

Abstract
Background and purpose:
In mouse tail arteries, selective α2-adrenoceptor antagonism with rauwolscine caused powerful dilation during constriction to the α1-adrenoceptor agonist phenylephrine. This study therefore assessed phenylephrine's selectivity at vascular α-adrenoceptors and the mechanism(s) underlying dilation to rauwolscine.
Experimental approach:
Mouse isolated tail arteries were assessed using a pressure myograph.
Key results:
The α2-adrenoceptor agonist UK14,304 caused low-maximum constriction that was inhibited by rauwolscine (3 × 10−8 M) but not by the selective α1-adrenoceptor antagonist prazosin (10−7 M). Concentration–effect curves to phenylephrine, cirazoline or noradrenaline were unaffected by rauwolscine but were inhibited by prazosin, which was more effective at high compared with low levels of constriction. In the presence of prazosin, rauwolscine inhibited the curves and was more effective at low compared with high levels of constriction. Although rauwolscine alone did not affect concentration–effect curves to phenylephrine, noradrenaline or cirazoline, it caused marked transient dilation when administered during constriction to these agonists. Dilation was mimicked by another α2-adrenoceptor antagonist (RX821002, 3 × 10−8 M), was dependent on agonist selectivity, and did not occur during adrenoceptor-independent constriction (U46619). During constriction to UK14,304 plus U46619, rauwolscine or rapid removal of UK14,304 caused transient dilation that virtually abolished the combined constriction. Endothelial denudation reduced these dilator responses.
Conclusions and implications:
Inhibition of α2-adrenoceptors caused transient dilation that was substantially greater than the contribution of α2-adrenoceptors to the constriction. This reflects a slowly reversing α2-adrenoceptor-mediated endothelium-dependent dilation and provides a rapid, sensitive test of α2-adrenoceptor activity. This approach also clearly emphasizes the poor selectivity of phenylephrine at vascular α-adrenoceptors.
Keywords: α1-adrenoceptors, α2-adrenoceptors, endothelium, phenylephrine, cirazoline
Introduction
α1- and α2-adrenoceptors (nomenclature follows Alexander et al., 2008) are expressed on vascular smooth muscle cells and initiate constriction in response to the endogenous catecholamines, adrenaline and noradrenaline (Flavahan and McGrath, 1980; Flavahan et al., 1984; Guimaraes and Moura, 2001; Philipp et al., 2002). Although α1-adrenoceptors are expressed by most blood vessels, functional constrictor α2-adrenoceptors have a more restricted distribution in the vascular system. Because of their role in mediating cold-induced cutaneous vasoconstriction, α2-adrenoceptors are more active in cutaneous compared with deep blood vessels (Flavahan et al., 1985; Flavahan, 2005). Furthermore, in contrast to α1-adrenoceptor activity, α2-adrenoceptor-mediated constriction increases in distal compared with proximal arteries; a pattern that continues in the venous system where α2-adrenoceptor activity is widely distributed (Flavahan et al., 1984; 1987; Flavahan and Vanhoutte, 1986b; Rajanayagam and Medgett, 1987; Guimaraes and Moura, 2001). However, even when the α2-adrenoceptor constrictor system is at its most powerful, it generally provides a lower maximum response compared with other receptor systems, including α1-adrenoceptors (Flavahan and McGrath, 1984; Flavahan et al., 1984; Chotani et al., 2000).
Phenylephrine is a commonly used nasal decongestant (Eccles, 2007; Kollar et al., 2007). It is generally considered to be a selective α1-adrenoceptor agonist and is widely used in animal and clinical studies to assess the functional activity of α1-adrenoceptors (Inoue et al., 2001; Arain et al., 2002; Mueed et al., 2004; Lewis et al., 2007; Kirby et al., 2008). While studying the agonist in mouse isolated tail arteries, we observed that selective α2-adrenoceptor antagonism with rauwolscine (3 × 10−8 M) caused powerful transient dilation of phenylephrine-induced constriction, suggesting that the agonist might cause constriction by activating α1- and α2-adrenoceptors. The present study was therefore performed to characterize the selectivity of phenylephrine at mouse vascular α1- and α2-adrenoceptors, and to investigate the mechanism underlying the relaxation to rauwolscine (using mouse isolated tail and mesenteric arteries). The results demonstrate that phenylephrine has limited selectivity at α-adrenoceptor subtypes and does indeed initiate vasoconstriction by activating α1- and α2-adrenoceptors. However, the transient dilator response evoked by α2-adrenoceptor antagonists is much greater than the contribution of α2-adrenoceptors to the constrictor response. This reflects the presence of a slowly reversing α2-adrenoceptor-mediated endothelium-dependent dilation, which transiently amplifies the dilator response to α2-adrenoceptor antagonism. Indeed, this provides a rapid and sensitive method of assessing agonist selectivity at vascular α-adrenoceptors.
Methods
Animals
All animal care and experimental procedures were approved by the Johns Hopkins University Institutional Animal Care and Use Committee. We used (C57BL6) mice from The Jackson Laboratory, Bar Harbor, ME, USA.
Blood vessel chamber
Eight- to fourteen-week-old male mice were killed by CO2 asphyxiation. Segments of mesenteric and proximal tail arteries were then rapidly and carefully isolated and placed in cold Krebs-Ringer-bicarbonate solution: 118.3 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25.0 mM NaHCO3 and 11.1 mM glucose (control solution). Arteries were cannulated at both ends with glass micropipettes, secured using 12-0 nylon monofilament suture, and placed in a microvascular chamber (Living Systems, Burlington, VT, USA) (Flavahan, 2005). They were studied in the absence of flow and maintained at a constant transmural pressure (PTM) of 60 mmHg (Flavahan, 2005). Under these conditions, the internal diameter of the arteries was approximately 300 µm. The chamber was placed on the stage of an inverted microscope (Nikon TMS-F; Nikon, Tokyo, Japan) connected to a video camera, and superfused with control solution (maintained at 37°C, gassed with 16% O2, 5% CO2, balance N2; pH 7.4). In general, as performed in our previous studies, the control solution filling the chamber and superfusing the arterial segments was re-circulated in a continuous fashion (total volume 90 mL, flow rate 25 mL·min−1). However, in one series of experiments, continuous perfusion of the chamber was achieved in an open, non-re-circulating, manner (rate 25 mL·min−1). The blood vessel image was projected onto a video monitor and internal diameters continuously monitored by a video dimension analyser (Living Systems) and BIOPAC data acquisition system (Santa Barbara, CA, USA).
Experimental protocols
Arterial segments were allowed to equilibrate for at least 30 min before starting experiments. Concentration–effect curves to the adrenergic agonists phenylephrine (10−8 to 3 × 10−5 M), cirazoline (10−9 to 10−6 M), UK14,304 [5-bromo-N-(4,5-dihydro-1H-imidazol-2-yl)-6-quinoxalinamine] (10−9 to 10−7 M), or noradrenaline (10−8 to 10−5 M), or to the thromboxane receptor agonist U46619 (10−9 to 10−7 M) were generated by increasing the concentration of the agonists in half-log increments, once the constriction to the previous concentration had peaked. Following completion of the concentration–effect curve, the response to the agonists was terminated by repeatedly exchanging the buffer solution and allowing the blood vessels to return to their stable baseline levels. In some experiments, concentration–effect curves to agonists were determined under control conditions or in the presence of the selective α2-adrenoceptor antagonists, rauwolscine (3 × 10−8 M) or RX821002 (3 × 10−8 M) (Flavahan et al., 1984; Devedjian et al., 1994), and/or the selective α1-adrenoceptor antagonist, prazosin (10−7 M) (Flavahan et al., 1984). At the concentrations used, these antagonists are potent and selective antagonists at their respective receptors. Antagonists were present 30 min before and during the concentration–effect curves to the agonists. In some experiments, arteries were stably constricted with phenylephrine (3 × 10−7 M), cirazoline (3 × 10−8 M), or noradrenaline (3 × 10−7 M), or to a combination of U46619 (to ∼60% of baseline diameter, 10−8 to 3 × 10−8 M) plus UK14,304 (3 × 10−8 M), and then vasodilator responses to rauwolscine (3 × 10−8 M) or RX821002 (3 × 10−8 M) determined. Time control studies were performed using paired arterial segments that were not exposed to the receptor antagonists. In some experiments, endothelium denudation was performed by inserting a 70 µm wire into the lumen of the artery and was confirmed by loss of the dilator response to acetylcholine (10−6 M, assessed during constriction to U46619 to approximately 60% of baseline diameter).
