

Abstract
Background and purpose:
The heart of the canine model of chronic atrioventricular block is known to have a ventricular electrical remodelling, which mimics the pathophysiology of long QT syndrome. Using this model, we explored a new pharmacological therapeutic strategy for the prevention of cardiac sudden death.
Experimental approach:
The L-type Ca2+ channel blocker amlodipine (2.5 mg·day−1), L/N-type Ca2+ channel blocker cilnidipine (5 mg·day−1), or the angiotensin II receptor blocker candesartan (12 mg·day−1) was administered orally to the dogs with chronic atrioventricular block for 4 weeks. Electropharmacological assessments with the monophasic action potential (MAP) recordings and blood sample analyses were performed before and 4 weeks after the start of drug administration.
Key results:
Amlodipine and cilnidipine decreased the blood pressure, while candesartan hardly affected it. The QT interval, MAP duration and beat-to-beat variability of the ventricular repolarization period were shortened only in the cilnidipine group, but such effects were not observed in the amlodipine or candesartan group. Plasma concentrations of adrenaline, angiotensin II and aldosterone decreased in the cilnidipine group. In contrast, plasma concentrations of angiotensin II and aldosterone were elevated in the amlodipine group, whereas in the candesartan group an increase in plasma levels of angiotensin II and a decrease in noradrenaline and adrenaline concentrations were observed.
Conclusions and implications:
Long-term blockade of L/N-type Ca2+ channels ameliorated the ventricular electrical remodelling in the hypertrophied heart which causes the prolongation of the QT interval. This could provide a novel therapeutic strategy for the treatment of cardiovascular diseases.
Keywords: cilnidipine, N-type Ca2+ channel, chronic atrioventricular block dog, hypertrophied heart, long QT interval
Introduction
Cardiac hypertrophy is a strong independent risk factor for cardiac sudden death. A hypertrophied heart generally has structural, functional and electrical remodelling that progressively leads to a decline in cardiac functions and predisposes the heart to arrhythmias (Tomaselli and Marbán, 1999). Abnormal repolarization and Ca2+ handling anomalies in the cardiomyocytes are thought to facilitate early afterdepolarization and triggered activity (Antzelevitch, 2007). Recently, inhibitors of cardiac ion channels, the ryanodine receptor or calmodulin kinase II have been evaluated in proarrhythmic animal models as a downstream therapy (Verduyn et al., 1995; Mazur et al., 1999; Antzelevitch et al., 2004; Lu et al., 2006).
The pathophysiology of ventricular repolarization delay has been extensively analysed in animals with chronic atrioventricular block, known as a volume-overloaded hypertrophy models (Vos et al., 1998; Volders et al., 1999; Verduyn et al., 2001; Sugiyama et al., 2002; Takahara et al., 2006; 2007a;). The long-term bradycardia in these models induces downregulation of cardiac K+ channels leading to electrical remodelling together with compensatory activation of sympathetic tone and the renin-angiotensin system (Volders et al., 1999; Verduyn et al., 2001; Takahara et al., 2007a). Modulation of the pathophysiology of the electrical remodelling may become a new pharmacological strategy, an upstream therapy, to reduce the risk of lethal arrhythmias. However, the electrical remodelling in the hypertrophied heart caused by volume overload has been found to be refractory to pharmacological therapy with an angiotensin AT1 receptor blocker and to cardiac pacing, a non-pharmacological therapy (Peschar et al., 2003; Reddy et al., 2003; Schoenmakers et al., 2003).
N-type Ca2+ channels have been demonstrated to play a pivotal role in neurotransmitter release from sympathetic nerve endings (Hirning et al., 1988; Molderings et al., 2000). A previous clinical study has demonstrated that the L/N-type Ca2+ channel blocker cilnidipine, which is used as an antihypertensive drug in Japan (Uneyama et al., 1997; 1999; Takahara etal., 2007b; Takahara, 2009), suppresses the overactivity of the cardiac sympathetic system in hypertensive patients more effectively than amlodipine (Sakata et al., 1999). Recent experimental data have shown that cilnidipine also inhibits the local renin-angiotensin system and aldosterone secretion from adrenocortical cells (Takemori et al., 2005; Konda et al., 2006; 2009; Aritomi et al., 2007). Thus, the pleiotropic effects of cilnidipine on neurohumoral factors may provide a new strategy for the treatment of cardiovascular diseases, as reported in hypertensive patients with chronic renal disease (Fujita et al., 2007). In this study, we assessed the potential utility of cilnidipine as an upstream treatment of ventricular repolarization delay in the hypertrophied heart, and compared its effects with those of the L-type Ca2+ channel blocker amlodipine and angiotensin AT1 receptor blocker candesartan, in a canine model of chronic atrioventricular block.
Methods
Animal model
The investigation was performed according to the Guidelines for Animal Experiments, University of Yamanashi and Ajinomoto Co., Inc., which are equivalent to those of the US National Institute of Health. The beagle dogs of either sex weighing about 10 kg were kept in individual cages on a 12 h light (6:00 h–18:00 h)–dark (18:00 h–6:00 h) cycle. The ventilation provided a total air exchange rate of 10–15 times per hour. The temperature was maintained at 23 ± 2°C, and relative humidity was 50 ± 30%. Each dog was fed 200 ± 10 g of standard diet for dogs (CD-5M, CLEA Japan, Tokyo, Japan) in the morning, and water was available ad libitum.
The surgical procedure using a catheter ablation technique was carried out as described previously (Sugiyama et al., 2002; Takahara et al., 2006; 2007a;). Briefly, the beagle dogs were anaesthetized with sodium pentobarbital (30 mg·kg−1, i.v.) and artificially ventilated with room air (SN-408-3; Shinano, Tokyo, Japan). Additional doses of sodium pentobarbital (3–6 mg·kg−1, i.v.) were given when necessary. The surface lead II electrocardiogram (ECG) was continuously monitored using a polygraph system (RM-6000; Nihon Kohden, Tokyo, Japan). A quad-polar electrodes catheter with a large tip of 4 mm (D7-DL-252; Cordis-Webster, Baldwin Park, CA, USA) was inserted through the right femoral vein using the standard percutaneous technique under sterile conditions and positioned at the tricuspid valve by watching the bipolar electrograms from the pair of distal electrodes. The optimal site for ablation of the atrioventricular node, namely the compact atrioventricular node, was determined on the basis of the intracardiac electrogram, of which a very small His deflection was recorded and the atrium/ventricular voltage ratio was >2. The power source for atrioventricular node ablation was an electrosurgical generator (MS-1500; Mera, Tokyo, Japan) delivering continuous unmodulated radiofrequency energy at a frequency of 500 kHz. After proper positioning, the radiofrequency energy of 20 W was delivered for 10 s from the tip electrode to an indifferent patch electrode positioned on the animal's back, which was continued for 30 s if junctional rhythm was induced. The end-point of this procedure was the development of complete atrioventricular block with the onset of stable idioventricular escaped rhythm.
