, neurones
Introduction
In the healthy brain, nitric oxide (NO) is produced mainly by neuronal nitric oxide synthase expressed in a subset of neurones, and endothelial nitric oxide synthase in endothelium, but during inflammation inducible nitric oxide synthase is expressed in astrocytes and/or microglia (Guix et al., 2005). High NO is toxic to neurones, and there are numerous studies demonstrating this with cultured neurones (Dawson et al., 1991; Lipton et al., 1993; Bonfoco et al., 1995; Leist et al., 1997; Almeida et al., 2001; Bal-Price and Brown, 2001; Takahata et al., 2003; Mander et al., 2005). The mechanisms by which NO is toxic to neurones are unclear, but may include inhibition of mitochondrial respiration (Almeida et al., 2001; Bal-Price and Brown, 2001; Mander et al., 2005), activation of glutamate release resulting in excitotoxicity (Leist et al., 1997; Bal-Price and Brown, 2001; Takahata et al., 2003) and/or conversion of NO to peroxynitrite, which results in the induction of oxidative stress (Lipton et al., 1993; Bonfoco et al., 1995).
NO has a relatively short half-life, because it reacts directly with oxygen and because NO is broken down by a variety of metabolic routes (Kelm, 1999). Therefore, in the laboratory, NO is usually supplied by an NO donor which maintains a sustained level of NO by its continuous release. Various types of NO donors exist including organic nitrates (such as glyceryl trinitrate), S-nitrosothiols [e.g. S-nitrosoglutathione (GSNO)], transition metal nitrosyls (e.g. sodium nitroprusside) and NONOates [e.g. (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate, also known as DETA-NONOate or NOC-18]. DETA-NONOate is one of the most commonly used NO donors because: (i) it has a half-life of about 20 h that maintains NO levels over an extended period; (ii) its spontaneous break-down is not affected by the presence of light, metals, thiols or cells; (iii) its break-down produces only NO and non-toxic diethylenetriamine (DETA) (Feelisch, 1998; Yamamoto and Bing, 2000; Wang et al., 2002).
We and others have previously shown that DETA-NONOate causes delayed cell death of cerebellar granule neurones that is accompanied by inhibition of mitochondrial respiration, decrease in neuronal ATP and release of glutamate (Bal-Price and Brown, 2001). The inhibition of mitochondrial respiration by NO and the subsequent neuronal death are greater at low oxygen levels (hypoxia) because of the competition between NO and oxygen for binding to mitochondrial cytochrome oxidase (Mander et al., 2005). The neuronal death induced by NO can be prevented by antagonists of the NMDA-type glutamate receptor, indicating that the death is excitotoxic (Leist et al., 1997; Bal-Price and Brown, 2001).
Using a voltage-sensitive fluorescent dye, we recently found that the NO donor DETA-NONOate caused a rapid, apparent depolarization of cerebellar granule neurones in culture (Jekabsone et al., 2007). Here, we set out to investigate the mechanism of this depolarization by patch-clamping these neurones and investigating the affects of NO donors on whole-cell currents. We found that the DETA-NONOate rapidly and reversibly induced an inward current in these cells, but this was induced by the DETA-NONOate molecule itself rather than NO release. In contrast, GSNO caused an outward current, which could be mimicked by its break-down products.
Methods
Cell culture
We used cerebellar granule cell (CGC) cultures prepared from 7-day-old Wistar rats as described in detail previously (Bal-Price and Brown, 2001). The use of animals in this study was in accordance with the UK Animals (Scientific Procedures) Act 1986. Pups were anaesthetized with 5% halothane in oxygen, followed by decapitation. The adequacy of anaesthesia was tested by pinching the skin between the toes of the rats, and they were then decapitated only if there was no response. Brains were removed under sterile conditions, and the cerebellum dissected and dissociated in Versene solution (1:5000; Gibco BRL, Paisley, UK). Cells were plated onto glass cover slips coated with 0.001% poly-l-lysine at 0.25 × 106 cells·cm−2 in 24-well plates and ara-C was added 24h later. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 under Dulbecco's modified Eagle's medium supplemented with 5% horse serum, 5% fetal calf serum, 38 mM glucose, 5 mM HEPES, 2 mM glutamine, 25 mM KCl and 10 µg·mL−1 gentamicin. In addition to neurones, CGC cultures contained 22 ± 4% astrocytes and 2 ± 1% microglia as assessed by immunocytochemistry using antibodies against glial fibrillary acidic protein (a marker for astrocytes; Autogen Bioclear, Calne, UK) and OX-42 (a marker for microglia; Autogen Bioclear).