Data analysis and statistics
Vasoconstriction was expressed as a percentage of the internal diameter of the blood vessel prior to administering the agonist(s). α2-Adrenoceptor constriction is generally a low-maximum response compared with other constrictor systems (including α1-adrenoceptors), and generates only ∼30% constriction of mouse tail arteries. This difference in maximal response can facilitate identification of α-adrenoceptor subtypes activated by non-selective agonists. This is achieved by analysing the effect of antagonists on agonist concentration–effect curves at response levels attainable by α2-adrenoceptor activation and at levels attainable only by α1-adrenoceptor activation (Flavahan and McGrath, 1984; Flavahan et al., 1984; 1987; Flavahan and Vanhoutte, 1987). Therefore, the effect of α-adrenoceptor antagonists was analysed at agonist concentrations causing 15% and 60% constriction (CC15 and CC60 respectively). The 15% level of constriction (i.e. to 85% of baseline diameter) was selected because it approximates 50% of the maximal response to α2-adrenoceptor stimulation in tail arteries whereas the 60% level of constriction (i.e. to 40% of baseline diameter) exceeds the contractile activity of α2-adrenoceptors (Flavahan, 2005). Statistical evaluation of the data was performed by Student's paired t-test. When more than two means were compared, analysis of variance was used. If a significant F value was found, Tukey's test for multiple comparisons was employed to identify differences among groups. Data are expressed as means ± SEM for n number of experiments, where n equals the number of animals from which blood vessels were studied. Values were considered to be statistically different when P < 0.05.
Materials
All drugs were obtained from Sigma-Aldrich (St Louis, MO, USA) and were dissolved in distilled water with the exception of UK14,304, which was dissolved in dimethyl sulphoxide (highest chamber concentration of 0.001%). Drug concentrations are described as final molar concentration (M) in the chamber superfusate.
Results
Characterization of α-adrenoceptors involved in constriction to phenylephrine, noradrenaline or cirazoline
The selective α1-adrenoceptor antagonist prazosin (10−7 M) did not affect constrictor responses to the prototypic α2-adrenoceptor agonist UK14,304 (10−9 to 10−7 M) confirming prazosin's selectivity for α1-adrenoceptors in the mouse tail artery (Figure 1). In contrast, the selective α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M) markedly depressed responses to UK14,304 (Figure 1).
Figure 1.
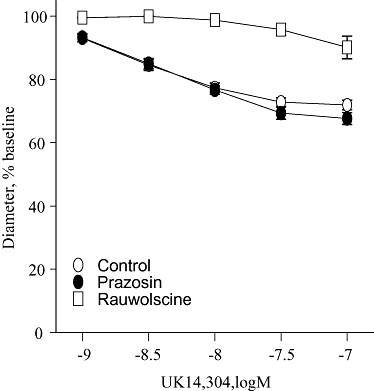
Effects of the selective α1-adrenoceptor antagonist prazosin (10−7 M) or the selective α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M) on constrictor responses to the selective α2-adrenoceptor agonist, UK14,304 (10−9 to 10−7 M) in mouse isolated tail arteries. Constriction was expressed as a percentage of the stable baseline diameter of the arteries (prior to agonist administration) and is presented as means ± SEM for n= 4–5.
Rauwolscine (3 × 10−8 M) did not significantly affect the concentration–effect curve to phenylephrine (Figure 2). Prazosin (10−7 M) caused a non-parallel shift in the concentration–effect curve to phenylephrine, having a greater inhibitory effect at a high (CC60) compared with a low level of constriction (CC15) (Table 1, Figure 2). In the presence of prazosin (10−7 M), rauwolscine (3 × 10−8 M) caused a significant rightward shift in the concentration–effect curve to phenylephrine, having a significantly greater inhibitory effect at a low (CC15) compared with a high level of constriction (CC60) (Table 1, Figure 2). Indeed, the combination of prazosin plus rauwolscine caused a parallel shift in the concentration–effect curve to phenylephrine (Table 1, Figure 2). Similar results were obtained with RX821002 (3 × 10−8 M), another selective α2-adrenoceptor antagonist (data not shown).
Figure 2.
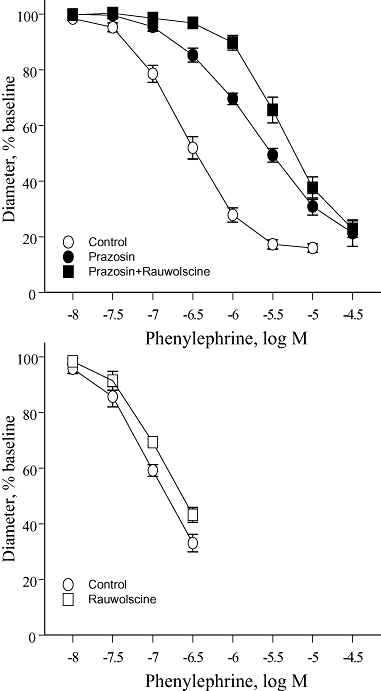
Effects of the selective α1-adrenoceptor antagonist prazosin (10−7 M), the selective α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M) or the combination of prazosin plus rauwolscine on constrictor responses to phenylephrine in mouse isolated tail arteries. Constriction was expressed as a percentage of the stable baseline diameter of the arteries (prior to agonist administration) and is presented as means ± SEM for n= 10 (upper panel) or 5 (lower panel).
Table 1.
Effects of antagonists on the concentration–effect curves to adrenoceptor agonistsa
| Agonist | Level |
Prazosin |
Rauwolsineb(after Prazosin) |
Prazosin+Rauwolscine |
|||
|---|---|---|---|---|---|---|---|
| Log shiftc | shift | Log shiftc | shift | Log shiftc | shift | ||
| Phenylephrine (10) | CC15 | 0.54 ± 0.13** | 3.5 | 0.66 ± 0.09*** | 4.5 | 1.31 ± 0.14*** | 20.4 |
| CC60 | 1.07 ± 0.15***## | 11.6 | 0.18 ± 0.03***### | 1.5 | 1.28 ± 0.14*** | 19.3 | |
| Noradrenaline (7) | CC15 | 0.52 ± 0.16* | 3.3 | 0.40 ± 0.14* | 2.5 | 1.06 ± 0.16*** | 11.5 |
| CC60 | 1.11 ± 0.08***## | 12.8 | No effect | – | 1.14 ± 0.16*** | 13.8 | |
| Cirazoline (4) | CC15 | 1.03 ± 0.06*** | 10.7 | 0.51 ± 0.06** | 3.2 | 1.60 ± 0.11*** | 40.2 |
| CC60 | 1.56 ± 0.18** | 36.4 | No effect | – | 1.59 ± 0.23** | 39.4 | |
Shifts in the concentration–effect curves were determined at the CC15 and CC60 levels of constriction. Data are expressed as means ± SEM. The numbers of experiments are indicated in parentheses after the name of the agonist.
Effect of rauwolscine refers to the effect of the antagonist in arteries in which α1-adrenoceptors have already been blocked with prazosin (10−7 M), i.e. the difference between the concentration–effect curves performed in the presence of ‘prazosin’ and ‘prazosin + rauwolscine’. The other comparisons were made to the control concentration–effect curves.
, indicates the log shift is significant at P < 0.05, P < 0.01 or P < 0.001 respectively.
, indicates the log shift is significantly different between the CC15 and CC60 levels of constriction at P < 0.01 or P < 0.001 respectively.