Holter ECG recording
A Holter recording and analysis system (QR2100 and HS1000, Fukuda ME Kogyo, Tokyo, Japan) was used to record and analyse ECG over 21 h. Ventricular premature contractions were defined as a premature depolarization of coupling interval ≤600 ms with prolonged, bizarre QRS complexes (Yoshida et al., 2002).
Plasma concentrations of neurohumoral factors and drugs
The plasma was obtained from the supernatant of blood containing EDTA after centrifugation at 1500×g for 15 min, and was stored at −80°C until the measurements. The concentrations of adrenaline, noradrenaline and dopamine in the plasma were measured using a high-performance liquid chromatographic technique, whereas those of angiotensin II, aldosterone and atrial natriuretic peptide were assessed by a radioimmunoassay, which were performed by SRL Co. (Tokyo, Japan). Plasma concentrations of amlodipine and cilnidipine were determined by liquid chromatography-tandem mass spectrometry, which were conducted at the Pharmaceutical Research Laboratories of Pharmaceuticals Company, Ajinomoto Co. Inc. (Kawasaki, Japan).
In vivo cardiovascular and electrophysiological measurements
Cardiovascular and electrophysiological variables were measured as described previously (Takahara et al., 2007a). The dogs were anaesthetized with pentobarbital sodium (30 mg·kg−1, i.v.) and artificially ventilated. Additional doses of sodium pentobarbital (3–6 mg·kg−1, i.v.) were given when necessary. A catheter containing heparin was placed in the aorta to measure the systemic blood pressure. A thermodilution catheter (TC-704; Nihon-Kohden) was positioned at the right side of the heart to monitor the pulmonary capillary wedge pressure. Cardiac output was measured by a standard thermodilution method with a cardiac output computer (MFC-1100; Nihon-Kohden). Total peripheral vascular resistance (TPR) was calculated using the basic equation: TPR = mean blood pressure/cardiac output. The surface lead II ECG was obtained from the limb electrodes. A bidirectional steerable monophasic action potential (MAP) recording/pacing combination catheter (1675P; EP Technologies, Sunnyvale, CA, USA) was positioned at the endocardium of the interventricular septum of the right ventricle. The duration of the MAP signal was measured at 90% repolarization level as MAP90. The heart was electrically driven using a cardiac stimulator (SEC-3102; Nihon-Kohden) with the pacing electrodes of the MAP recording/pacing combination catheter in the right ventricle. Stimulation pulses were rectangular in shape, 1–2 V (about twice the threshold voltage) and 1 ms duration. MAP90 was measured during the ventricular rhythm and at each pacing cycle length of 300–1000 ms. The effective refractory period (ERP) was assessed by programmed electrical stimulation to the right ventricle. The pacing protocol consisted of five beats of basal stimuli in each cycle length followed by an extra stimulus of various coupling intervals. The parameters were continuously monitored using a polygraph system (RM-6000; Nihon-Kohden), and analysed with a real-time full automatic data analysis system (MP/VAS 3 for Macintosh, ver 1.0; Physio-Tech, Tokyo, Japan).
Beat-to-beat analysis
MAP duration (MAP90) of 31 consecutive beats under stable idioventricular automaticity was measured before and after the drug administration. Poincaré plots with MAP90(n) versus MAP90(n+ 1) were prepared for each analysis time point. The mean orthogonal distance from the diagonal to the points of the Poincaré plot was determined as short-term variability (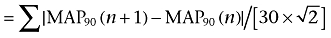 ). Whereas, the mean distance to the mean of the parameter parallel to the diagonal of the Poincaré plot was determined as long-term variability (
). Whereas, the mean distance to the mean of the parameter parallel to the diagonal of the Poincaré plot was determined as long-term variability (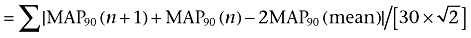 ). Similarly, the short-term and long-term variability of the RR interval was also calculated. These nomenclatures are adopted from investigations of heart rate variability using Holter monitoring in humans (Brennan et al., 2001), which have been applied to canine models of chronic atrioventricular block (Thomsen et al., 2004; 2006; Takahara et al., 2006; 2008;).
). Similarly, the short-term and long-term variability of the RR interval was also calculated. These nomenclatures are adopted from investigations of heart rate variability using Holter monitoring in humans (Brennan et al., 2001), which have been applied to canine models of chronic atrioventricular block (Thomsen et al., 2004; 2006; Takahara et al., 2006; 2008;).
Study protocol
The anatomical and electrophysiological remodellings have been shown to be completed within 4 weeks after the onset of atrioventricular block, and no further prolongation of QT interval was detected thereafter (Vos et al., 1998; Sugiyama et al., 2002; Peschar et al., 2003; Takahara et al., 2006). Thus, the present experiment was started ≥4 weeks after the surgery. The animals were divided into three groups and received either amlodipine (n= 8), cilnidipine (n= 7) or candesartan (n= 7).
At pre-drug control, venous blood was withdrawn from the brachial vein in a stable condition without anaesthesia, and the ECG was recorded for >21 h. After the Holter ECG recording, cardiovascular and electrophysiological parameters were monitored under pentobarbital anaesthesia. After the basal assessment, a commercially available tablet of amlodipine (2.5 mg), cilnidipine (5 mg) or candesartan (12 mg) was orally administered to the dogs every day in the morning. In the amlodipine and cilnidipine groups, 2 and 4 weeks after the start of drug administration, the blood sampling, Holter ECG recording and cardiovascular and electrophysiological analyses were similarly performed. In the candesartan group, the assessments were performed only 4 weeks after the start of drug administration.
Data analysis
Data are expressed as the mean ± SEM. The statistical comparisons within a parameter were evaluated by one-way, repeated-measures analysis of variance followed by Contrasts for mean values comparison, whereas those of paired data within a parameter were evaluated by unpaired t-test. The statistical differences of unpaired data between the groups were evaluated by unpaired t-test. A P value <0.05 was considered statistically significant. Drug/molecular target nomenclature conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2008).
Drugs
Amlodipine was obtained from Norvasc (Pfizer, Tokyo, Japan), cilnidipine from Atelec (Mochida, Tokyo, Japan) and candesartan from Blopress (Takeda, Osaka, Japan).