Electrophysiology
Following 6–8 days in vitro, cover slips were transferred to a recording chamber, and electrophysiological measurements were performed in the whole-cell configuration using an Axopatch 200 amplifier (Axon Instruments, Union City, CA, USA), Lab-PC+ A/D board (National Instruments, Inc., Austin, TX, USA) and the Strathclyde Electrophysiology Software Package (v.3.2.9). All experiments were performed in voltage-clamp mode. Currents were filtered at a frequency of 2 kHz (−3 dB), using the four-pole, low-pass Bessel filter provided on the amplifier, and acquired at a sampling frequency of 5 kHz. The cell membrane potential was routinely held at –60 mV. Patch electrodes were pulled with a Sutter P87 (Novato, CA 94949, USA) and type GC120TF-10 borosilicate glass (Harvard Apparatus, Edenbridge, Kent, UK). Pipette resistances ranged from 2.0 to 3.5 MΩ. Series resistance was always less than 5 MΩ and was not compensated for. Cells were perfused in a gravity fed bath with a constant laminar flow of saline at a rate of 4–5 mL·min−1, and rapid application of solutions (<100 ms) was achieved using a ValveBank 8II (Automate Scientific, Inc., San Francisco, CA, USA). After entering the whole-cell configuration, the membrane current was allowed to stabilize for at least 2 min before recordings were made. Maintaining the stability of the patches was technically challenging, and they would typically last for only 6–10 min. Patch pipettes were filled with filtered (0.2 µm, Millipore, Billerica, MA, USA) intracellular saline containing (mM): 140, CsCl; 1.0, MgCl2; 1.0, CaCl2; 10.0, EGTA; and 10, HEPES (pH 7.2) with CsOH. The cells were continuously perfused with an extracellular solution containing 140, NaCl; 5.4, KCl; 1.0, MgCl2; 1.0, CaCl2; and 10, HEPES (pH 7.2) with NaOH. Test solutions were dissolved in extracellular saline. Both test solutions and salines were prepared each day. All responses presented here were consistently reproduced in at least 10 cells derived from five or more independent neuronal cultures. Recordings were performed in the dark, at room temperature.
All measurements are shown as mean ± SEM.
Materials
All cell culture reagents were obtained from Gibco BRL, except fetal calf serum, which was from Labtech International (Ringmer, UK). DETA-NONOate, spermine-NONOate and 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO) were from Alexis Biochemicals (AXXORA Ltd., Nottingham, UK). All other reagents were of the highest obtainable grade. Receptor nomenclature conforms to the BJP Guide to Receptors and Channels (Alexander et al., 2008).
Results
In order to understand how NO donor drugs induce depolarization and cell death of neurones, we determined how the drugs affected whole-cell currents of rat cerebellar granule neurones in culture. Voltage-clamp experiments were performed on neuronal cell bodies that were physically isolated from other cell bodies. This was achieved by only patching cell bodies that were located at the centre of long axonal bundles. A total of 389 cerebellar granule neurones were examined during the course of these studies, and of these, 22 (5.6%) displayed spontaneous bursting activity following the rupturing of the cell membrane.
NONOate-induced effects
Application of DETA-NONOate to voltage-clamped neurones caused a rapid and sustained inward current (Figure 1A), which was associated with a large increase in the baseline noise (Figure 1A, inset). The amplitude of the response increased with increasing concentrations of drug (Figure 1B). To limit the quantities of drug used, all further experiments were performed at 3 mM DETA-NONOate. At this concentration, currents (average amplitude 238.0 ± 48.5 pA, n= 10) displayed a fairly rapid (10–90% rise time was 1026 ± 74 ms, n= 10) inward current that was far slower than the response time of the perfusion system (<100 ms). In the continued presence of DETA-NONOate, the currents reduced to a new baseline and developed a steady-state current (64 ± 4% of max, n= 10) that persisted until the drug was removed (Figure 1A). Removal of the drug caused a rapid and full reversal of the inward current and associated baseline noise (Figures 1 and 2).