The effects of antagonists on concentration–effect curves to noradrenaline or cirazoline were qualitatively similar to those observed with phenylephrine. Prazosin (10−7 M) caused non-parallel shifts in the concentration–effect curve to the agonists, with greater inhibitory effects at a high (CC60) compared with a low level of constriction (CC15) (Table 1, Figures 3 and 4). Rauwolscine (3 × 10−8 M), when administered alone, did not significantly affect the concentration–effect curves to noradrenaline or cirazoline. However, in the presence of prazosin (10−7 M), rauwolscine (3 × 10−8 M) inhibited the concentration–effect curves, having a greater inhibitory effect at a low (CC15) compared with high level of constriction (CC60) (Table 1, Figures 3 and 4). As with phenylephrine, the combination of prazosin plus rauwolscine caused parallel shifts in the concentration–effect curve to noradrenaline and cirazoline (Table 1, Figures 3 and 4).
Figure 3.
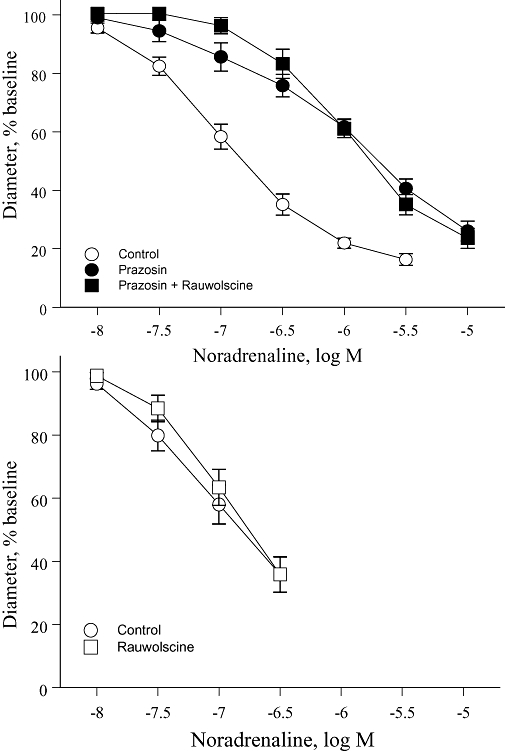
Effects of the selective α1-adrenoceptor antagonist prazosin (10−7 M), the selective α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M) or the combination of prazosin plus rauwolscine on constrictor responses to noradrenaline in mouse isolated tail arteries. Constriction was expressed as a percentage of the stable baseline diameter of the arteries (prior to agonist administration) and is presented as means ± SEM for n= 7 (upper panel) or 6 (lower panel).
Figure 4.
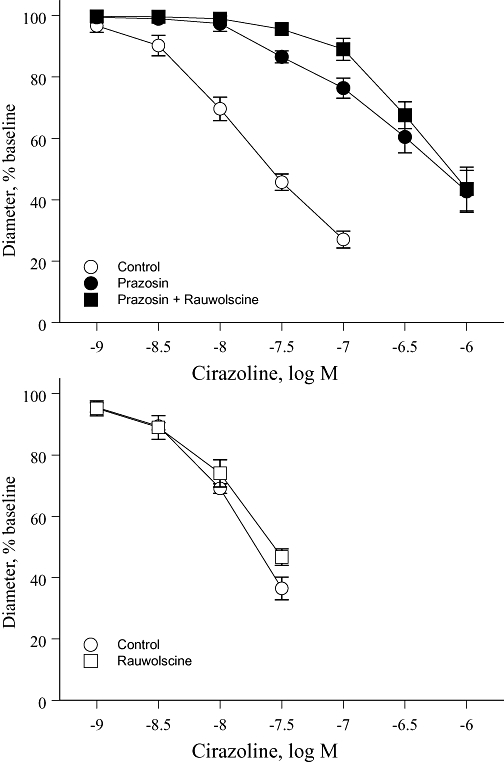
Effects of the selective α1-adrenoceptor antagonist prazosin (10−7 M), the selective α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M) or the combination of prazosin plus rauwolscine on constrictor responses to cirazoline in mouse isolated tail arteries. Constriction was expressed as a percentage of the stable baseline diameter of the arteries (prior to agonist administration) and is presented as means ± SEM for n= 4 (upper panel) or 5 (lower panel).
Reversal of agonist-induced constriction by α2-adrenoceptor antagonists
α-Adrenoceptor activation in quiescent arteries
During stable constriction to phenylephrine (3 × 10−7 M), noradrenaline (3 × 10−7 M) or cirazoline (3 × 10−8 M), rauwolscine (3 × 10−8 M) caused an impressive transient dilation that diminished over time and was significantly decreased 15 min after the peak relaxation (Table 2, Figure 5). RX821002 (3 × 10−8 M) had a similar dilator effect to rauwolscine (Table 2, Figure 5). In paired arterial segments not treated with α2-adrenoceptor antagonists, there was no time-dependent change in constriction to phenylephrine or cirazoline during the exposure period to the antagonists (Table 2, Figure 5). There was however a small time-dependent decrease in constriction to noradrenaline (Table 2, Figure 5), which may reflect oxidation and inactivation of this agonist.
Table 2.
Antagonist-induced dilation during responses to constrictor agonistsa
| Agonist | Treatment | Constrictionb |
Dilationb,c |
|
|---|---|---|---|---|
| Peak | After 15 min | |||
| Phenylephrine (12) | Rauwolscine | 49.5 ± 2.5% | 76.2 ± 2.6%*** | 64.5 ± 2.5%***### |
| Phenylephrine (5) | RX821002 | 56.2 ± 5.4% | 83.4 ± 3.9%*** | 68.0 ± 5.0%**### |
| Phenylephrine (12) | Time Control | 55.6 ± 3.0% | 55.5 ± 3.1% | 54.5 ± 3.2% |
| Noradrenaline (4) | Rauwolscine | 37.5 ± 5.4% | 76.9 ± 2.7%*** | 64.4 ± 4.1%***# |
| Noradrenaline (4) | Time Control | 45.3 ± 3.9% | 47.4 ± 3.2% | 52.6 ± 3.7%***## |
| Cirazoline (5) | Rauwolscine | 50.6 ± 4.5% | 67.5 ± 2.8%*** | 63.4 ± 4.6%*** |
| Cirazoline (5) | Time Control | 56.5 ± 3.7% | 57.1 ± 3.5% | 57.6 ± 4.6% |
| U46619 (3) | Rauwolscine | 62.4 ± 1.0% | 63.8 ± 1.8% | 63.0 ± 1.9% |
Dilations to rauwolscine (3 × 10−8 M) or RX821002 (3 × 10−8 M) were evaluated during constrictor responses to adrenoceptor agonists phenylephrine (3 × 10−7 M), noradrenaline (3 × 10−7 M), or cirazoline (3 × 10−8 M), or to the non-adrenoceptor agonist U46619 (3 × 10−9 M). Time-dependent changes in constriction to adrenoceptor agonists (in the absence of antagonists) were evaluated in paired arterial segments (time control).
Diameters associated with constriction to the agonists and dilation to the antagonists (or time-dependent changes in agonist constrictions) are all presented as a percentage of the baseline diameter prior to administration of the agonist, and expressed as means ± SEM. The numbers of experiments are provided in parenthesis after the name of the agonist.
indicates the level of constriction is significant different from the initial response to the constrictor agonist at P < 0.01 or P < 0.001 respectively.
indicates the diameter is significantly different from that occurring during the peak response to the antagonist at P < 0.05, P < 0.01 or P < 0.001, respectively.
Figure 5.
