

Abstract
Accounting for the genetic substructure of human populations has become a major practical issue for studying complex genetic disorders. Allele frequency differences among ethnic groups and subgroups and admixture between different ethnic groups can result in frequent false-positive results or reduced power in genetic studies. Here, we review the problems and progress in defining population differences and the application of statistical methods to improve association studies. It is now possible to take into account the confounding effects of population stratification using thousands of unselected genome-wide single-nucleotide polymorphisms or, alternatively, selected panels of ancestry informative markers. These methods do not require any demographic information and therefore can be widely applied to genotypes available from multiple sources. We further suggest that it will be important to explore results in homogeneous population subsets as we seek to define the extent to which genomic variation influences complex phenotypes.
INTRODUCTION
Over the last 2 years a large number of allelic variants have been associated with susceptibility to a wide variety of common complex diseases. This progress has mainly resulted from the use of genotyping arrays containing several hundred thousand single-nucleotide polymorphisms (SNPs) that capture a significant amount of the variation defined by the HapMap. However, because of the generally modest nature of these new associations, robust conclusions from these genome-wide association (GWA) studies have also required very careful analysis to exclude false-positive results that are the consequence of stratification differences between cases and controls (1–3). This stratification is most often due to differences in population genetic structure and substructure that cannot be accounted for by demographic information derived from the subjects. Nevertheless, statistical methods can be applied to discern and correct for these differences. It has now become imperative to carry out such corrections, both for whole genome association studies as well as in the context of follow-up studies and other candidate gene studies to explore suggestive, but not proven associations.
It should be noted that alternative approaches using family-based controls has long been advocated to exclude false-positives due to population stratification and can be applied to GWA (4,5). These methods most commonly apply variants of the transmission disequilibrium test (TDT) (6) are effective but require first-degree relatives and at a minimum necessitate genotyping of three individuals (the proband and both parents) to achieve similar power to the case–control method. The TDT methodology has the additional advantage of being able to determine whether maternally or paternally inherited alleles have differential effects. However, successful recruitment of such families is often difficult, especially for diseases of adult onset. Therefore, the majority of GWA studies and replication studies are dependent on the case–control design. Also, as discussed below, the ability to use publically available population controls may increase the power and efficiency of association testing if the issues of population stratification and genotyping differences between studies are appropriately addressed.
THE GENERAL PROBLEM: ALLELE FREQUENCY DIFFERENCES IN DIFFERENT DATA SETS
Both in theory and practice, allele frequency differences between two sample sets (cases and controls) may be unrelated to the genetics of the particular phenotype under study. This can result in false associations of particular SNPs or markers. There are four general causes for false-positive results in this situation. (i) Inappropriate statistical thresholds including the fail to account for multiple comparisons. This problem is usually addressed by either the Bonferroni correction or by the less conservative false discovery rate methods (7–9). However, in situations where the power is low and the expected number of true positives is therefore also low, false discovery rate may not be appropriate. This issue has been the subject of intense discussion and the several recommendations including the use of Bayesian methods have been suggested (1,10,11). (i) Genotype artifact (12,13); genotyping artifacts can be largely addressed by quality filters. These include exclusions based on completeness of genotyping data and large deviations from Hardy–Weinberg equilibrium. Similar DNA template preparation and genotyping of both sample sets can of course also militate against such an artifact. (iii) Environmental factors; environmental factors could potentially lead to false associations but require extreme circumstances to result in changes in allele frequency in either the case or control population sets. Excluding a recent epidemic, it is very unlikely that differences in environmental factors will cause a major shift in the SNP frequency unless the environmental factor itself determines the selection of cases or controls. While this is not generally an issue, certain control population groups have been selected for specific demographic factors (e.g. smokers in lung cancer studies), where allele frequencies could be skewed by selection for nicotine addiction or lung cancer protection. (iv) Unrecognized ancestry differences; once genotyping artifacts and statistical thresholds have been addressed, this is probably the most common reason for false-positive findings. In a set of 300K SNPs there may be thousands of SNPs with substantial differences in allele frequency among population subgroups. Even when studies are restricted to a single continental origin, many false-positive results may still be observed due to subtle differences in the ethnic make-up of case and control participants (12,14–19). While careful matching for demographic factors can reduce this problem, statistical methods can now be applied to address this issue and in our opinion reduce the need in GWA studies to obtain the ideally matched case and control groups selected using population-based sampling.