Figure 1.
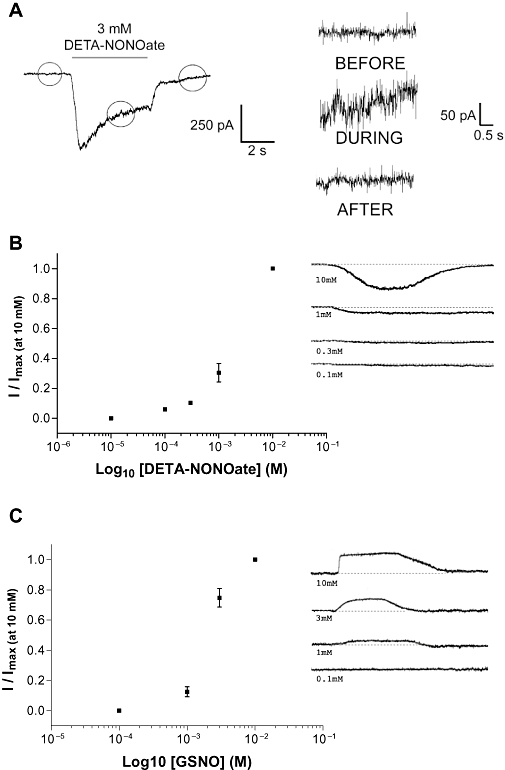
Whole-cell patch-clamp of cerebellar granule cells. (A) Typical response to (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA-NONOate), showing the increase in baseline noise associated with the inward current. KCl depolarizations of similar magnitude did not elicit changes in baseline noise. (B) DETA-NONOate (n= 3) concentration response. The mean ± SEM values were: 10 µM, 0.0 ± 0; 100 µM, 0.06 ± 0.01; 300 µM, 0.103 ± 0.01; 1 mM, 0.30 ± 0.06; 10 mM, 1.0 ± 0.0. (C) S-nitrosoglutathione (n= 6) concentration response. The mean ± SEM values were: 100 µM, 0.0 ± 0.0; 1 mM, 0.12 ± 0.03; 3 mM 0.75 ± 0.06; 10 mM, 1.0 ± 0.0. All values were normalized to the response at 10 mM. Representative responses are shown to the right of each graph.
Figure 2.
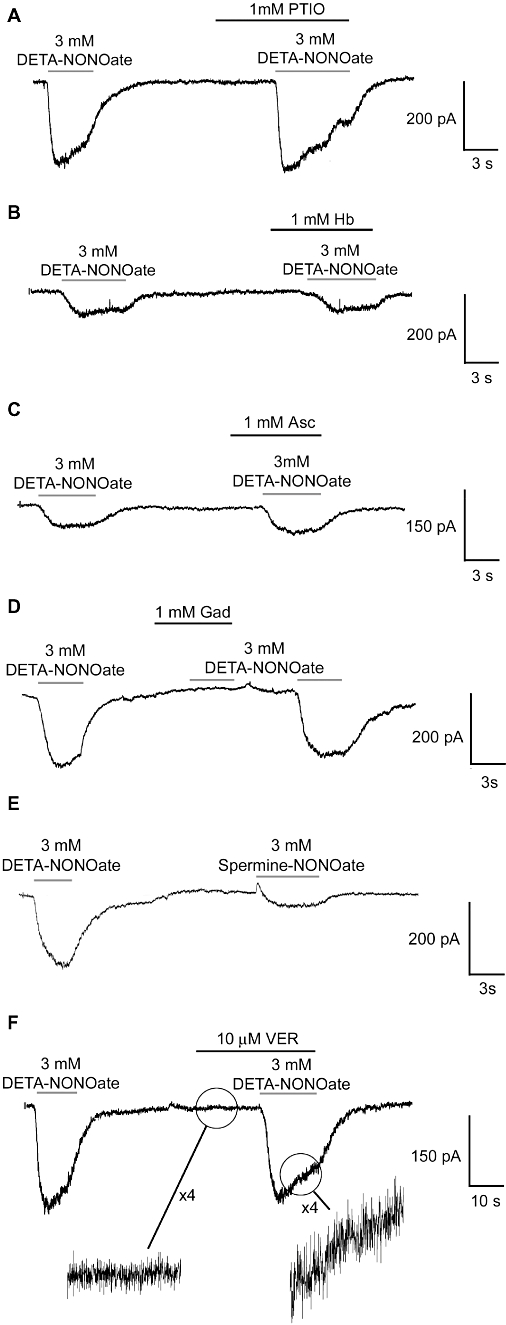
Typical responses to (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA-NONOate) and effects of various antagonists on these responses. Each trace is representative of at least 10 similar applications performed on five or more independent neuronal preparations. PTIO, 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide; Hb, haemoglobin; Asc, ascorbate; Gad, gadolinium; VER, verapamil.