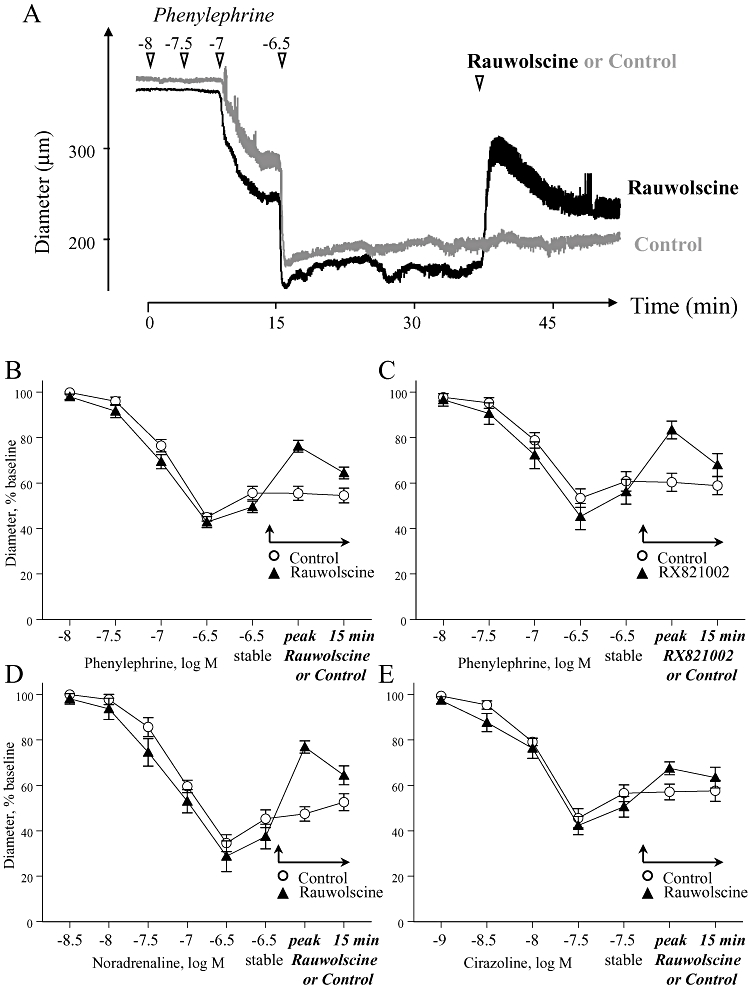
The acute dilator effect of selective α2-adrenoceptor antagonists rauwolscine (3 × 10−8 M) or RX821009 (3 × 10−8 M) during stable constriction of mouse isolated tail arteries with phenylephrine, cirazoline or noradrenaline. Paired arterial segments were exposed to increasing concentrations of an α-adrenoceptor agonist and constriction allowed to stabilize (‘stable’, at 3 × 10−7 M for noradrenaline or phenylephrine, and 3 × 10−8 M for cirazoline). Administration of the antagonists then caused dilation, which was assessed at the peak response and 15 min later. In all experiments, one of each paired arterial segments served as a time control and was not exposed to the antagonists (Control). The upper trace (A) presents a typical recording from one of the experiments with phenylephrine and rauwolscine. The lower graphs represent the means ± SEM for n= 12 (B), n= 5 (C), n= 4 (D) and n= 5 (E). All responses were expressed as a percentage of the stable baseline diameter of the arteries prior to agonist administration.
Therefore, although pretreatment with rauwolscine (when administered alone) did not inhibit the concentration–effect curves to noradrenaline, phenylephrine or cirazoline (Figures 2B, 3B and 4B), it caused impressive transient dilation when administered during sustained constriction to these agonists (Figure 5). Concentration–effect curves were generated using initial peak constrictions, whereas the antagonist dilator responses were assessed during more prolonged constrictor responses to the agonists. Therefore, the difference in antagonist activity could reflect distinct temporal characteristics of α1-adrenoceptor and α2-adrenoceptor constrictor components, with α1-adrenoceptors contributing predominantly to the initial response and α2-adrenoceptors contributing to maintained constriction. However, pretreatment with rauwolscine (3 × 10−8 M) did not affect the time course of constriction to phenylephrine (3 × 10−7 M) (Figure 6).
Figure 6.
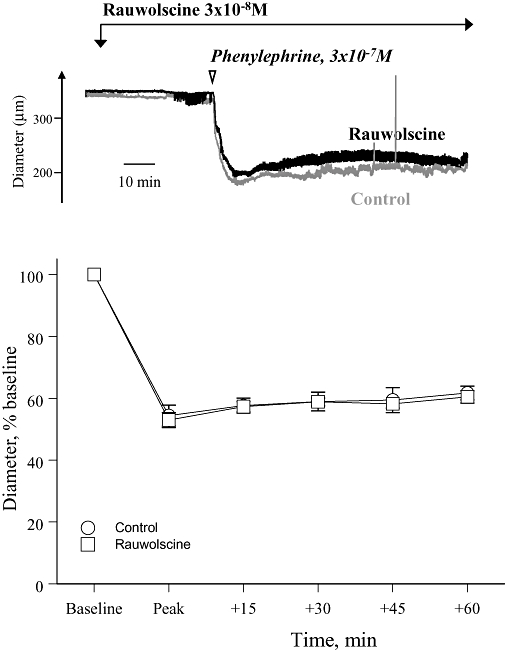
Effect of the selective α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M) on the time course of the constrictor response to phenylephrine (3 × 10−7 M) in mouse isolated tail arteries. The upper panel presents a recording from a typical experiment, whereas the lower panel presents the means ± SEM for n= 4. In the recording, internal diameter is presented in microns, whereas in the graph, internal diameter is expressed as a percentage of the baseline prior to agonist administration. The response to phenylephrine was assessed at the peak of constriction and then 15, 30, 45 and 60 min later.
The dilator response to α2-adrenoceptor antagonists was dependent on α2-adrenoceptor activity. Mouse mesenteric arteries do not express functional constrictor α2-adrenoceptors, and do not constrict in response to UK14,304 (Figure 7) (Flavahan, 2005). When administered during a sustained constriction to phenylephrine in mouse mesenteric artery, rauwolscine (3 × 10−8 M) did not cause dilation (Figure 7). Similarly, during constriction of tail arteries to the thromboxane receptor agonist U46619, rauwolscine (3 × 10−8 M) did not cause dilation (Table 2, Figure 8).
Figure 7.
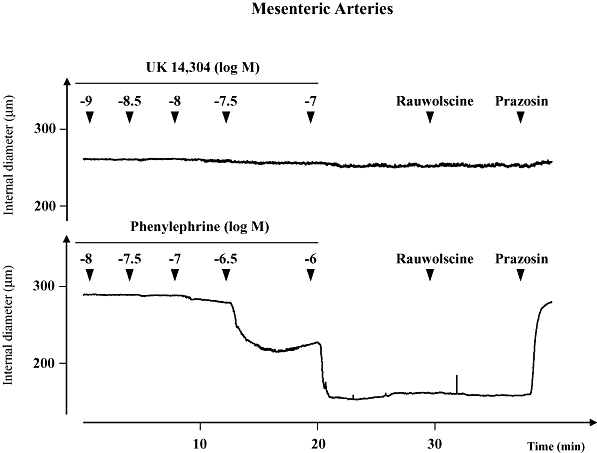
Representative recording demonstrating the activity of the α-adrenoceptor agonists UK14,304 (UK, 10−9 to 10−7 M) or phenylephrine (PE, 10−8 to 10−6 M) in mouse mesenteric arteries. UK14,304 failed to constrict the artery. Constriction to phenylephrine was not affected by acute administration of the selective α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M), but was reversed by the selective α1-adrenoceptor antagonist prazosin (10−7 M). The recording is representative of four experiments.
Figure 8.
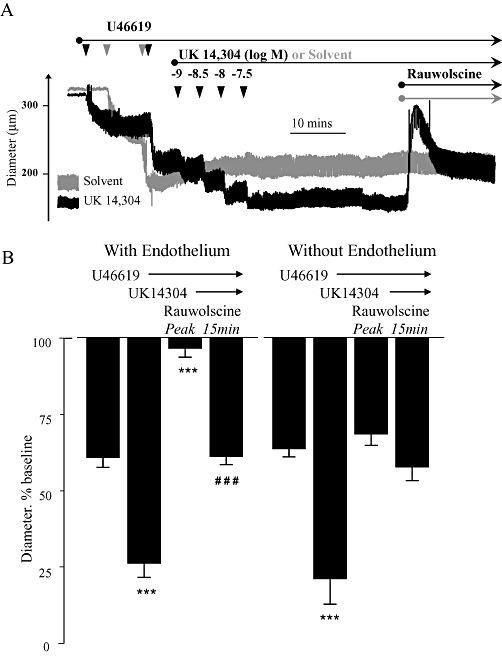
Effects of acute administration of rauwolscine (3 × 10−8 M) on combined constriction to the thromboxane receptor agonist U46619 and the α2-adrenoceptor agonist UK14,304 in mouse isolated tail arteries. Arteries (with and without endothelium) were initially constricted with U44619 to approximately 60% of resting baseline diameter, and then further constricted with UK14,304 (up to 3 × 10−8 M). Once the constriction stabilized, the dilator effect of the selective α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M) was determined. (A) Representative recording from a typical experiment. In arteries constricted only with U46619 (grey trace), rauwolscine did not cause dilation (Table 2). (B) Means ± SEM for n= 5. Responses were expressed as a percentage of the baseline internal diameter prior to administration of the agonists. The influence of rauwolscine was assessed at the peak of dilation and 15 min later. * indicates that the diameter is significantly different from the initial constriction to U46619 (***P < 0.001). # indicates the effect of rauwolscine after 15 min is significantly different from the peak effect (###P < 0.001).