Results
ECG and survival rate
Body weights of the dogs subjected to chronic atrioventricular block at pre-drug control in the amlodipine, cilnidipine and candesartan groups were 9.6 ± 0.6, 10.1 ± 0.1 and 10.1 ± 0.4 kg respectively, and no significant change was detected during the study. In the cilnidipine and candesartan groups, all animals survived during the study, whereas one animal died suddenly on the 18th day in the amlodipine group. The number of ventricular premature contractions at pre-drug control in the amlodipine, cilnidipine and candesartan group was 63 ± 31, 284 ± 177 and 156 ± 94 beats per 21 h respectively. In the cilnidipine group, the number of the premature ventricular contraction was reduced to 25 ± 16 beats per 21 h at 2 weeks, whereas no significant change was detected in the amlodipine or candesartan group. Ventricular tachycardia or torsade de pointes arrhythmia was not detected during the Holter ECG study.
After the assessment at 4 weeks, we additionally administered a torsadogenic dose of cisapride (10 mg·kg−1, p.o., Sugiyama et al., 2002) to one dog with chronic atrioventricular block in the cilnidipine group, and torsade de pointes arrhythmia was detected in this animal.
Cardiovascular effects
The effects of amlodipine, cilnidipine and candesartan on the cardiovascular parameters are summarized in Figure 1. In the amlodipine and cilnidipine groups, the mean blood pressure and TPR decreased and cardiac output and cardiac index increased at 2 and 4 weeks. In the candesartan group, no significant changes in any of cardiovascular parameters were detected.
Figure 1.
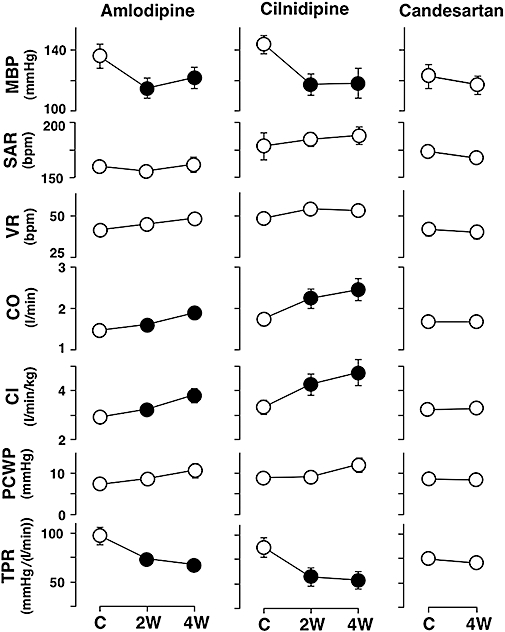
Cardiovascular effects of amlodipine, cilnidipine and candesartan in the canine model of chronic atrioventricular block. Cardiovascular variables in the amlodipine (n= 8) and cilnidipine (n= 7) groups were obtained at pre-drug control (C) and 2 weeks (2W) and 4 weeks (4W) after the start of drug administration, whereas those in the candesartan group (n= 7) were obtained at pre-drug control and 4 weeks after the start of drug administration. Data are presented as the mean ± SEM. Solid symbols represent the significant differences from each pre-drug control (C) value, P < 0.05. MBP, mean blood pressure; SAR, sinoatrial rate; VR, ventricular rate; CO, cardiac output; CI, cardiac index; PCWP, pulmonary capillary wedge pressure; TPR, total peripheral vascular resistance.
Electrophysiological effects
Typical tracings of the effects of cilnidipine on the ECG and MAP signal during the idioventricular rhythm are depicted in Figure 2A. The effects of amlodipine, cilnidipine and candesartan on the ventricular repolarization phase are presented in Figure 2B. The QT interval and MAP90 were abbreviated at 2 and 4 weeks only in the cilnidipine group, whereas no significant changes in these parameters were detected in the amlodipine and candesartan groups.
Figure 2.
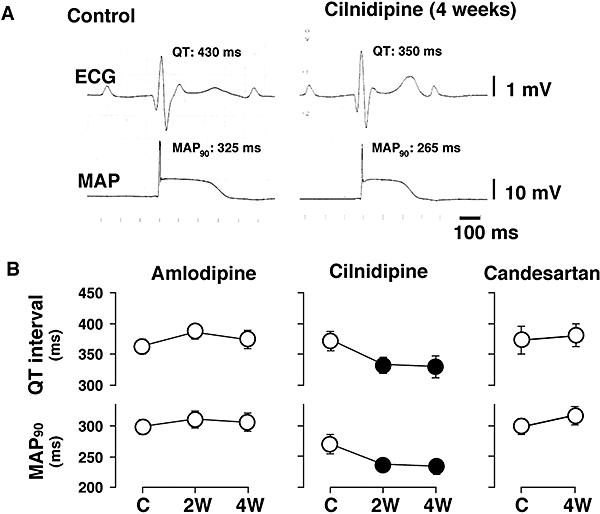
Effects of the different drugs on the electrocardiogram (ECG) and monophasic action potential (MAP) signal during idioventricular rhythm. (A) Typical tracings of effects of cilnidipine on ECG and MAP signal. (B) Effects of amlodipine (n= 8), cilnidipine (n= 7) and candesartan (n= 7) on the QT interval and MAP duration. These parameters were obtained at pre-drug control (C) and 2 weeks (2W) and 4 weeks (4W) after the start of drug administration. Data are presented as the mean ± SEM. Solid symbols represent the significant differences from each pre-drug control (C) value, P < 0.05.
Typical results of Poincaré plots of the MAP90, in which cilnidipine decreased the MAP90 and short-term variability, are shown in Figure 3. The effects of the drugs on the short-term and long-term variability of MAP90 are shown in Table 1. Cilnidipine significantly decreased the short-term variability without affecting the long-term variability of the MAP duration, whereas amlodipine and candesartan had no significant effects on either variable. Pre-drug control values for the short-term variability of the RR interval in the amlodipine, cilnidipine and candesartan group were 18.3 ± 4.2, 6.2 ± 1.7 and 6.5 ± 1.2 ms respectively, whereas those for the long-term variability were 15.6 ± 4.8, 7.5 ± 2.0 and 10.5 ± 1.6 ms respectively. No significant change was detected in the variability of the RR interval in either the amlodipine, cilnidipine or candesartan group.
Figure 3.