To illustrate how population substructure and genotyping errors can result in false-positive association tests, we provide simple examples (Fig. 1A). First, differences in population stratification in cases and controls are shown for a hypothetical SNP in which the allele frequency difference between northern and southern European populations is 10%. This magnitude of allele frequency difference is observed for hundreds of SNPs (∼3% of SNPs) when northern European and southern European subjects are genotyped with several hundred thousand SNPs. In this particular scenario, the failure to account for the differences in subject origin (north versus south) resulted in a highly significant false-positive test. Second, the potential problem of genotyping error is similarly illustrated for a relatively small difference in allele calls (5% difference). Differential allele calling between genotyping platforms or between different laboratories has been noted as a major source of false-positives (12). Thus, these examples emphasize the importance of accounting for population structure and measures to ensure that there are no systematic differences in SNP genotyping results.
Figure 1.

Examples of how stratification and ancestry can affect case–control association tests. In the top panel, examples of type 1 errors are shown. Population substructure can result in false-positive associations when the regional origin/ancestry of cases and controls are not matched. In the example shown, a 10% allele frequency difference in northern European compared with southern European results in a highly significant P-value (Armitage’s χ2 test) when the numbers of cases and controls derived from these regions are different. The top panel also shows an example of genotyping error that can result from genotyping cases and controls using different array chips. The bottom panel illustrates how type 2 errors, false-negative results may result from heterogeneous sample sets. In this example, a true positive result may not reach an appropriate threshold for significance when the signal is diluted by a population in which the causative SNP is absent.
Another issue of potential importance to finding disease-associated alleles is that certain allelic variations will only be important in specific ethnic or continental subgroups. For complex genetic disease, one such example is PTPN22, where an allelic variant associated with autoimmune disease is only present with substantial frequency only in European populations (20,21). If association tests are not performed in specific population groups then a substantial decrease in power can be observed (Fig. 1B). Thus, definition of ethnic group subsets or homogeneous groups (see later section) is another potentially valuable application of defining population structure and substructure.
USING STRUCTURED ASSOCIATION AND PRINCIPAL COMPONENT ANALYSES TO CONTROL FOR TYPE 1 ERRORS
Several different methods have been developed to address issues of population structure and substructure in the context of whole genome association studies (16,22–27). Solutions such as genomic control address the general inflation of the χ2 (or other test for significance) globally but do not account for differences that are specific to each SNP (15,16). Importantly, as has been shown by multiple studies, it is critical to evaluate each SNP based on how it is affected by ‘ancestry’ differences in each individual in the sample sets being studied (16,22,25). Some SNPs show very different allele frequencies in different continental or ethnic groups, whereas other SNPs do not. The potential for stratification based on differences in population structure in case and control groups is different for different SNPs. Thus, the general application of genomic control can result in over-correction for some SNPs and under-correction for other SNPs. Adjustments for general inflation of the statistical tests should be performed after addressing the issue of population structure and substructure.
Generally, there are three approaches that have achieved some measure of application to large data sets: (i) structured association tests; (ii) principal components analyses (PCA) and (iii) multidimensional scaling (MDS).
Structured association depends on applying information from model-based or distance-based clustering algorithms. One popular version, (STRAT) (22), uses the output from the model-based STRUCTURE program (28,29) to perform tests conditional on the group membership. The group membership is determined using a Bayesian clustering algorithm that fits the data to the number of cluster groups (K) that is specified. The association test is testing the null hypothesis is that there is no dependence of allele frequencies on phenotypes within each group. However, this is computational intensive and thus difficult to apply when large numbers of SNPs and large numbers of subjects are being considered. In addition, it requires estimation of the number of groups (K) and this may have some uncertainty. Another popular program, PLINK (26), uses identical by state distance to do hierarchical clustering and then performs Cochran–Mantel–Haenszel tests of association conditional on clusters. This approach, like PCA and MDS, can be performed with very large data sets. Some of critical concepts/methods are defined and illustrated in Figure 2.