α2-Adrenoceptor activation during constriction with U46619
The ability of rauwolscine (3 × 10−8 M) to reverse (or dilate) an α2-adrenoceptor-mediated constriction to UK14,304 was evaluated during simultaneous constriction with U46619. In tail arteries constricted with U46619, the subsequent addition of the α2-adrenoceptor agonist UK14,304 (3 × 10−8 M) caused a significant further constriction (Figure 8). When administered during this combined constriction, the α2-adrenoceptor antagonist rauwolscine (3 × 10−8 M) caused an initial dilation that completely reversed the α2-adrenoceptor component of the constriction and also almost abolished the U46619-dependent component of constriction (Figure 8). After 15 min, the dilation to rauwolscine was significantly attenuated and the new stable level of constriction was not significantly different from the original constriction to U46619, although it was still significantly less than the combined constrictor response to U46619 plus UK14,304 (Figure 8). In the absence of UK14,304, rauwolscine (3 × 10−8 M) did not cause dilation during constriction to U46619 (Figure 8).
The dilation associated with antagonist-induced termination of α2-adrenoceptor activity was also observed following rapid removal of UK14,304 during a combined constriction to U44619 plus UK14,304. For these studies, constriction of tail arteries was analysed in an open perfusion system rather than the standard re-circulating perfusion system used in our experiments. In tail arteries constricted with U46619, the α2-adrenoceptor agonist UK14,304 (3 × 10−8 M) caused a significant further constriction (Figure 9). Abrupt removal of UK14,304 from the perfusate (in the continued presence of U46619) caused an initial dilation that completely reversed the α2-adrenoceptor component of the constriction and almost abolished the U46619-dependent component of constriction (Figure 9). After 15 min, the dilation was significantly diminished and the new stable level of constriction was not significantly different from the original constriction to U46619, although it was still significantly less than the combined constrictor response to UK14,304 plus U46619 (Figure 9). The constriction and dilation associated with introduction and removal, respectively, of UK14,304 (3 × 10−8 M) in the open perfusion system were dramatically reduced when the experiment was repeated in the presence of the α2-adrenoceptor antagonist, rauwolscine (3 × 10−8 M). Following pretreatment with rauwolscine, the introduction of UK14,304 (3 × 10−8 M) to U46619-constricted arteries caused constriction equivalent to 6.4 ± 1.5% of baseline diameter (n= 4) (was 26.5 ± 0.9% under control conditions, n= 4, Figure 9), whereas removal of UK14,304 was associated with simple reversal of this constriction and a dilation equivalent to 5.4 ± 1.6% of baseline diameter (n= 4) (was 51.2 ± 2.8% under control conditions, n= 4, Figure 9).
Figure 9.
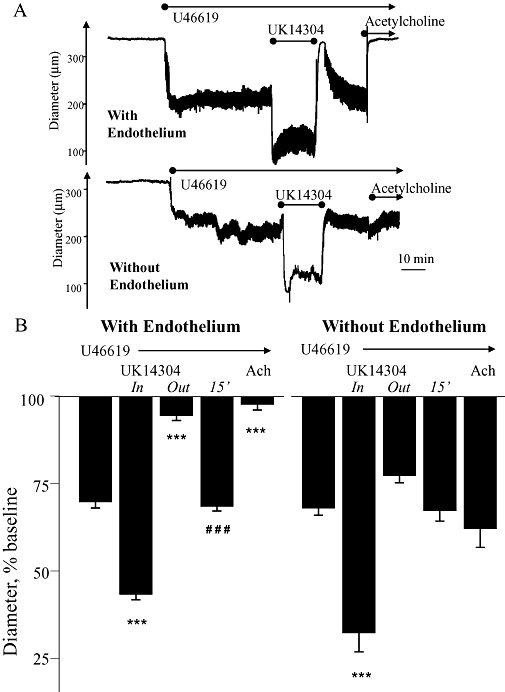
Effects of rapid withdrawal of the selective α2-adrenoceptor agonist UK14,304 (3 × 10−8 M) during combined constriction of mouse isolated tail arteries to UK14,304 and the thromboxane receptor agonist U46619. In an open perfusion system, arteries (with and without endothelium) were initially constricted with U44619 to approximately 60% of resting baseline diameter, and then further constricted with UK14,304 (3 × 10−8 M). Once the constriction had stabilized, UK14,304 was removed from the perfusate and constriction was maintained only by the continued presence of U46619. (A) Representative recording from a typical experiment in arteries with and without endothelium. (B) Means ± SEM for n= 4. Responses were expressed as a percentage of the baseline internal diameter prior to administration of the agonists. The effect of removing UK14,304 was assessed at the peak of dilation (‘out’) and 15 min later (15'). * indicates that the diameter is significantly different from the initial constriction to U46619 (***P < 0.001). # indicates the effect of removing UK14,304 is significantly different between the peak effect (‘out’) and 15 min later (15') (###P < 0.001).
Role of the endothelium
Endothelial denudation significantly reduced the dilator response evoked by administration of rauwolscine (3 × 10−8 M) (Figure 8) or abrupt removal of UK14,304 (Figure 9) in tail arteries constricted with U46619 plus UK14,304. In endothelium-denuded arteries, the dilation in each case was sufficient to abolish the α2-adrenoceptor component of the combined constriction but it did not significantly affect the component of constriction evoked by U46619 (Figures 8 and 9).
Endothelium denudation did not significantly affect the constrictor response to activation of α2-adrenoceptors with UK14,304 either during combined constriction with U46619 (e.g. Figures 8 and 9) or under basal conditions (i.e. absence of U46619). Under basal conditions, concentration–effect curves to UK14,304 were characterized by log CC15 values of –8.07 ± 0.21 and –7.88 ± 0.13 (n= 5; P= NS, not significant) and maximal constriction to 74.9 ± 3.5% and 77.6 ± 1.3% of baseline diameter (n= 5, P= NS) for endothelium-containing and endothelium-denuded arteries respectively. Likewise, endothelium denudation did not significantly affect constriction to U46619 (log CC15 values of –8.17 ± 0.04 and –8.12 ± 0.07, n= 5, P= NS and maximal-attained constriction, at 10−7 M, to 25.7 ± 2.6% and 17.7 ± 3.6% of baseline diameter, n= 5, P= NS, for endothelium-containing and endothelium-denuded arteries respectively).