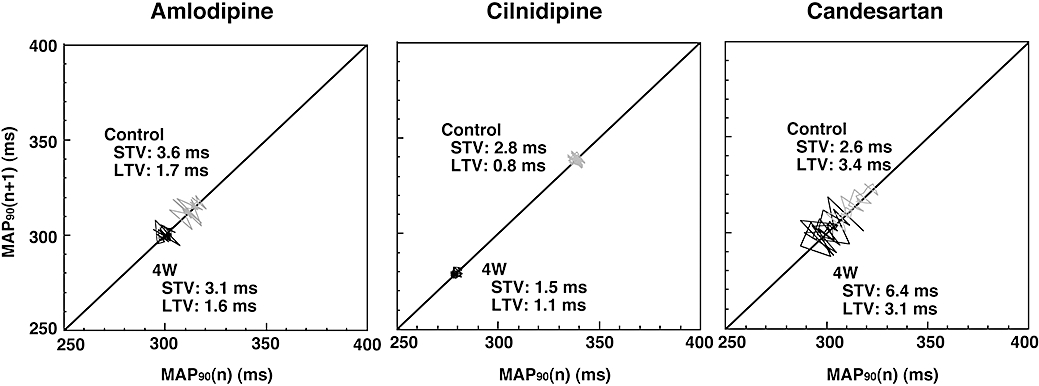
Effects of amlodipine, cilnidipine and candesartan on the Poincaré plots of the duration of the monophasic action potential (MAP90) in the canine model of chronic atrioventricular block. Thirty-one beats were plotted for each of two analysis time points; before (Control) and 4 weeks after the drug administration (4W). STV, short-term variability; LTV, long-term variability.
Table 1.
Effects of amlodipine, cilnidipine and candesartan on the beat-to-beat variability of the monophasic action potential (MAP) duration in the chronic atrioventricular block dogs
|
Amlodipine |
Cilnidipine |
Candesartan |
||||||
|---|---|---|---|---|---|---|---|---|
| C | 2W | 4W | C | 2W | 4W | C | 4W | |
| STV (ms) | 3.8 ± 0.6 | 4.3 ± 1.1 | 5.0 ± 0.7 | 4.2 ± 1.2 | 1.8 ± 0.4* | 2.2 ± 0.4* | 4.1 ± 0.8 | 5.2 ± 0.9 |
| LTV (ms) | 2.4 ± 0.4 | 3.1 ± 1.2 | 3.3 ± 0.8 | 2.0 ± 0.5 | 1.4 ± 0.3 | 1.7 ± 0.3 | 2.6 ± 0.6 | 3.3 ± 0.6 |
Short-term variability (STV) and long-term variability (LTV) of the MAP duration in the amlodipine and cilnidipine groups were obtained at pre-drug control (C) and 2 weeks (2W) and 4 weeks (4W) after the start of drug administration, whereas those in the candesartan group were obtained at pre-drug control and 4 weeks after the start of drug administration. Data are presented as the mean ± SEM.
P < 0.05, compared with corresponding pre-drug control value.
Effects of amlodipine, cilnidipine and candesartan on the MAP90 during the ventricular pacing and ERP are shown in Figure 4. In the cilnidipine group, the MAP90 and ERP were reduced at various pacing cycle lengths between 300 and 1000 ms, whereas no significant changes in these parameters were detected in the amlodipine and candesartan groups.
Figure 4.
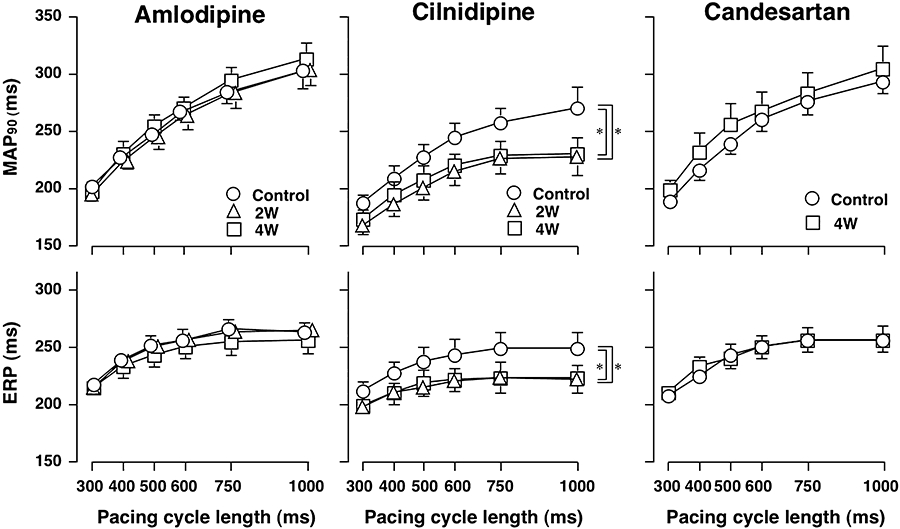
Electrophysiological effects of amlodipine, cilnidipine and candesartan on the ventricular repolarization phase of the canine model of chronic atrioventricular block. MAP duration (MAP90) and effective refractory period (ERP) of various basic pacing cycle length of 300, 400, 500, 600, 750 and 1000 ms in the amlodipine (n= 8) and cilnidipine (n= 7) groups were obtained at pre-drug control (C) and 2 weeks (2W) and 4 weeks (4W) after the start of drug administration, whereas those in the candesartan group (n= 7) were obtained at pre-drug control and 4 weeks after the start of drug administration. Data are presented as the mean ± SEM. *P < 0.05 compared with corresponding pre-drug control value (C).
Plasma concentrations of neurohormonal factors and drugs
The effects of amlodipine, cilnidipine and candesartan on the plasma levels of neurohumoral factors are shown in Figure 5. In the amlodipine group, the plasma concentrations of angiotensin II and aldosterone increased at 4 weeks. The plasma concentration of noradrenaline in the animal that died on the 18th day was 810 ng·mL−1 at 2 weeks, which was >1.4 times higher than that at control (569 ng·mL−1). In the cilnidipine group, the plasma concentrations of adrenaline, angiotensin II and aldosterone were decreased at 2 and 4 weeks. In the candesartan group, the plasma concentrations of noradrenaline and adrenaline were decreased, and that of angiotensin II increased, at 4 weeks. The maximum plasma concentrations of amlodipine and cilnidipine on the first day were 24.22 ± 3.99 and 9.16 ± 1.44 ng·mL−1 respectively. An i.v. injection of angiotensin II (0.1 µg·kg−1) elevated the mean blood pressure by +33 ± 2 mmHg at control and this increase was completely abolished at 4 weeks in the candesartan group.
Figure 5.