Similar to 3 mM DETA-NONOate, 3 mM spermine-NONOate also resulted in an inward current, suggesting that this response might be common to NONOates (Figure 2E). This response to 3 mM spermine-NONOate (49.1 pA ± 21.5, n= 10) was of reduced amplitude when compared to that elicited by DETA-NONOate.
GSNO-induced effects
The GSNO response was dose dependent (Figure 1C). Perfusion with 3 mM GSNO was slower activating (10–90% rise time was 3545 ± 246 ms, n= 10) than the response to DETA-NONOate, and was a sustained outward current (average amplitude 88.7 ± 12.4 pA, n= 10) that returned to the resting state following the removal of the compound (Figure 3A). In 19% (20 of 104) of the experiments, the outward current in response to GSNO was preceded by a fast inward current spike (Figure 3A). In contrast to the DETA-NONOate response, the outward currents elicited by GSNO were not associated with an increase in the baseline noise (Figure 3A).
Figure 3.
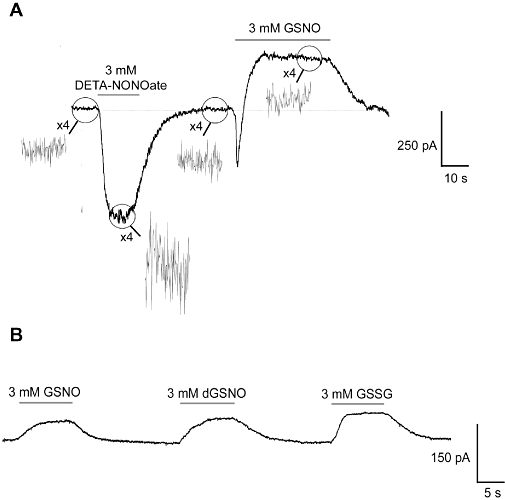
Typical responses to S-nitrosoglutathione (GSNO). Application of decomposed GSNO (dGSNO) and oxidized glutathione (GSSG) elicited responses of similar amplitude to fresh GSNO stocks. Each trace is representative of at least 10 similar applications performed on five or more independent neuronal preparations.
Inhibition of the DETA-NONOate response
Application of decomposed 3 mM DETA-NONOate (drug in solution left to break down to DETA and nitrite for 2 days), 3 mM nitrite or saline alone caused no change in the recorded current. The response to 3 mM DETA-NONOate was unaltered by the microscope illumination (data not shown), suggesting the response was not a consequence of S-nitrosation (which is broken down by visible light).
The response to 3 mM DETA-NONOate was not inhibited by the NO scavengers, haemoglobin (1 mM), or PTIO (1 mM) or, the NO2 scavenger ascorbate (1 mM), and none of these compounds had effects when applied alone (Figure 2A–C). In contrast, 1 mM gadolinium (a broad-spectrum cation-channel antagonist) had no effect when applied alone, but completely inhibited the response to DETA-NONOate and associated increase in baseline noise (Figure 2D). This block was enhanced by a 5 s pre-application, which is indicative of a closed-channel block. Reversal potential measurements were attempted for this gadolinium-sensitive channel, but because of the fragility of the patches and the small amplitude of the currents under bi-ionic conditions, this was not possible. With the saline compositions and holding potential used here, an inward current could have resulted from Cl− efflux or Na+/K+ influx, but if the block by gadolinium is taken into account, it is likely that the channel is cationic.