Discussion
Because of its role in thermoregulation, the mouse tail artery is somewhat unusual by possessing a prominent smooth muscle α2-adrenoceptor constrictor response (Chotani et al., 2000; Flavahan, 2005). However, even at its most powerful, α2-adrenoceptor constriction evoked by high-efficacy agonists such as UK14,304 is a low-maximum response compared with α1-adrenoceptor activation (Flavahan and McGrath, 1984; Flavahan et al., 1984), causing only ∼ 30% constriction of the tail artery (Chotani et al., 2000; Flavahan, 2005). Because of this disparity in maximal responses to activation of α1- and α2-adrenoceptor subtypes, the selective α1-adrenoceptor antagonist prazosin causes non-parallel shifts in the concentration–effect curve to non-selective agonists (e.g. noradrenaline) being more potent at high compared with low levels of constriction (Flavahan and McGrath, 1984; Flavahan et al., 1984; 1987; Flavahan and Vanhoutte, 1986b). At high levels of constriction, the inhibitory potency of prazosin is consistent with its antagonism of α1-adrenoceptors whereas at low levels of constriction, the presence of the low-maximum α2-adrenoceptor response restricts the inhibitory effect of prazosin resulting in ‘prazosin-resistance’ (Flavahan and McGrath, 1984; Flavahan et al., 1984; 1987; Flavahan and Vanhoutte, 1986b). Indeed, in the present study, prazosin caused a non-parallel shift in the concentration–effect curve to phenylephrine, being more potent at a high level of constriction (60%, which is outside the range of the low-maximum α2-adrenoceptor response) compared with a lower intensity of constriction (15%, which is approximately 50% of the maximal α2-adrenoceptor response). The potency of prazosin at the high level of constriction (11.6-fold shift, CC60) is consistent with an antagonist dissociation constant (KB) of 9 × 10−9 M and α1-adrenoceptor activity in cutaneous blood vessels (Flavahan et al., 1984). However, at the low level of constriction, the potency of prazosin (3.5-fold shift, CC15) was significantly lower and not consistent with simple α1-adrenoceptor activity (Flavahan et al., 1984; Flavahan and Vanhoutte, 1986a). In the presence of prazosin, the selective α2-adrenoceptor antagonist rauwolscine caused inhibition of the concentration–effect curve at the low but not at the high level of constriction, and therefore removed this prazosin-resistant component. The demonstration that the prazosin-resistant component is rauwolscine-sensitive confirms that it represents α2-adrenoceptor constriction to the agonist. In the absence of prazosin, rauwolscine did not significantly affect the concentration–effect curve to phenylephrine, which confirms that rauwolscine acts selectively to inhibit α2-adrenoceptors and indicates that phenylephrine acts predominantly through α1-adrenoceptors. If one compares the concentration–effect curve to phenylephrine after rauwolscine with the curve after prazosin plus rauwolscine, the inclusion of prazosin caused a parallel shift in the curve to phenylephrine and there was no indication of resistance to prazosin. This again demonstrates that rauwolscine inhibits the prazosin-resistant, α2-adrenoceptor-mediated constriction to phenylephrine. However, loss of the α2-adrenoceptor component did not influence the control concentration–effect curve to phenylephrine and its influence was only evident following inhibition of the α1-adrenoceptor response. We have previously demonstrated that there is minimal interaction between α1- and α2-adrenoceptor components of vasoconstriction in cutaneous blood vessels (Flavahan et al., 1984; Flavahan and Vanhoutte, 1987). The control concentration–effect curve to phenylephrine therefore approximates the α1-adrenoceptor component of the response, and the curve after prazosin (i.e. the prazosin-resistant component at the CC15 level of constriction) identifies the α2-adrenoceptor component. The magnitude of the difference between these components characterizes the functional selectivity of phenylephrine at α1- and α2-adrenoceptors in the tail artery, which is 3.5-fold (Table 1).
The effects of prazosin and rauwolscine on responses to cirazoline or the physiological agonist noradrenaline were qualitatively similar to those observed with phenylephrine and were also consistent with activation of α1-adrenoceptors and α2-adrenoceptors by these agonists. As with phenylephrine, rauwolscine did not inhibit the concentration–effect curves indicating that the control curve approximates the α1-adrenoceptor component of the response. The difference between the control curve and the prazosin-resistant component of the response (at the CC15 level of constriction), which provides an indication of the functional selectivity of the agonists at α1- and α2-adrenoceptors, was 3.3-fold and 10.7 for noradrenaline and cirazoline respectively. Therefore, in the mouse tail artery, all three agonists act as preferential α1-adrenoceptor agonists. However, phenylephrine and noradrenaline exhibit similar low selectivity at α1-adrenoceptor and α2-adrenoceptors, and cirazoline was the most selective α1-adrenoceptor agonist tested.
Phenylephrine is generally considered to be a highly selective α1-adrenoceptor agonist. Although previous studies have challenged this assumption, reporting selectivity ratios at α1-and α2-adrenoceptors of ≥10-fold (Flavahan and McGrath, 1981; Guimaraes et al., 1987; Brown et al., 1988), the relative lack of selectivity for phenylephrine observed in the present study was still surprising. There are theoretical conditions when selective activation of one receptor could provide an erroneous appearance of non-selectivity. For example, constitutively active α2-adrenoceptors (Jansson et al., 1998; Murrin et al., 2000) could theoretically amplify an α1-adrenoceptor response to phenylephrine. Likewise, the possible existence of α1/α2-adrenoceptor oligomers could theoretically enable α1-adrenoceptor-dependent activation of α2-adrenoceptor function. Under these conditions, phenylephrine could function as a selective α1-adrenoceptor agonist, but the resulting vasoconstriction would be inhibited by selective α2-adrenoceptor antagonists acting as inverse agonists (Jansson et al., 1998; Murrin et al., 2000; Vilardaga et al., 2005). However, these scenarios are unlikely to have occurred in the mouse tail artery. There was no evidence of constitutively active α2-adrenoceptors: rauwolscine did not inhibit constriction to U46619 or to the α-adrenoceptor agonists (when administered in the absence of prazosin). Furthermore, these scenarios require α1-adrenoceptor activation to enable visualization of the α2-adrenoceptor constrictor response, which would cause the α2-adrenoceptor response to be sensitive to inhibition of α1-adrenoceptors, i.e. prazosin-sensitive. The presence of a prazosin-resistant, rauwolscine-sensitive constriction to phenylephrine suggests that the α2-adrenoceptor constrictor component of the response to the agonist is not dependent on α1-adrenoceptor activation. One caveat to this interpretation is that α1/α2-adrenoceptor oligomerization might change the binding affinity of prazosin for α1-adrenoceptors. However, at least for α1/α1-adrenoceptor dimers, the oligomerization process does not alter the ability of prazosin to inhibit receptor activation (Uberti et al., 2003). Furthermore, oligomerization of α1- and α2-adrenoceptors has never been documented. Therefore, the pharmacological analysis indicates that phenylephrine is acting as a poorly selective agonist at α1-adrenoceptors in this blood vessel.
Although rauwolscine did not affect the control concentration–effect curves to the α-adrenoceptor agonists, it caused a rapid and marked relaxation when administered during stable constriction to the agonists. The magnitude of the dilation to rauwolscine was dependent on the selectivity of the agonists (most powerful with phenylephrine and noradrenaline, less effective with cirazoline) suggesting that the effect was mediated by inhibition of α2-adrenoceptors. Indeed, rauwolscine did not cause relaxation during constriction of mouse tail artery to U46619 or during phenylephrine-induced constriction of mouse mesenteric artery, which does not express functional smooth muscle constrictor α2-adrenoceptors (Flavahan, 2005). Furthermore, the dilator response to rauwolscine in tail arteries was mimicked by another α2-adrenoceptor antagonist, RX821002. Indeed, the acute administration of α2-adrenoceptor antagonists appears to be an efficient and reliable approach to evaluate agonist selectivity. However, the ability of α2-adrenoceptor antagonists to cause powerful relaxation was perplexing. For example, with phenylephrine or noradrenaline, the magnitude of the peak dilation to rauwolscine or RX821002 would be equivalent to an approximate threefold shift in the concentration–effect curve to the agonist (Figure 5). Surprisingly, this inhibitory effect was not evident from directly analysing the effects of the α2-adrenoceptor antagonists (when given alone) on the concentration–effect curves. Because these curves were constructed using the initial peak response to the agonists, we considered that the dilator responses might result from temporal differences in the α1-adrenoceptor and α2-adrenoceptor components of the response: the initial peak response could be dominated by the α1-adrenoceptor response, whereas α2-adrenoceptors may play a more important role to maintain vasoconstriction (McGrath et al., 1982; Fukui et al., 2005). However, there was no difference in the time course to phenylephrine when the tissues were pretreated with the α2-adrenoceptor antagonist. Indeed, we found no evidence for temporal differences in α1-adrenoceptor and α2-adrenoceptor vasoconstriction.