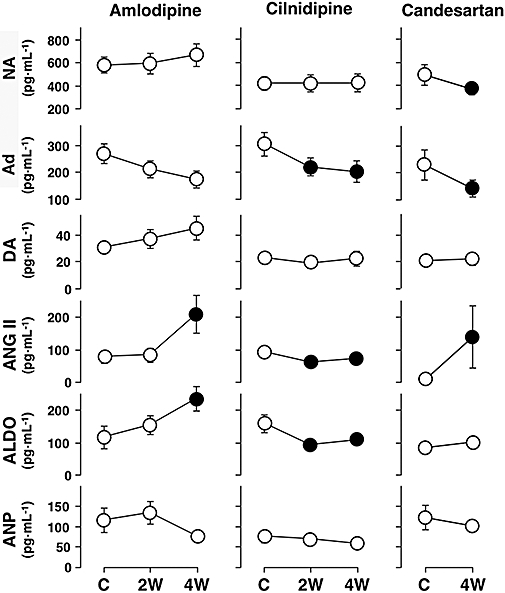
Effects of amlodipine, cilnidipine and candesartan on neurohormones in the canine model of chronic atrioventricular block. Plasma concentrations of each neurohumoral factor in the amlodipine (n= 8) and cilnidipine (n= 7) groups were obtained at pre-drug control (C) and 2 weeks (2W) and 4 weeks (4W) after the start of drug administration, whereas those in the candesartan group (n= 7) were obtained at pre-drug control and 4 weeks after the start of drug administration. Data are presented as the mean ± SEM. Solid symbols represent significant differences from each pre-drug control (C) value, P < 0.05. NA, noradrenaline; Ad, adrenaline; DA, dopamine; ANG II, angiotensin II; ALDO, aldosterone; ANP, atrial natriuretic peptide.
Discussion
The results of the present study clearly show that the L/N-type Ca2+ channel blocker cilnidipine shortened the ventricular repolarization period of the chronic atrioventricular block in dogs, and this effect was not observed with the L-type Ca2+ channel blocker amlodipine or the angiotensin AT1 receptor blocker candesartan. So, this effect may be closely associated with long-term blockade of L/N-type Ca2+ channels, because the dose of each drug corresponded to the therapeutic level based on experimental data from hypertensive dogs (Yamanaka et al., 1991; Yoshimoto et al., 1992; Ito et al., 1995).
Effects on haemodynamics and neurohumoral factors
Our previous cardiovascular and neurohumoral studies have shown that in the canine model of chronic atrioventricular block the heart compensates by becoming hypertrophied (Takahara et al., 2007a). The optimal doses of amlodipine (2.5 mg per dog) and cilnidipine (5 mg per dog) used in this study were determined on the basis of previous results obtained in reno-hypertensive dogs; these drugs effectively lowered the blood pressure at doses of 0.2 mg·kg−1 and 0.3–1.0 mg·kg−1 respectively (Yamanaka et al., 1991; Yoshimoto et al., 1992). In this study, both amlodipine and cilnidipine decreased blood pressure and peripheral vascular resistance to a similar extent, which reflects the fundamental pharmacological profile of dihydropyridine Ca2+ channel blockers. In the amlodipine group, one animal died on the 18th day; this might be associated with increased sympathetic tone, which could trigger a lethal arrhythmia. Candesartan failed to affect blood pressure in this study. However, we confirmed that the dose of candesartan used effectively blocks angiotensin AT1 receptors in vivo, and also this dose has been shown to lower blood pressure in reno-vascular hypertensive dogs (Ito et al., 1995). In a previous study, angiotensin II and aldosterone were demonstrated to play an important role in the development of ventricular remodelling in this animal model (Vos et al., 1998). Thus, the finding that cilnidipine reduced plasma angiotensin II and aldosterone levels may be important when analysing the present results. Although the precise mechanism by which cilnidipine causes this effect is not clear at present, N-type Ca2+ channel blockade of cilnidipine may inhibit catecholamine release from the sympathetic nerve ending and adrenal gland (Takahara et al., 1997; Nagayama et al., 1998), leading to suppression of the renin-angiotensin-aldosterone system (Karlberg, 1983), and suppression of aldosterone secretion from adrenocortical cells (Aritomi et al., 2007). In contrast, amlodipine might have activated the renin-angiotensin-aldosterone system by increasing the sympathetic tone in this animal model, because it decreased mean blood pressure to a similar extent to that of cilnidipine.
Effects on ventricular repolarization
Disease-mediated reduction in net repolarizing current of cardiac cells, known as reduced repolarization reserve (Roden, 1998), can prolong the QT interval, which in some cases may amplify any electrical imbalances in the ventricular myocardium. This could result in the development of early afterdepolarization-induced triggered activity, leading to the generation of life-threatening cardiac arrhythmias including torsade de pointes (Antzelevitch, 2007). Volders et al. (1999) have already demonstrated the significant down-regulation of the slow component of the delayed rectifier K+ currents (IKs) and a small decrease in the rapid component of delayed rectifier K+ currents (IKr) in hearts with chronic atrioventricular block. We have also confirmed that mRNA levels of KvLQT1 and MiRP1 were significantly lower in hearts with chronic atrioventricular block than in normal hearts (data not shown), which suggests that our canine model possesses similar subcellular adaptations to those in the model used by Volders et al. (1999). The electrical remodelling in this animal model has been demonstrated to remain unchanged for at least for 14 weeks, once it is completed (Peschar et al., 2003).
As clearly shown in Figure 2, cilnidipine shortens the ventricular repolarization period of the hypertrophied heart, suggesting that it may increase the repolarization reserve. This observation does not accord with results from our previous study that demonstrated the ventricular repolarization process was little affected by acutely administered cilnidipine in this animal model (Takahara et al., 2004), indicating that it cannot be simply explained by its immediate effects on sympathetic N-type and vascular L-type Ca2+ channels. In our previous study, which used the same animal model, the QT interval of the remodelled dog was about 80 ms longer than that of the dog with acute atrioventricular block (Takahara et al., 2006), whereas in the present study the shortening of the QT interval by cilnidipine was about 40 ms. Also, in one experiment, a torsadogenic dose of cisapride ‘paradoxically’ induced torsade de pointes arrhythmia in a dog with chronic atrioventricular block after 4 weeks of cilnidipine treatment. Thus, the electrical remodelling caused by chronic atrioventricular block may be partially reversed by cilnidipine. The important observation in this study is that cilnidipine also decreased the beat-to-beat variability of ventricular repolarization, as shown in Figure 3. As it has been suggested that the greater the extent of the beat-to-beat variability of ventricular repolarization the higher the risk of sudden cardiac death (Thomsen et al., 2005), the present results may imply that cilnidipine can lower the risk for lethal arrhythmias in the remodelled heart.