Figure 2.
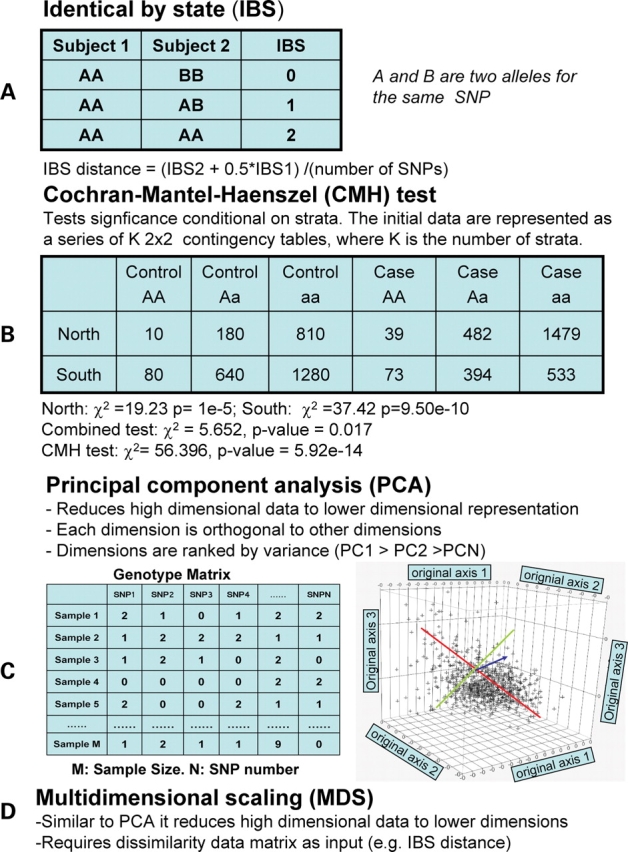
Definition of statistical terms and tests. (A) Schematic of how identical by state distance is measured. (B) An example of the application of the Cochran–Mantel–Haenszel (CMH) test. In this example, two strata (North and South) are defined. These strata (K) can be determined using some methodology (e.g. clustering algorithm). The method assumes that each stratum has the same odds ratio. In this example the A allele has an odds ratio of 1.5. The substantial gain in power is illustrated by comparing CMH test result with the combined data. (C) The features of PCA; the high dimensional data shown in the genotype matrix (M × N) is reduced to orthogonal dimensions with the largest variance in PC1 represented by the red line. (D) The features of MDS are shown.
Both PCA and MDS can infer a continuous axis of genetic variation that does not depend on assignment of individuals to various subpopulations. When Euclidean distance is used, classical metric MDS is the same as PCA. In general, both approaches reduce high-dimensional data (number of SNPs) to smaller numbers of dimensions that group ‘patterns’ together based on the observed data. The popular program, EIGENSTRAT (16), calculates ancestry-adjusted genotypes and phenotypes using the continuous axis of variation from PCA to compute the association statistic (The adjusted genotypes are the residuals from the regression of the original genotypes against the continuous axis of variation. The adjusted phenotypes are similarly determined).
For the most effective application of PCA it is important to remove small regions of the genome that by themselves cause specific groupings of subjects. The inclusion of these regions will result in the grouping of individuals that is not based on genome-wide population structure or substructure but on the specific patterns that are unique to these particular chromosomal segments. These are regions that contain large numbers of markers in high-linkage disequilibrium will provide multiple contributions to the overall data patterns. Not controlling for these regions does not affect false-positives since these regions do not by themselves define the genome-wide substructure. With current SNP arrays this requires exclusion of large genomic inversions (30) and the HLA region for the initial analysis of population structure. These regions can be added back when statistical tests are performed for association. This procedure will then enable the assessment of association within these intervals and has been demonstrated for both HLA and a gene(s) within the chromosome 8 inversion in studies of systemic lupus erythematosus (3).