The basis for the surprising dilation to α2-adrenoceptor antagonists became evident when rauwolscine's ability to reverse an α2-adrenoceptor-dependent constriction to UK14,304 was assessed during simultaneous constriction to U46619. Rauwolscine caused dilation that not only completely reversed the α2-adrenoceptor-mediated component of constriction; it virtually abolished the constriction to U46619. This profound dilation, however, was only transient: after 15 min the dilator effect of rauwolscine was restricted to elimination of the α2-adrenoceptor constriction without any effect on the U46619 component of the constriction. The large transient dilation did not result from non-selective activity of the antagonist because rauwolscine did not affect constriction to U46619 in the absence of the α2-adrenoceptor agonist. Therefore, the dilation appeared to be dependent on the termination of α2-adrenoceptor agonist activity. Indeed, abrupt removal of UK14,304 (during combined constriction to UK14,304 and U46619) also caused marked dilation of the U46619 component of constriction. As with rauwolscine, this dilation was transient and after 15 min, the original U46619 constriction was regained. These results indicate that α2-adrenoceptor activation initiates two responses in this blood vessel: constriction that normally predominates and reverses quickly following interruption of receptor stimulation, and dilation, which is slower to reverse than the constrictor response. By rapidly terminating agonist–receptor interaction, acute administration of an α2-adrenoceptor antagonist leads to immediate cessation of the constrictor response and a slower reversal of the dilator response. The peak transient dilation therefore reflects the combined loss of the α2-adrenoceptor constrictor component and the uncovering of the α2-adrenoceptor dilator response. With the slower decay of the α2-adrenoceptor-mediated dilator response, the peak dilation gradually recedes (within 15 min) to reveal the true magnitude of the underlying α2-adrenoceptor-independent constriction. Endothelial denudation abolished the transient dilator responses associated with rapid termination of the α2-adrenoceptor constrictor response (caused either by administration of an antagonist or abrupt removal of the agonist) indicating that the slowly reversing dilation is mediated by activation of endothelial α2-adrenoceptors (Cocks and Angus, 1983; Bockman et al., 1993; Shafaroudi et al., 2005). Endothelial α2-adrenoceptors were considered to be most prominent in the coronary system and to play a limited role in blood vessels (including cutaneous arteries) where smooth muscle α2-adrenoceptors play a prominent constrictor role (Miller and Vanhoutte, 1985; Angus et al., 1986; Flavahan et al., 1989; Ohgushi et al., 1993; Flavahan, 2005). The results of the present study indicate that α2-adrenoceptors can initiate powerful endothelium-dependent dilation and smooth muscle constriction in cutaneous arteries. Endothelium denudation did not significantly affect the vasoconstrictor response to α2-adrenoceptor activation in the mouse tail artery. This is consistent with our previous studies (Flavahan, 2005) and indicates that the α2-adrenoceptor endothelium-dependent dilator response is normally dominated by the smooth muscle constrictor response.
At high concentrations, rauwolscine and other α2-adrenoceptor antagonists have been reported to relax contractions evoked by phenylephrine in the rat isolated aorta and mesenteric arteries (Kim et al., 1999; Artigues-Varin et al., 2002). These rat arteries do not express functional constrictor α2-adrenoceptors, and the contraction to phenylephrine is mediated by α1-adrenoceptors (Macia et al., 1984; Flavahan and Vanhoutte, 1986a; Nielsen et al., 1991; Flavahan, 2005). Indeed, because of the high concentrations of rauwolscine (IC50∼10−6 M) and other α2-adrenoceptor antagonists employed by Kim et al. (1999) and Artigues-Varin et al. (2002), these agents no longer function as selective α2-adrenoceptor antagonists and inhibit α1-adrenoceptors (Digges and Summers, 1983). Therefore, in contrast to the results of the present study, the sustained relaxation to rauwolscine in rat aorta and mesenteric artery is mediated by antagonism of α1-adrenoceptors (Artigues-Varin et al., 2002).
In conclusion, the results of the present study demonstrate that the prototypic α1-adrenoceptor agonist phenylephrine has limited selectivity at α-adrenoceptor subtypes and is a powerful agonist at vascular smooth muscle α2-adrenoceptors. However, the transient dilator response evoked by α2-adrenoceptor antagonists is much greater than the contribution of α2-adrenoceptors to the constrictor response. This reflects the presence of a slowly-reversing α2-adrenoceptor-mediated endothelium-dependent dilation, which transiently amplifies the dilator response to α2-adrenoceptor antagonism. Indeed, this provides a novel, rapid and sensitive approach to assess agonist activity at vascular α-adrenoceptors.
Acknowledgments
This research was supported by grants from the NIH (HL080119 and OH008531).
Statement of conflicts of interest
None.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus JA, Cocks TM, Satoh K. The alpha adrenoceptors on endothelial cells. Fed Proc. 1986;45:2355–2359. [PubMed] [Google Scholar]
- Arain SR, Williams DJ, Robinson BJ, Uhrich TD, Ebert TJ. Vascular responsiveness to brachial artery infusions of phenylephrine during isoflurane and desflurane anesthesia. Anesth Analg. 2002;94:1137–1140. doi: 10.1097/00000539-200205000-00014. table of contents. [DOI] [PubMed] [Google Scholar]
- Artigues-Varin C, Richard V, Varin R, Mulder P, Thuillez C. Alpha2-adrenoceptor ligands inhibit alpha1-adrenoceptor-mediated contraction of isolated rat arteries. Fundam Clin Pharmacol. 2002;16:281–287. doi: 10.1046/j.1472-8206.2002.00091.x. [DOI] [PubMed] [Google Scholar]
- Bockman CS, Jeffries WB, Abel PW. Binding and functional characterization of alpha-2 adrenergic receptor subtypes on pig vascular endothelium. J Pharmacol Exp Ther. 1993;267:1126–1133. [PubMed] [Google Scholar]
- Brown CM, McGrath JC, Midgley JM, Muir AG, O'Brien JW, Thonoor CM, et al. Activities of octopamine and synephrine stereoisomers on alpha-adrenoceptors. Br J Pharmacol. 1988;93:417–429. doi: 10.1111/j.1476-5381.1988.tb11449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotani MA, Flavahan S, Mitra S, Daunt D, Flavahan NA. Silent alpha(2C)-adrenergic receptors enable cold-induced vasoconstriction in cutaneous arteries. Am J Physiol Heart Circ Physiol. 2000;278:H1075–1083. doi: 10.1152/ajpheart.2000.278.4.H1075. [DOI] [PubMed] [Google Scholar]
- Cocks TM, Angus JA. Endothelium-dependent relaxation of coronary arteries by noradrenaline and serotonin. Nature. 1983;305:627–630. doi: 10.1038/305627a0. [DOI] [PubMed] [Google Scholar]
- Devedjian JC, Esclapez F, Denis-Pouxviel C, Paris H. Further characterization of human alpha 2-adrenoceptor subtypes: [3H]RX821002 binding and definition of additional selective drugs. Eur J Pharmacol. 1994;252:43–49. doi: 10.1016/0014-2999(94)90573-8. [DOI] [PubMed] [Google Scholar]
- Digges KG, Summers RJ. Effects of yohimbine stereoisomers on contractions of rat aortic strips produced by agonists with different selectivity for alpha 1- and alpha 2-adrenoceptors. Eur J Pharmacol. 1983;96:95–99. doi: 10.1016/0014-2999(83)90533-2. [DOI] [PubMed] [Google Scholar]