The cellular mechanism(s) by which cilnidipine shortens the ventricular repolarization has not been fully elucidated at present. Previous in vitro electrophysiological studies have demonstrated that angiotensin II decreases IKr, transient outward K+ currents (Ito) and inward rectifier K+ currents (IK1) of the cardiomyocytes (Yu et al., 2000; Domenighetti et al., 2007; Wang et al., 2008) and that aldosterone decreases Ito (Bénitah et al., 2001). Based on the differences in the effects on the neurohumoral factors between cilnidipine and other drugs, we speculate that the inhibitory effect of cilnidipine on the renin-angiotensin-aldosterone system (Takemori et al., 2005; Konda et al., 2006; Aritomi et al., 2007) may have decreased the suppression of the K+ channels. In contrast, candesartan has been shown to prolong the ventricular action potential duration via suppression of IKs and Ito (Caballero et al., 2001), which may partly explain why candesartan did not affect the ventricular repolarization period in this animal model.
Possible clinical applications
A previous report has suggested that regression of structural and electrical remodelling should be considered clinically as an independent process for the prevention of the onset of lethal arrhythmias in the future (Peschar et al., 2003), because regression of ventricular hypertrophy did not necessarily improve the electrical remodelling in the canine model of chronic atrioventricular block (Reddy et al., 2003). Although the electrical remodelling caused by volume overload is thought to be an irreversible phenomenon (Peschar et al., 2003), the present study successfully demonstrated that cilnidipine can shorten the ventricular repolarization period of the chronic atrioventricular block in this canine model. This is the first study to show a recovery from electrical remodelling in the volume-overloaded hypertrophied heart (Peschar et al., 2003; Reddy et al., 2003; Schoenmakers et al., 2003).
Based on the results from the present study, cilnidipine could be useful for the treatment of patients whose prolonged QT interval is a strong risk factor for torsade de pointes arrhythmias (Topilski et al., 2007). Cilnidipine is expected to have a similar restorative effect on the electrical remodelling process of chronic atrioventricular block in humans. Indeed, a recent electrophysiological study has demonstrated that IKs and Ito are down-regulated in the diabetic canine heart, leading to QT interval prolongation (Lengyel et al., 2007). A long QT interval has also been reported in patients with various cardiovascular diseases including hypertension with hypertrophy, hypertrophic cardiomyopathy and end-stage renal failure (Dritsas et al., 1992; Singh et al., 1997; Covic et al., 2002; Swynghedauw et al., 2003; Raizada et al., 2005; Wong et al., 2005). Thus, further analysis of the function of cardiac K+ channel subtypes in such pathological conditions will provide important information on the effectiveness of cilnidipine as a therapy for this ventricular repolarization abnormality.
Conclusion
Long-term blockade of L/N-type Ca2+ channels may become a new upstream therapy to reduce the risk of lethal arrhythmias.
Acknowledgments
The authors thank Dr Hiroshi Miyano (Ajinomoto Co. Inc.) for helpful advice on measurement of plasma drug concentrations. The authors also thank Dr Kiyotaka Hoshiai and Miss Yukiko Sakurai for analysing the ECG. This study was supported in part by Grant-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (#19590532) and The Pharmacological Research Foundation (Tokyo).
Glossary
Abbreviations:
- ERP
effective refractory period
- IK1
inward rectifier K+ currents
- IKr
rapid component of delayed rectifier K+ currents
- IKs
slow component of delayed rectifier K+ currents
- Ito
transient outward K+ currents
- MAP
monophasic action potential
- MAP90
duration of MAP signal at 90% repolarization level
- TPR
total peripheral vascular resistance
Conflict of interest
The authors state no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd ed. Br J Pharmacol. 2008;153(Suppl 2):S1, S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C. Ionic, molecular, and cellular bases of QT-interval prolongation and torsade de pointes. Europace. 2007;9(Suppl 4):iv4–15. doi: 10.1093/europace/eum166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aritomi S, Nogi Y, Koganei H, Mitsui A, Konda T, Yoshimura M. The role of N-type calcium channels in regulating aldosterone secretion in H295R human adrenocortical cells. J Am Soc Nephrol. 2007;18:155A. [Google Scholar]
- Bénitah JP, Perrier E, Gómez AM, Vassort G. Effects of aldosterone on transient outward K+ current density in rat ventricular myocytes. J Physiol. 2001;537:151–160. doi: 10.1111/j.1469-7793.2001.0151k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan M, Palaniswami M, Kamen P. Do existing measures of Poincaré plot geometry reflect nonlinear features of heart rate variability? IEEE Trans Biomed Eng. 2001;48:1342–1347. doi: 10.1109/10.959330. [DOI] [PubMed] [Google Scholar]
- Caballero R, Delpón E, Valenzuela C, Longobardo M, González T, Tamargo J. Direct effects of candesartan and eprosartan on human cloned potassium channels involved in cardiac repolarization. Mol Pharmacol. 2001;59:825–836. doi: 10.1124/mol.59.4.825. [DOI] [PubMed] [Google Scholar]
- Covic A, Diaconita M, Gusbeth-Tatomir P, Covic M, Botezan A, Ungureanu G, et al. Haemodialysis increases QTc interval but not QTc dispersion in ESRD patients without manifest cardiac disease. Nephrol Dial Transplant. 2002;17:2170–2177. doi: 10.1093/ndt/17.12.2170. [DOI] [PubMed] [Google Scholar]
- Domenighetti AA, Boixel C, Cefai D, Abriel H, Pedrazzini T. Chronic angiotensin II stimulation in the heart produces an acquired long QT syndrome associated with IK1 potassium current downregulation. J Mol Cell Cardiol. 2007;42:63–70. doi: 10.1016/j.yjmcc.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Dritsas A, Sbarouni E, Gilligan D, Nihoyannopoulos P, Oakley CM. QT-interval abnormalities in hypertrophic cardiomyopathy. Clin Cardiol. 1992;15:739–742. doi: 10.1002/clc.4960151010. [DOI] [PubMed] [Google Scholar]