Previous studies have proposed that NO neurotoxicity is the result of the release of glutamate induced by NO that can be inhibited by NMDA antagonists. Here, perfusion of the antagonists MK-801 (10 µM, NMDA receptor antagonist) or CNQX (60 µM, kainate/quisqualate receptor antagonists) had no effects when applied alone or in the presence of 3 mM DETA-NONOate (data not shown). This suggests that the response to DETA-NONOate is not mediated by such channels. Application of 10 µM verapamil, a voltage-activated calcium-channel blocker, had no measurable effect on the amplitude of the DETA-NONOate response or associated increase in baseline noise, suggesting the increase in noise was a result of localized channel openings rather than increased EPSP activity from neighbouring neurones (Figure 2F).
Inhibition of the GSNO response
Decomposed 3 mM GSNO caused an outward current response that was of similar amplitude to that induced by fresh 3 mM GSNO stock solution, and responses of similar profile and amplitude were seen with oxidized glutathione (GSSG) at the same concentration (Figure 3B). Similar to DETA-NONOate, application of 1 mM haemoglobin, 1 mM PTIO or 1 mM ascorbate had no effect alone, or in the presence of 3 mM GSNO (data not shown). The response to GSNO was also not altered by bright light (microscope illumination), which releases NO from GSNO (and other S-nitrosothiols). MK-801 (10 µM), CNQX (60 µM), ZD7288 (100 µM; a K+ channel blocker) and ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one, 25 µM; a guanylate cyclase inhibitor) had no effect when applied alone or in the presence of 3 mM GSNO (data not shown).
Discussion
DETA-NONOate and GSNO are commonly used NO donor drugs (Feelisch, 1998; Yamamoto and Bing, 2000; Wang et al., 2002). We have previously measured steady-state concentrations of NO by use of an NO electrode which gave 0.9–1.5 µM for 1 mM DETA/NO, and 1.6–1.9 µM NO for 1 mM GSNO (Borutaite and Brown, 2003). Here, we showed that, at high concentrations, these two compounds elicit distinct effects on the electrical properties of cerebellar granule neurones that were not mediated by NO. Both responses were sustained for the full-time course of the application and were characterized by either an inward current (DETA-NONOate) or an outward current (GSNO). Our investigations indicate that the DETA-NONOate response is due to the DETA-NONOate molecule itself (rather than NO or DETA), and is mediated by a cation-selective channel. In contrast, the effects of GSNO may be due to GSSG or the glutathione moiety of GSNO, but the target that is responsible for these effects remains unknown.
The finding that the NO scavengers haemoglobin and PTIO did not decrease the responses to DETA-NONOate and GSNO, indicates that the responses are not due to NO production from these compounds or to species derivative of that NO. As both NO donors and scavengers are hydrophilic and were added at the same time, they should go into the same compartments/domains. Also, as NO passes rapidly through membranes, removal of NO from one compartment will deplete NO in other compartments.
The effects of DETA-NONOate on CGCs are probably mediated by a cation-selective channel, but the specific channel type remains unknown. Gadolinium (Gd3+) is a broad-spectrum blocker of a variety of cation channels, but over the last few years many of the gadolinium-blocked channels have turned out to be transient receptor potential (TRP) channels (Aarts et al., 2003; Venkatachalam and Montell, 2007). Gadolinium-sensitive TRP channels of various kinds are present in cerebellar granule neurones, and can affect cell death (Pinilla et al., 2005; Jia et al., 2007). In cortical neurones, high concentrations of DETA-NONOate were found to activate a gadolinium-sensitive cation channel, which was attributed to NO (or peroxynitrite) activation of TRPM7 channels, but it was not checked whether this activation was mediated by NO or DETA-NONOate itself (Aarts et al., 2003). In astrocytes, we have previously found that high concentrations of DETA-NONOate caused gadolinium-sensitive calcium influx, but again we did not check whether this was mediated by NO or DETA-NONOate itself (Bal-Price et al., 2002).