- Eccles R. Substitution of phenylephrine for pseudoephedrine as a nasal decongeststant. An illogical way to control methamphetamine abuse. Br J Clin Pharmacol. 2007;63:10–14. doi: 10.1111/j.1365-2125.2006.02833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan NA. Phenylpropanolamine constricts mouse and human blood vessels by preferentially activating alpha2-adrenoceptors. J Pharmacol Exp Ther. 2005;313:432–439. doi: 10.1124/jpet.104.076653. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, McGrath JC. Blockage by yohimbine of prazosin-resistant pressor effects of adrenaline in the pithed rat. Br J Pharmacol. 1980;69:355–357. doi: 10.1111/j.1476-5381.1980.tb07021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan NA, McGrath JC. Demonstration of simultaneous α1-adrenoceptor, α2-adrenoceptor, α1-adrenoceptor and α2-adrenoceptor mediated effects of phenylephrine in the cardiovascular system of the pithed rat. Br J Pharmacol. 1981;72:P585. [Google Scholar]
- Flavahan NA, McGrath JC. Are human vascular alpha-adrenoceptors atypical? [letter. J Cardiovasc Pharmacol. 1984;6:208–210. [PubMed] [Google Scholar]
- Flavahan NA, Vanhoutte PM. Alpha1-adrenoceptor subclassification in vascular smooth muscle. Trends Pharmacol Sci. 1986a;7:347–349. [Google Scholar]
- Flavahan NA, Vanhoutte PM. Alpha-1 and alpha-2 adrenoceptor: response coupling in canine saphenous and femoral veins. J Pharmacol Exp Ther. 1986b;238:131–138. [PubMed] [Google Scholar]
- Flavahan NA, Vanhoutte PM. Heterogeneity of alpha-adrenergic responses. In: Ruffolo RR Jr, editor. Vascular Smooth Muscle: Role of Receptor Subtypes and Receptor-Reserve, in the Alpha1-Adrenoceptor. New Jersey, USA: Humana Press; 1987. pp. 351–403. [Google Scholar]
- Flavahan NA, Rimele TJ, Cooke JP, Vanhoutte PM. Characterization of postjunctional alpha-1 and alpha-2 adrenoceptors activated by exogenous or nerve-released norepinephrine in the canine saphenous vein. J Pharmacol Exp Ther. 1984;230:699–705. [PubMed] [Google Scholar]
- Flavahan NA, Lindblad LE, Verbeuren TJ, Shepherd JT, Vanhoutte PM. Cooling and alpha 1- and alpha 2-adrenergic responses in cutaneous veins: role of receptor reserve. Am J Physiol. 1985;249:H950–955. doi: 10.1152/ajpheart.1985.249.5.H950. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Cooke JP, Shepherd JT, Vanhoutte PM. Human postjunctional alpha-1 and alpha-2 adrenoceptors: differential distribution in arteries of the limbs. J Pharmacol Exp Ther. 1987;241:361–365. [PubMed] [Google Scholar]
- Flavahan NA, Shimokawa H, Vanhoutte PM. Pertussis toxin inhibits endothelium-dependent relaxations to certain agonists in porcine coronary arteries. J Physiol (Lond) 1989;408:549–560. doi: 10.1113/jphysiol.1989.sp017475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui D, Yang XP, Chiba S. Neurogenic double-peaked vasoconstriction of human gastroepiploic artery is mediated by both alpha1- and alpha2-adrenoceptors. Br J Pharmacol. 2005;144:737–742. doi: 10.1038/sj.bjp.0705975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev. 2001;53:319–356. [PubMed] [Google Scholar]
- Guimaraes S, Paiva MQ, Moura D. Alpha 2-adrenoceptor-mediated responses to so-called selective alpha 1-adrenoceptor agonists after partial blockade of alpha 1-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol. 1987;335:397–402. doi: 10.1007/BF00165554. [DOI] [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, et al. The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)-adrenoceptor-activated Ca(2+)-permeable cation channel. Circ Res. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- Jansson CC, Kukkonen JP, Nasman J, Huifang G, Wurster S, Virtanen R, et al. Protean agonism at alpha2A-adrenoceptors. Mol Pharmacol. 1998;53:963–968. [PubMed] [Google Scholar]
- Kim ND, Kang KW, Kang SY, Vanhoutte PM. Alpha2-adrenoceptor antagonists evoke endothelium-dependent and -independent relaxations in the isolated rat aorta. J Cardiovasc Pharmacol. 1999;34:148–152. doi: 10.1097/00005344-199907000-00023. [DOI] [PubMed] [Google Scholar]
- Kirby BS, Voyles WF, Carlson RE, Dinenno FA. Graded sympatholytic effect of exogenous ATP on postjunctional alpha-adrenergic vasoconstriction in the human forearm: implications for vascular control in contracting muscle. J Physiol. 2008;586:4305–4316. doi: 10.1113/jphysiol.2008.154252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollar C, Schneider H, Waksman J, Krusinska E. Meta-analysis of the efficacy of a single dose of phenylephrine 10 mg compared with placebo in adults with acute nasal congestion due to the common cold. Clin Ther. 2007;29:1057–1070. doi: 10.1016/j.clinthera.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Lewis SJ, Hoque A, Sandock K, Robertson TP, Bates JN, Kooy NW. Differential effects of peroxynitrite on the function of arginine vasopressin V(1a) receptors and alpha(1)-adrenoceptors in vivo. Vascul Pharmacol. 2007;46:24–34. doi: 10.1016/j.vph.2006.06.004. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Flavahan NA, McKean CE. alpha 1- and alpha 2-adrenoceptor-mediated pressor and chronotropic effects in the rat and rabbit. J Cardiovasc Pharmacol. 1982;4(Suppl. 1):S101–107. doi: 10.1097/00005344-198200041-00021. [DOI] [PubMed] [Google Scholar]
- Macia RA, Matthews WD, Lafferty J, DeMarinis RM. Assessment of alpha-adrenergic receptor subtypes in isolated rat aortic segments. Naunyn Schmiedebergs Arch Pharmacol. 1984;325:306–309. doi: 10.1007/BF00504373. [DOI] [PubMed] [Google Scholar]
- Miller VM, Vanhoutte PM. Endothelial alpha 2-adrenoceptors in canine pulmonary and systemic blood vessels. Eur J Pharmacol. 1985;118:123–129. doi: 10.1016/0014-2999(85)90670-3. [DOI] [PubMed] [Google Scholar]
- Mueed I, Bains P, Zhang L, Macleod KM. Differential participation of protein kinase C and Rho kinase in alpha 1-adrenoceptor mediated contraction in rat arteries. Can J Physiol Pharmacol. 2004;82:895–902. doi: 10.1139/y04-086. [DOI] [PubMed] [Google Scholar]
- Murrin LC, Gerety ME, Happe HK, Bylund DB. Inverse agonism at alpha(2)-adrenoceptors in native tissue. Eur J Pharmacol. 2000;398:185–191. doi: 10.1016/s0014-2999(00)00317-4. [DOI] [PubMed] [Google Scholar]
- Nielsen H, Pilegaard HK, Hasenkam JM, Mortensen FV, Mulvany MJ. Heterogeneity of postjunctional alpha-adrenoceptors in isolated mesenteric resistance arteries from rats, rabbits, pigs, and humans. J Cardiovasc Pharmacol. 1991;18:4–10. doi: 10.1097/00005344-199107000-00002. [DOI] [PubMed] [Google Scholar]
- Ohgushi M, Yasue H, Kugiyama K, Murohara T, Sakaino N. Contraction and endothelium dependent relaxation via alpha adrenoceptors are variable in various pig arteries. Cardiovasc Res. 1993;27:779–784. doi: 10.1093/cvr/27.5.779. [DOI] [PubMed] [Google Scholar]
- Philipp M, Brede M, Hein L. Physiological significance of alpha(2)-adrenergic receptor subtype diversity: one receptor is not enough. Am J Physiol Regul Integr Comp Physiol. 2002;283:R287–295. doi: 10.1152/ajpregu.00123.2002. [DOI] [PubMed] [Google Scholar]
- Rajanayagam MA, Medgett IC. Greater activation of smooth muscle alpha-2 adrenoceptors by epinephrine in distal than in proximal segments of rat tail artery. J Pharmacol Exp Ther. 1987;240:989–997. [PubMed] [Google Scholar]
- Shafaroudi MM, McBride M, Deighan C, Wokoma A, Macmillan J, Daly CJ, et al. Two ‘knockout’ mouse models demonstrate that aortic vasodilatation is mediated via alpha2a-adrenoceptors located on the endothelium. J Pharmacol Exp Ther. 2005;314:804–810. doi: 10.1124/jpet.105.085944. [DOI] [PubMed] [Google Scholar]
- Uberti MA, Hall RA, Minneman KP. Subtype-specific dimerization of alpha 1-adrenoceptors: effects on receptor expression and pharmacological properties. Mol Pharmacol. 2003;64:1379–1390. doi: 10.1124/mol.64.6.1379. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Steinmeyer R, Harms GS, Lohse MJ. Molecular basis of inverse agonism in a G protein-coupled receptor. Nat Chem Biol. 2005;1:25–28. doi: 10.1038/nchembio705. [DOI] [PubMed] [Google Scholar]