- Fujita T, Ando K, Nishimura H, Ideura T, Yasuda G, Isshiki M, et al. Cilnidipine versus Amlodipine Randomised Trial for Evaluation in Renal Disease (CARTER) Study Investigators Antiproteinuric effect of the calcium channel blocker cilnidipine added to renin-angiotensin inhibition in hypertensive patients with chronic renal disease. Kidney Int. 2007;72:1543–1549. doi: 10.1038/sj.ki.5002623. [DOI] [PubMed] [Google Scholar]
- Hirning LD, Fox AP, McCleskey EW, Olivera BM, Thayer SA, Miller RJ, et al. Dominant role of N-type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science. 1988;239:57–61. doi: 10.1126/science.2447647. [DOI] [PubMed] [Google Scholar]
- Ito K, Shiomi M, Kito G. Effects of the non-peptide angiotensin II receptor antagonist TCV-116 on systemic and renal hemodynamics in dogs with renal hypertension. Hypertens Res. 1995;18:69–75. doi: 10.1291/hypres.18.69. [DOI] [PubMed] [Google Scholar]
- Karlberg BE. Adrenergic regulation of renin release and effects on angiotensin and aldosterone. Acta Med Scand Suppl. 1983;672:33–40. doi: 10.1111/j.0954-6820.1983.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Konda T, Enomoto A, Takahara A, Yamamoto H. Effects of L/N-type calcium channel antagonist, cilnidipine on progressive renal injuries in Dahl salt-sensitive rats. Biol Pharm Bull. 2006;29:933–937. doi: 10.1248/bpb.29.933. [DOI] [PubMed] [Google Scholar]
- Konda T, Enomoto A, Aritomi S, Koganei H, Ogawa T, Nitta K. Different effects of L/N-type and L-type calcium channel blockers on the renin-angiotensin-aldosterone system in SHR/Izm. Am J Nephrol. 2009;30:155–161. doi: 10.1159/000210396. [DOI] [PubMed] [Google Scholar]
- Lengyel C, Virág L, Bíró T, Jost N, Magyar J, Biliczki P, et al. Diabetes mellitus attenuates the repolarization reserve in mammalian heart. Cardiovasc Res. 2007;73:512–520. doi: 10.1016/j.cardiores.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Lu HR, Vlaminckx E, Van de Water A, Gallacher DJ. Calmodulin antagonist W-7 prevents sparfloxacin-induced early afterdepolarizations (EADs) in isolated rabbit purkinje fibers: importance of beat-to-beat instability of the repolarization. J Cardiovasc Electrophysiol. 2006;17:415–422. doi: 10.1111/j.1540-8167.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- Mazur A, Roden DM, Anderson ME. Systemic administration of calmodulin antagonist W-7 or protein kinase A inhibitor H-8 prevents torsade de pointes in rabbits. Circulation. 1999;100:2437–2442. doi: 10.1161/01.cir.100.24.2437. [DOI] [PubMed] [Google Scholar]
- Molderings GJ, Likungu J, Göthert M. N-type calcium channels control sympathetic neurotransmission in human heart atrium. Circulation. 2000;101:403–407. doi: 10.1161/01.cir.101.4.403. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Yoshida M, Suzuki-Kusaba M, Hisa H, Kimura T, Satoh S. Effect of cilnidipine, a novel dihydropyridine Ca2+ channel blocker, on adrenal catecholamine secretion in anesthetized dogs. J Cardiovasc Pharmacol. 1998;32:479–484. doi: 10.1097/00005344-199809000-00020. [DOI] [PubMed] [Google Scholar]
- Peschar M, Vernooy K, Vanagt WY, Reneman RS, Vos MA, Prinzen FW. Absence of reverse electrical remodeling during regression of volume overload hypertrophy in canine ventricles. Cardiovasc Res. 2003;58:510–517. doi: 10.1016/s0008-6363(03)00331-6. [DOI] [PubMed] [Google Scholar]
- Raizada V, Skipper B, Luo W, Garza L, Hines CW, Harford AA, et al. Renin-angiotensin polymorphisms and QTc interval prolongation in end-stage renal disease. Kidney Int. 2005;68:1186–1189. doi: 10.1111/j.1523-1755.2005.00510.x. [DOI] [PubMed] [Google Scholar]
- Reddy HK, Wasson S, Koshy SK, Komatireddy R. Structural correlates of electrical remodeling in ventricular hypertrophy. Cardiovasc Res. 2003;58:495–497. doi: 10.1016/s0008-6363(03)00369-9. [DOI] [PubMed] [Google Scholar]
- Roden DM. Taking the ‘idio’ out of ‘idiosyncratic’: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- Sakata K, Shirotani M, Yoshida H, Nawada R, Obayashi K, Togi K, et al. Effects of amlodipine and cilnidipine on cardiac sympathetic nervous system and neurohormonal status in essential hypertension. Hypertension. 1999;33:1447–1452. doi: 10.1161/01.hyp.33.6.1447. [DOI] [PubMed] [Google Scholar]
- Schoenmakers M, Ramakers C, van Opstal JM, Leunissen JD, Londoño C, Vos MA. Asynchronous development of electrical remodeling and cardiac hypertrophy in the complete AV block dog. Cardiovasc Res. 2003;59:351–359. doi: 10.1016/s0008-6363(03)00430-9. [DOI] [PubMed] [Google Scholar]
- Singh JP, Johnston J, Sleight P, Bird R, Ryder K, Hart G. Left ventricular hypertrophy in hypertensive patients is associated with abnormal rate adaptation of QT interval. J Am Coll Cardiol. 1997;29:778–784. doi: 10.1016/s0735-1097(96)00576-1. [DOI] [PubMed] [Google Scholar]
- Sugiyama A, Ishida Y, Satoh Y, Aoki S, Hori M, Akie Y, et al. Electrophysiological, anatomical and histological remodeling of the heart to AV block enhances susceptibility to arrhythmogenic effects of QT-prolonging drugs. Jpn J Pharmacol. 2002;88:341–350. doi: 10.1254/jjp.88.341. [DOI] [PubMed] [Google Scholar]
- Swynghedauw B, Baillard C, Milliez P. The long QT interval is not only inherited but is also linked to cardiac hypertrophy. J Mol Med. 2003;81:336–345. doi: 10.1007/s00109-003-0437-8. [DOI] [PubMed] [Google Scholar]
- Takahara A. Cilnidipine: a new generation Ca2+ channel blocker with inhibitory action on sympathetic neurotransmitter release. Cardiovasc Ther. 2009;27:124–139. doi: 10.1111/j.1755-5922.2009.00079.x. [DOI] [PubMed] [Google Scholar]
- Takahara A, Dohmoto H, Hisa H, Satoh S, Yoshimoto R. Cilnidipine attenuates renal nerve stimulation-induced renal vasoconstriction and antinatriuresis in anesthetized dogs. Jpn J Pharmacol. 1997;75:27–32. doi: 10.1254/jjp.75.27. [DOI] [PubMed] [Google Scholar]
- Takahara A, Sugiyama A, Satoh Y, Nakamura Y, Hashimoto K. Cardiovascular effects of an L/N-type Ca2+ channel blocker cilnidipine assessed in the chronic atrioventricular conduction block dogs. J Pharmacol Sci. 2004;96:219–223. doi: 10.1254/jphs.scj04007x. [DOI] [PubMed] [Google Scholar]