We wish to point out some additional caveats for the use of PCA. First, it is useful to examine the actual distribution of samples in multiple principal components (PCs). Genotyping artifacts can be revealed by tight grouping of particular samples and correlation with particular genotype array chips or plate grouping of samples. Second, in our experience, ∼40 000 random markers can resolve European substructure in diverse European population sets (30). Third, the number of PCs that need to be examined will vary from data set to data set. In general, it is useful to monitor the residual inflation of the median χ2 distribution using the genomic control parameter (λgc) to determine the number of PCs that should be considered. We suggest that the λgc plateau is a reasonable guide to number of PCs needed to adjust for population substructure. This λgc plateau also corresponds to the number of PCs showing population substructure that can be estimated using a split half test (30). The final χ2 statistic should be divided by the residual λgc before multiple comparison corrections are applied to determine the appropriate P-value.
Other issues that may need to be addressed for individual data sets include familial relationships among cases and/or controls. This can result in apparent population substructure in which these individuals appear to have their own ancestry group. Thus using PCA or model-based methods, this ‘substructure’ should be defined. However, since in general the underlying association tests that will be applied depend on analyzing unrelated individuals, we suggest excluding first-degree relatives identified by descent methods in quality filters. In addition, the appropriate threshold for excluding various subjects as outliers may vary from study to study. While specific criteria can be specified in the context of a particular data set, there are no generally accepted rules or established reference subjects. Furthermore, it is useful to emphasize that PCA and MDS results reflect data structure and not ancestry per se. Therefore, as suggested earlier, investigators should carefully explore data sets to determine whether the results correlate with potential artifacts caused by systematic differences in genotyping. Unusual patterns in particular PCs may also provide clues to potential problems caused by an extensive number of SNPs in tight linkage disequilibrium (30).
CONTINENTAL VERSUS SUB-CONTINENTAL DIFFERENCES IN ALLELIC VARIATION
Studies over the last 6 years have shown substantial differences in allele frequencies within different continental populations (31). There is controversy with regard to whether there are discrete divisions between the continents (32,33). However, in general the number of SNPs showing large allele frequency differences between major continental populations (the fraction of SNPs with Fst’s (34) >0.25 or allele frequency differences >40%) are an order of magnitude greater than that seen within continental populations. Therefore, in theory the largest source of type 1 errors will be caused by differences in the distribution of ancestry from major continental populations in case and control sets. In practice, self-identification of ancestry substantially reduces this problem. However, we have observed that admixture as well as sample handing and database errors still necessitate addressing this problem first, even with good demographic matching. In the context of whole genome studies examining primarily a single population group (e.g. European ancestry), both PCA and MDS will distinguish (under typical parameters) individuals with different continental origins and most individuals with substantial admixture. This can also be readily accomplished using small numbers (96 recommended) of ancestry informative markers (AIMs) (35) with the application of model-based clustering algorithms such as those used in STRUCTURE (29) and ADMIXMAP (25).
Allele frequency differences within continental subgroups, although much smaller than continental differences (e.g. Italian/Swedish mean Fst = 0.006; Europe/Amerindian mean Fst = 0.126), can also result in false-positive results (17). As shown in both modeling studies as well as in multiple studies of complex diseases, these differences are large enough to result in false-positive associations. This is true not only for the largest gradient (north/south) but also for more subtle differences in Northern European populations (30). Figure 3 shows the first two PCs and clustering of population subgroups for over 3000 subjects of European descent. These clustering patterns show a strong correlation with grandparental identity, when known. There are also strong regional differences depending on the site of collection for European American participants.
Figure 3.

European Population Substructure. The first two principal components are shown for a diverse group of >3000 European and European American subjects. The clustering of different groups within Europe is shown for each individual designated by a symbol. The subjects from specific countries of origin or with four grandparental defined European countries of origin are color coded as shown in the legend with grey symbols indicated European Americans (EURA) with insufficient country of origin information. The groups from the following countries or regions are included: Sweden (SWED), Ireland (IRISH), Italy (ITN), Greece (GRK), Germany (GERM), Eastern Europe (EEUR), Hungary (HUN), United Kingdom (UK), Scandinavia (SCAN), and Spain (SPN). Although not included in this figure, additional studies indicate that the cluster in the right upper quadrant corresponds to an Ashkenazi Jewish grouping.