We do not know the nature of the conductance affected by GSNO and GSSG (a break-down product of GSNO), although the findings that GSSG and decomposed GSNO gave the same effect suggests that GSSG alone is capable of inducing the response. A possible explanation involves changes in the behaviour of cell surface receptors. Redox modulation of a range of receptors has been reported in the literature, and in particular, extracellular GSSG has been shown to reversibly inhibit the NMDA (Sucher and Lipton, 1991) and GABAA receptors (Amato et al., 1999; Calero and Calvo, 2008), as well as voltage-activated sodium channels (Wang et al., 2007). Any channel with vicinal thiols on the extracellular side is potentially susceptible to such redox regulation. Another possibility is that GSNO and/or GSSG block a ‘GSH receptor’ or glutamate receptor that causes depolarization (Hermann et al., 2000; Janáky et al., 2000). With the saline compositions and holding potential used in our study, the outward current could result from the block of a leak Cl− or Na+/K+ selective channel, due to allosteric mechanisms or channel block by GSNO and GSSG (Rego et al., 2001; Han et al., 2002). Insensitivity of the baseline current or GSNO-induced response to gadolinium suggests that it is not a cationic channel. However, the actions of GSNO might also be mediated by S-nitrosation of a channel or regulatory proteins, as shown with L-type calcium channels, although the rate of activation and reversal of the GSNO response appears too fast (>3 s) for S-nitrosation and its reversal (Poteser et al., 2001); the S-nitrosation-mediated response described by Poteser et al. (2001) required at least several minutes to reverse. Our finding that ascorbate did not affect the response to GSNO also suggests that neither NO, NO2 nor N2O3 are involved in the response, as ascorbate causes NO release from GSNO (Gorren et al., 1996), and reacts rapidly with NO2 and N2O3. The finding that bright field illumination did not affect the response to GSNO suggests that S-nitrosation is not involved in the response as S-nitrosothiols (including GSNO) are broken down by optical light.
DETA-NONOate and GSNO have been widely used in NO research. The data presented here urge caution in the interpretation of results when using high concentrations of these donors, as not all of their effects may be mediated by NO alone.
Acknowledgments
AJT is funded by the Wellcome Trust. PKM was funded by the BBSRC during these studies, and now works for GSK.
Glossary
Abbreviations:
- CGC
cerebellar granule cell
- DETA
diethylenetriamine
- eNOS
endothelial nitric oxide synthase
- GSNO
S-nitrosoglutathione
- GSSG
oxidized glutathione
- iNOS
inducible nitric oxide synthase
- NO
nitric oxide
- nNOS
neuronal nitric oxide synthase
- PTIO
2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide
Conflict of interest
None.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edn (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Almeida J, Bolaños JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci USA. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato A, Connolly CN, Moss SJ, Smart TG. Modulation of neuronal and recombinant GABAA receptors by redox reagents. J Physiol. 1999;517:35–50. doi: 10.1111/j.1469-7793.1999.0035z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6649. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal-Price A, Moneer Z, Brown GC. Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia. 2002;40:312–323. doi: 10.1002/glia.10124. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borutaite V, Brown GC. Nitric oxide induces apoptosis via hydrogen peroxide, but necrosis via energy and thiol depletion. Free Radic Biol Med. 2003;35:1457–1468. doi: 10.1016/j.freeradbiomed.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Calero CI, Calvo DJ. Redox modulation of homomeric ρ1 GABAC receptors. J Neurochem. 2008;105:2367–2374. doi: 10.1111/j.1471-4159.2008.05319.x. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feelisch M. The use of nitric oxide donors in pharmacological studies. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:113–122. doi: 10.1007/pl00005231. [DOI] [PubMed] [Google Scholar]