- Takahara A, Sugiyama A, Ishida Y, Satoh Y, Wang K, Nakamura Y, et al. Long-term bradycardia caused by atrioventricular block can remodel the canine heart to detect the histamine H1 blocker terfenadine-induced torsades de pointes arrhythmias. Br J Pharmacol. 2006;147:634–641. doi: 10.1038/sj.bjp.0706493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara A, Sugiyama A, Satoh Y, Iwasaki H, Nakamura Y, Hashimoto K. Cardiovascular profile of the canine torsades de pointes arrhythmia model assessed by echocardiographic and haemodynamic methods. Basic Clin Pharmacol Toxicol. 2007a;101:35–40. doi: 10.1111/j.1742-7843.2007.00071.x. [DOI] [PubMed] [Google Scholar]
- Takahara A, Iwasaki H, Nakamura Y, Sugiyama A. Cardiac effects of L/N-type Ca2+ channel blocker cilnidipine in anesthetized dogs. Eur J Pharmacol. 2007b;565:166–170. doi: 10.1016/j.ejphar.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Takahara A, Nakamura Y, Sugiyama A. Beat-to-beat variability of repolarization differentiates the extent of torsadogenic potential of multi ion channel-blockers bepridil and amiodarone. Eur J Pharmacol. 2008;596:127–131. doi: 10.1016/j.ejphar.2008.08.018. [DOI] [PubMed] [Google Scholar]
- Takemori K, Ishida H, Dote K, Yamamoto K, Ito H. Prophylactic effects of an N- and L-type Ca2+ antagonist, cilnidipine, against cardiac hypertrophy and dysfunction in stroke-prone, spontaneously hypertensive rats. Can J Physiol Pharmacol. 2005;83:785–790. doi: 10.1139/y05-067. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J, et al. Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation. 2004;110:2453–2459. doi: 10.1161/01.CIR.0000145162.64183.C8. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Truin M, van Opstal JM, Beekman JD, Volders PG, Stengl M, et al. Sudden cardiac death in dogs with remodeled hearts is associated with larger beat-to-beat variability of repolarization. Basic Res Cardiol. 2005;100:279–287. doi: 10.1007/s00395-005-0519-6. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Volders PG, Beekman JD, Matz J, Vos MA. Beat-to-beat variability of repolarization determines proarrhythmic outcome in dogs susceptible to drug-induced torsades de pointes. J Am Coll Cardiol. 2006;48:1268–1276. doi: 10.1016/j.jacc.2006.05.048. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- Topilski I, Rogowski O, Rosso R, Justo D, Copperman Y, Glikson M, et al. The morphology of the QT interval predicts torsade de pointes during acquired bradyarrhythmias. J Am Coll Cardiol. 2007;49:320–328. doi: 10.1016/j.jacc.2006.08.058. [DOI] [PubMed] [Google Scholar]
- Uneyama H, Takahara A, Dohmoto H, Yoshimoto R, Inoue K, Akaike N. Blockade of N-type Ca2+ current by cilnidipine (FRC-8653) in acutely dissociated rat sympathetic neurones. Br J Pharmacol. 1997;122:37–42. doi: 10.1038/sj.bjp.0701342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uneyama H, Uchida H, Konda T, Yoshimoto R. Cilnidipine: preclinical profile and clinical evaluation. Cardiovasc Drug Rev. 1999;17:341–357. [Google Scholar]
- Verduyn SC, Ramakers C, Snoep G, Leunissen JD, Wellens HJ, Vos MA. Time course of structural adaptations in chronic AV block dogs: evidence for differential ventricular remodeling. Am J Physiol. 2001;280:H2882, H2890. doi: 10.1152/ajpheart.2001.280.6.H2882. [DOI] [PubMed] [Google Scholar]
- Verduyn SC, Vos MA, Gorgels AP, van der Zande J, Leunissen JD, Wellens HJ. The effect of flunarizine and ryanodine on acquired torsades de pointes arrhythmias in the intact canine heart. J Cardiovasc Electrophysiol. 1995;6:189–200. doi: 10.1111/j.1540-8167.1995.tb00770.x. [DOI] [PubMed] [Google Scholar]
- Volders PG, Sipido KR, Vos MA, Spätjens RL, Leunissen JD, Carmeliet E, et al. Downregulation of delayed rectifier K+ currents in dogs with chronic complete atrioventricular block and acquired torsades de pointes. Circulation. 1999;100:2455–2461. doi: 10.1161/01.cir.100.24.2455. [DOI] [PubMed] [Google Scholar]
- Vos MA, de Groot SH, Verduyn SC, van der Zande J, Leunissen HD, Cleutjens JP, et al. Enhanced susceptibility for acquired torsade de pointes arrhythmias in the dog with chronic, complete AV block is related to cardiac hypertrophy and electrical remodeling. Circulation. 1998;98:1125–1135. doi: 10.1161/01.cir.98.11.1125. [DOI] [PubMed] [Google Scholar]
- Wang YH, Shi CX, Dong F, Sheng JW, Xu YF. Inhibition of the rapid component of the delayed rectifier potassium current in ventricular myocytes by angiotensin II via the AT1 receptor. Br J Pharmacol. 2008;154:429–439. doi: 10.1038/bjp.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KY, McSwiggan S, Kennedy NS, Wong SY, Gavin A, MacWalter RS, et al. Spectrum of cardiac abnormalities associated with long QT in stroke survivors. Heart. 2005;91:1306–1310. doi: 10.1136/hrt.2004.045187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Suzuki M, Munehasu S, Ishiko J. Antihypertensive effects of amlodipine, a new calcium antagonist. Folia Pharmacol Jpn. 1991;97:115–126. doi: 10.1254/fpj.97.2_115. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Sugiyama A, Satoh Y, Ishida Y, Yoneyama M, Kugiyama K, et al. Comparison of the in vivo electrophysiological and proarrhythmic effects of amiodarone with those of a selective class III drug, sematilide, using a canine chronic atrioventricular block model. Circ J. 2002;66:758–762. doi: 10.1253/circj.66.758. [DOI] [PubMed] [Google Scholar]
- Yoshimoto R, Hirasawa A, Dohmoto H, Iwata S. Antihypertensive effects of 2-methoxyethyl (E)-3-phenyl-2-propen-1-yl(±)-1,4-dihydro-2,6 dimethyl-4-(3-nitrophenyl) pyridine-3,5-dicar boxylate (FRC-8653), a novel dihydropyridine derivative, in renovascular hypertensive dogs. Pharmacometrics. 1992;44:45–51. [Google Scholar]
- Yu H, Gao J, Wang H, Wymore R, Steinberg S, McKinnon D, et al. Effects of the renin-angiotensin system on the current Ito in epicardial and endocardial ventricular myocytes from the canine heart. Circ Res. 2000;86:1062–1068. doi: 10.1161/01.res.86.10.1062. [DOI] [PubMed] [Google Scholar]