Similar to controlling for differences between continental populations, both model-dependent and model-independent statistical methods can be applied to adjust for substructure within a continental population. For WGA studies, the previously discussed computational programs, PLINK and EIGENSTRAT, have been used extensively in association studies of European ancestry. In our experience, EIGENSTRAT that uses PCA has been most effective in ascertaining subtle differences in substructure that can be present in various European American sample sets. However, additional studies will be necessary to compare both the ability to minimize type 1 errors while maximizing the ability, i.e. statistical power, to discern true positives. For candidate gene studies, AIMs can largely provide the substructure information [(17,30,36–38) and see section Application of ancestry informative markers].
USE OF COMMON CONTROLS FOR GENOTYPING STUDIES
The ability to adjust for population stratification raises the possibility of utilizing large common population control group(s) for GWA studies. Of course, carefully matched control data sets do have advantages beyond balancing demographic differences in ancestry. For example, minimizing differences in environmental exposure or age at censorship may increase power for specific analyses. However, it is unclear whether adjustment for these factors in the study of complex diseases will have large effects that may alter type 1 or type 2 error. Regardless, the common practical application of control matching by geography and age does not usually enable close matching of the myriad of potential modifiers. Furthermore, the use of geography may also be problematic in our mobile societies. If statistical methods adjusting for population structure can effectively match ancestry in diverse population sets, then the major objection to using common control data sets can be overcome. Other issues, including differences in genotyping platforms are also of potential concern; however, we anticipate that large numbers of ‘common controls’ will be available on the two current platforms (Illumina and Affymetrix) that are currently being utilized. Theoretically, imputation can also be utilized to allow the combining of information from different platforms using different SNP arrays. However, the combination of genotypes from different platforms is likely to accentuate the problem of false-positive associations caused by genotyping differences.
The ability to use large sample sizes to increase power is both attractive and we believe practical. The availability of iControlDB (http://www.illumina.com/pages.ilmn?ID=231) and other large datasets (e.g. dbGaP, http://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?login=&page=login) can provide large numbers of genotyped control population groups. We have applied this general approach in our own studies of rheumatoid arthritis and other diseases. These studies have successfully identified allelic variants that have been confirmed in replication studies (2,3). It should be noted that some care and statistical adjustment may be necessary when using common population control groups, since multiple studies of the same disease may contain overlapping control subjects.
USE OF HOMOGENEOUS SUBJECT SETS
It is also potentially valuable to apply population substructure information to selecting subsets of subjects with very similar or nearly homogeneous ancestry. Such an approach can limit ancestral diversity in a subject group and may be analogous to studying a disease in a single ethnic group. This could be a major asset in the study of particular traits since genetic heterogeneity may limit power in studying complex genetic disease in much the same fashion as has been discussed for single-gene diseases. Thus we would suggest that selecting and matching homogeneous cases and controls may decrease type 2 error rates. PCA analysis is well-suited to the application of robust distance methods that can be applied to multiple PCs. Adapting this statistical approach, we have used a diverse European data set to illustrate this type of method. In this example, we have utilized a Swedish cohort with Myasthenia Gravis as the case set and have derived controls from both Swedish subjects and a diverse European American data set (unpublished data). The case cohort was first selected by setting parameters to remove multivariate outliers. After removing case outliers, a second phase repeating the process was applied to the case–control dataset, where the controls included both Swedish and European American subjects. This method dramatically reduced the residual inflation of the median χ2 distribution (Swedish only, λgc = 1.039, combined Swedish and European American, λgc = 2.19, homogeneous subject set, λgc = 1.018 genomic inflation) and is graphically depicted in Figure 4.
Figure 4.