- Gorren AC, Astrid S, Schmidt K, Mayer B. Decomposition of S-nitrosoglutathione in the presence of copper ions and glutathione. Arch Biochem Biophys. 1996;330:219–228. doi: 10.1006/abbi.1996.0247. [DOI] [PubMed] [Google Scholar]
- Guix FX, Uribesalgo I, Coma M, Muñoz FJ. The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol. 2005;76:126–152. doi: 10.1016/j.pneurobio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Han J, Truell J, Gnatenco C, Kim D. Characterization of four types of background potassium channels in rat cerebellar granule neurons. J Physiol. 2002;542:431–444. doi: 10.1113/jphysiol.2002.017590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann A, Varga V, Janáky R, Dohovics R, Saransaari P, Oja SS. Interference of S-nitrosoglutathione with the binding of ligands to ionotropic glutamate receptors in pig cerebral cortical synaptic membranes. Neurochem Res. 2000;251:119–1124. doi: 10.1023/a:1007626230278. [DOI] [PubMed] [Google Scholar]
- Janáky R, Shaw CA, Varga V, Hermann A, Dohovics R, Saransaari P, et al. Specific glutathione binding sites in pig cerebral cortical synaptic membranes. Neuroscience. 2000;95:617–624. doi: 10.1016/s0306-4522(99)00442-x. [DOI] [PubMed] [Google Scholar]
- Jekabsone A, Neher JJ, Borutaite V, Brown GC. Nitric oxide from neuronal nitric oxide synthase sensitises neurons to hypoxia-induced death via competitive inhibition of cytochrome oxidase. J Neurochem. 2007;103:346–356. doi: 10.1111/j.1471-4159.2007.04765.x. [DOI] [PubMed] [Google Scholar]
- Jia Y, Zhou J, Tai Y, Wang Y. TRPC channels promote cerebellar granule neuron survival. Nat Neurosci. 2007;10:559–567. doi: 10.1038/nn1870. [DOI] [PubMed] [Google Scholar]
- Kelm M. Nitric oxide metabolism and breakdown. Biochim Biophys Acta. 1999;1411:273–289. doi: 10.1016/s0005-2728(99)00020-1. [DOI] [PubMed] [Google Scholar]
- Leist M, Fava E, Montecucco C, Nicotera P. Peroxynitrite and nitric oxide donors induce neuronal apoptosis by eliciting autocrine excitotoxicity. Eur J Neurosci. 1997;9:1488–1498. doi: 10.1111/j.1460-9568.1997.tb01503.x. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, et al. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Mander P, Borutaite V, Moncada S, Brown GC. Nitric oxide from inflammatory-activated glia synergizes with hypoxia to induce neuronal death. J Neurosci Res. 2005;79:208–215. doi: 10.1002/jnr.20285. [DOI] [PubMed] [Google Scholar]
- Pinilla PJ, Hernández AT, Camello MC, Pozo MJ, Toescu EC, Camello PJ. Non-stimulated Ca2+ leak pathway in cerebellar granule neurones. Biochem Pharmacol. 2005;70:786–793. doi: 10.1016/j.bcp.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Poteser M, Romanin C, Schreibmayer W, Mayer B, Groschner K. S-nitrosation controls gating and conductance of the α1 subunit of class C L-type Ca2+ channels. J Biol Chem. 2001;276:14797–14803. doi: 10.1074/jbc.M008244200. [DOI] [PubMed] [Google Scholar]
- Rego AC, Lambert JJ, Nicholls DG. Developmental profile of excitatory GABAA responses in cultured rat cerebellar granule cells. Neuroreport. 2001;12:477–482. doi: 10.1097/00001756-200103050-00011. [DOI] [PubMed] [Google Scholar]
- Sucher NJ, Lipton SA. Redox modulatory site of the NMDA receptor-channel complex: regulation by oxidized glutathione. J Neurosci Res. 1991;30:582–591. doi: 10.1002/jnr.490300316. [DOI] [PubMed] [Google Scholar]
- Takahata K, Katsuki H, Kume T, Ito K, Tochikawa Y, Muraoka S, et al. Retinal neurotoxicity of nitric oxide donors with different half-life of nitric oxide release: involvement of N-methyl-d-aspartate receptor. J Pharmacol Sci. 2003;92:428–432. doi: 10.1254/jphs.92.428. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, et al. Nitric oxide donors: chemical activities and biological applications. Chem Rev. 2002;102:1091–1134. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- Wang W, Ma J, Zhang P, Luo A. Redox reaction modulates transient and persistent sodium current during hypoxia in guinea pig ventricular myocytes. Pflugers Arch. 2007;454:461–475. doi: 10.1007/s00424-007-0219-1. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Bing RJ. Nitric oxide donors. Proc Soc Exp Biol Med. 2000;225:200–206. doi: 10.1046/j.1525-1373.2000.22525.x. [DOI] [PubMed] [Google Scholar]