Illustration of the use of PCA to select homogeneous sample sets. In this example, cases derived from a Swedish population and the controls were from both Sweden and European Americans. (A) The first and second principal component (PC1 and PC2) for the Swedish cases and control subjects. (B) Homogeneous subject set selected from Swedish cases and controls and 3447 European American subjects (same set as shown in Fig. 1). Color code indicates the origin of the subjects. The basic procedure was to remove Multivariate outliers based on Mahalanobis distance. The minimum covariance determinant (MCD) estimators of location and scatter of PCA scores of the entire dataset were calculated using R. The Mahalanobis distances were then calculated using the robust estimators, leading to robust distance (RD). For multivariate normally distributed data the RD values are approximately χ2 distributed with p degree-of-freedom (p is the number of dimensions). The procedure was applied in two steps. For the first phase of selection we removed case outliers using robust distance measurements. The significance level was set at α = 0.001 to remove the case outliers. A second phase repeating the same process was applied to the case–control dataset. This was based on the case-only robust estimators of location and scatter in order to define a more homogeneous case–control sample set. The significance level was set at α = 0.05 for this phase of the procedure.
APPLICATION OF ANCESTRY INFORMATIVE MARKERS
We and others have also developed sets of AIMs to facilitate genetic studies when sample sets have not been typed with genome-wide arrays of more than hundred thousand SNPs (17,30,35–37,39,40). Such sets of AIMs are designed to provide most of the ancestry information using smaller cost-effective arrays and are valuable in follow-up studies to confirm associations and in fine-mapping analyses. First, a small number of AIMs can be used to provide continental ancestry information (35,39,40). One such set of 128 AIMs and subsets of these AIMs were recently demonstrated to be effective in modeling studies. Reference genotypes for these AIMs and a commercial panel of 96 AIMs are available (35). Additional panels that can be used for examining substructure within continental populations are under development. For European populations, several sets of markers have been defined for distinguishing the largest gradient (north/south) (30,36,37) and at least one set of markers for distinguishing an east/west gradient within northern European populations (30). Relatively small numbers of SNPs are necessary for the north/south gradient (e.g. 192 north–south European substructure AIMs) and will control for many SNPs including those linked to the lactase gene (LCT) that follow this pattern. LCT is particularly noteworthy since it has been used as a beacon for substructure differences within European populations; a variant within the LCT is associated with lactase persistence in many northern European populations and has been demonstrated to be under strong positive selection in Europeans (41,42). A large number of SNPs in linkage disequilibrium with LCT have been used in modeling studies (16,17) and can be used as one measure of whether or not substructure has been controlled in studies using diverse European subjects.
For other differences in European population substructure, larger numbers of SNPs (e.g. ∼1200 North European substructure AIMs) are necessary. The potential value and use of these SNP sets has recently been discussed (38) and depends in part on the sample sets being examined. We anticipate that arrays for European substructure will become available on a cost–effective platform. Ongoing studies are also examining other continental populations.
It is also worth noting that panels of AIMs could be used as an initial screen to determine which samples should be used in GWA tests. This should enhance efficiency since it will minimize the loss of power that may result from controlling for substructure by pre-selecting matched cases and controls. Alternatively, AIMs can be used to appropriately match control subjects to pre-existing cases. As discussed earlier (see section Use of homogeneous mapping sets), a modification of this approach can also be used to identify more homogeneous matched cases and controls. This will decrease genetic heterogeneity as well as residual genomic control inflation. The feasibility of this approach and the general application of selected AIMs for genotyping will clearly depend on the availability of standardized ancestry marker panels that can be run at reasonable cost.
CONCLUDING REMARKS
In this brief review, we have highlighted the importance and potential value of examining and applying population genetic structure and substructure in studies of complex genetic disease. This applies to both candidate gene studies as well as GWA studies. We believe that ancestral differences should also be examined in the context of any clinical epidemiological study. For genetic studies, in addition to addressing type 1 errors, we have emphasized the possibility that these methods may also enable increased power by the ability to reduce genetic heterogeneity: different ancestry subgroups may have unique disease modifiers or unique epistatic effects. Although not discussed here, there is mounting evidence that natural selection has shaped a considerable part of the differentiation observed between different ethnic subsets. If this is a major factor in our genomic evolution then it will be of even more importance to examine a variety of different ethnic groups in future genetic studies for many common diseases.
ELECTRONIC DATABASE AND SOFTWARE INFORMATION
Software
STRUCTURE/STRAT (http://pritch.bsd.uchicago.edu/software.html).
EIGENSTRAT/EIGENSOFT (http://genepath.med.harvard.edu/~reich/Software.htm).
PLINK (http://pngu.mgh.harvard.edu/~purcell/plink/summary.shtml).
Control genotypes
iControlDB (http://www.illumina.com/pages.ilmn?ID=231).
dbGaP (http://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?login=&page=login).
FUNDING
These studies were supported by NIH grants: R01-DK071185 and R01-AR44422.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plenge R.M., Seielstad M., Padyukov L., Lee A.T., Remmers E.F., Ding B., Liew A., Khalili H., Chandrasekaran A., Davies L.R., et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N. Engl. J. Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hom G., Graham R.R., Modrek B., Taylor K.E., Ortmann W., Garnier S., Lee A.T., Chung S.A., Ferreira R.C., Pant P.V., et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 2008;358:900–909. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- 4.Laird N.M., Lange C. Family-based designs in the age of large-scale gene-association studies. Nat. Rev. Genet. 2006;7:385–394. doi: 10.1038/nrg1839. [DOI] [PubMed] [Google Scholar]
- 5.Chen W.M., Abecasis G.R. Family-based association tests for genomewide association scans. Am. J. Hum. Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spielman R.S., McGinnis R.E., Ewens W.J. The transmission/disequilibrium test detects cosegregation and linkage. Am. J. Hum. Genet. 1994;54:559–560. author reply 560–553. [PMC free article] [PubMed] [Google Scholar]
- 7.Fernando R.L., Nettleton D., Southey B.R., Dekkers J.C., Rothschild M.F., Soller M. Controlling the proportion of false positives in multiple dependent tests. Genetics. 2004;166:611–619. doi: 10.1534/genetics.166.1.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 9.Benjamini Y., Yekutili D. Technical Report, Tel Aviv University; 1999. The Control of the False Discovery Rate in Multiple Testing under Dependency. [Google Scholar]
- 10.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 11.Chanock S.J., Manolio T., Boehnke M., Boerwinkle E., Hunter D.J., Thomas G., Hirschhorn J.N., Abecasis G., Altshuler D., Bailey-Wilson J.E., et al. Replicating genotype-phenotype associations. Nature. 2007;447:655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 12.Clayton D.G., Walker N.M., Smyth D.J., Pask R., Cooper J.D., Maier L.M., Smink L.J., Lam A.C., Ovington N.R., Stevens H.E., et al. Population structure, differential bias and genomic control in a large-scale, case–control association study. Nat. Genet. 2005;37:1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- 13.Hirschhorn J.N., Daly M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 14.Marchini J., Cardon L.R., Phillips M.S., Donnelly P. The effects of human population structure on large genetic association studies. Nat. Genet. 2004;36:512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- 15.Campbell C.D., Ogburn E.L., Lunetta K.L., Lyon H.N., Freedman M.L., Groop L.C., Altshuler D., Ardlie K.G., Hirschhorn J.N. Demonstrating stratification in a European American population. Nat. Genet. 2005;37:868–872. doi: 10.1038/ng1607. [DOI] [PubMed] [Google Scholar]
- 16.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 17.Seldin M.F., Shigeta R., Villoslada P., Selmi C., Tuomilehto J., Silva G., Belmont J.W., Klareskog L., Gregersen P.K. European population substructure: clustering of Northern and Southern populations. PLoS Genet. 2006;2:1339–1351. doi: 10.1371/journal.pgen.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freedman M.L., Reich D., Penney K.L., McDonald G.J., Mignault A.A., Patterson N., Gabriel S.B., Topol E.J., Smoller J.W., Pato C.N., et al. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 19.Helgason A., Yngvadottir B., Hrafnkelsson B., Gulcher J., Stefansson K. An Icelandic example of the impact of population structure on association studies. Nat. Genet. 2005;37:90–95. doi: 10.1038/ng1492. [DOI] [PubMed] [Google Scholar]
- 20.Bottini N., Musumeci L., Alonso A., Rahmouni S., Nika K., Rostamkhani M., MacMurray J., Meloni G.F., Lucarelli P., Pellecchia M., et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 2004;36:337–338. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 21.Begovich A.B., Carlton V.E., Honigberg L.A., Schrodi S.J., Chokkalingam A.P., Alexander H.C., Ardlie K.G., Huang Q., Smith A.M., Spoerke J.M., et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pritchard J.K., Stephens M., Rosenberg N.A., Donnelly P. Association mapping in structured populations. Am. J. Hum. Genet. 2000;67:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawson K.J., Belkhir K. A Bayesian approach to the identification of panmictic populations and the assignment of individuals. Genet. Res. 2001;78:59–77. doi: 10.1017/s001667230100502x. [DOI] [PubMed] [Google Scholar]
- 24.Satten G.A., Flanders W.D., Yang Q. Accounting for unmeasured population substructure in case–control studies of genetic association using a novel latent-class model. Am. J. Hum. Genet. 2001;68:466–477. doi: 10.1086/318195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoggart C.J., Parra E.J., Shriver M.D., Bonilla C., Kittles R.A., Clayton D.G., McKeigue P.M. Control of confounding of genetic associations in stratified populations. Am. J. Hum. Genet. 2003;72:1492–1504. doi: 10.1086/375613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epstein M.P., Allen A.S., Satten G.A. A simple and improved correction for population stratification in case–control studies. Am. J. Hum. Genet. 2007;80:921–930. doi: 10.1086/516842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pritchard J.K., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian C., Plenge R.M., Ransom M., Lee A., Villoslada P., Selmi C., Klareskog L., Pulver A.E., Qi L., Gregersen P.K., et al. Analysis and application of European genetic substructure using 300 K SNP information. PLoS Genet. 2008;4:e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenberg N.A., Pritchard J.K., Weber J.L., Cann H.M., Kidd K.K., Zhivotovsky L.A., Feldman M.W. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 32.Rosenberg N.A., Mahajan S., Ramachandran S., Zhao C., Pritchard J.K., Feldman M.W. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 2005;1:e70. doi: 10.1371/journal.pgen.0010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serre D., Paabo S. Evidence for gradients of human genetic diversity within and among continents. Genome Res. 2004;14:1679–1685. doi: 10.1101/gr.2529604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weir B., Cockerham C. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 35.Kosoy R., Nassir R., Tian C., White P.A., Butler L.M., Silva G., Kittles R., Alarcon-Riquelme M.E., Gregersen P.K., Belmont J.W., et al. Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America. Hum. Mutat. doi: 10.1002/humu.20822. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price A.L., Butler J., Patterson N., Capelli C., Pascali V.L., Scarnicci F., Ruiz-Linares A., Groop L., Saetta A.A., Korkolopoulou P., et al. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet. 2008;4:e236. doi: 10.1371/journal.pgen.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bauchet M., McEvoy B., Pearson L.N., Quillen E.E., Sarkisian T., Hovhannesyan K., Deka R., Bradley D.G., Shriver M.D. Measuring European population stratification with microarray genotype data. Am. J. Hum. Genet. 2007;80:948–956. doi: 10.1086/513477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seldin M.F., Price A.L. Application of ancestry informative markers to association studies in European Americans. PLoS Genet. 2008;4:e5. doi: 10.1371/journal.pgen.0040005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang N., Li H., Criswell L.A., Gregersen P.K., Alarcon-Riquelme M.E., Kittles R., Shigeta R., Silva G., Patel P.I., Belmont J.W., et al. Examination of ancestry and ethnic affiliation using highly informative diallelic DNA markers: application to diverse and admixed populations and implications for clinical epidemiology and forensic medicine. Hum. Genet. 2005;118:382–392. doi: 10.1007/s00439-005-0012-1. [DOI] [PubMed] [Google Scholar]
- 40.Halder I., Shriver M., Thomas M., Fernandez J.R., Frudakis T. A panel of ancestry informative markers for estimating individual biogeographical ancestry and admixture from four continents: utility and applications. Hum. Mutat. 2008;29:648–658. doi: 10.1002/humu.20695. [DOI] [PubMed] [Google Scholar]
- 41.Hamblin M.T., Di Rienzo A. Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am. J. Hum. Genet. 2000;66:1669–1679. doi: 10.1086/302879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bersaglieri T., Sabeti P.C., Patterson N., Vanderploeg T., Schaffner S.F., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]