

Abstract
With large amounts of public expression microrray data being generated by mulitple laboratories, it is a significant task for the bench researcher to routinely identify available datasets, then to evaluate the collective evidence across these datasets for regulation of a specific gene in a given system. 17β-estradiol stimulation of MCF-7 cells is a widely used model in the growth of breast cancer. While myriad independent studies have profiled the global effects of this hormone on gene expression in these cells, disparate experimental variables and the limited power of the individual studies have combined to restrict the agreement between them as to the specific gene expression signature elicited by this hormone. To address these issues, we have developed a freely-accessible web resource, Gene Expression MetaSignatures (GEMS, www.nursa.org/gems) that provides the user a consensus for each gene in the system. We conducted a weighted meta-analysis encompassing over 13,000 genes across ten independent published datasets addressing the effect of 17β-estradiol on MCF-7 cells at early (3-4h) and late (24h) time points. In a literature survey of 58 genes previously shown to be regulated by 17β-estradiol in MCF-7 cells, the meta-analysis combined the statistical power of the underlying datasets to call regulation of these genes with nearly 85% accuracy (FDR-corrected p-value < 0.05). We anticipate that with future expression microarray dataset contributions from investigators GEMS will evolve into an important resource for the cancer and nuclear receptor signaling communities.
Introduction
17β-estradiol (17βE2) exerts profound effects on gene expression and cellular function in a wide variety of reproductive, musculoskeletal, neuronal and metabolic tissues. The mechanism of action of 17βE2 encompasses (i) genomic actions through estrogen receptors (ERs) bound to cognate response elements in their target genes, (ii) trans-interactions of 17βE2-bound ERs with other transcription factors, and (iii) cross-talk of the ligand with rapid signaling cascades originating in the cytoplasm with endpoints in both the cytoplasm and nucleus 1. The action of 17βE2 in the mammary gland is understood to be an important factor in the development of breast cancer.
The advent of techniques enabling genome-wide interrogation of regulation of gene expression has given rise to a proliferation of studies profiling the transcriptional response to 17βE2 in cultured cell model systems, most notably MCF-7 cells 2-11. While these studies have identified large numbers of regulated genes across a variety of ligand treatment time points, substantial divergence exists among them as to the specific core gene expression signature generated by 17βE2 in this cell type 12. This highlights the importance of developing methods to integrate findings across studies, such as meta-analysis. One of the goals of the Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource is to supplement traditional models of distribution of scientific information with freely-accessible web-based resources designed to allow the bench researcher to intuitively and routinely integrate information across existing high content datasets. Here we describe a weighted meta-analysis that harnesses the combined statistical power of existing public, independent microarray datasets to arrive at a consensus gene list – a metasignature - for 17βE2 treatment of MCF-7 cells. We also describe an accompanying web resource, Gene Expression MetaSignatures (GEMS), which makes the results of our meta-analysis available to the community as a convenient, intuitive interface.
Materials and Methods
Differential gene expression analysis
Differential gene expression was determined at two time points: “early” (3-4h 17βE2 treatment) and “late” (24h 17βE2 treatment). Each dataset was processed separately using the linear modeling functions from the limma BioC software package 13. Empirical Bayes moderated t-tests were used to compare 17βE2 treated samples to their vehicle controls as described 14. P-values from each dataset were corrected for multiple testing by controlling the false discovery rate (FDR) 15.
Meta-analysis of the 17β-estradiol expression signature
For the meta-analysis, each dataset was reduced to the probe sets held in common between the Affymetrix Human Genome U133 Plus 2.0 and Human Genome U133A GeneChip® arrays. In order to obtain a single cumulative measure for each probe set indicative of differential gene expression surveyed across all the datasets, we applied the weighted Z-method developed by Mosteller and Bush 16 and by Liptak 17. Briefly, for each probeset, the set of observed t-statistics from each dataset are converted to corresponding standard normal deviates (same percentile) and combined using the following formula:
where Zp• is the combined Z-score for the pth probeset, k is the number of datasets, and wi is the degrees of freedom in the ith dataset. The combined Z-scores for each probe set are then converted to two-tailed p-values and corrected for multiple testing by controlling the FDR as done for differential gene expression.
Results
Time point selection and dataset normalization
Extensive MEDLINE and GEO surveys identified two time points most commonly selected by investigators in the field studying the effects of 17βE2 on gene expression in MCF-7 cells: 3-4hrs of treatment with 17βE2 (here designated “early”, and 24hrs of treatment, here designated “late”. Since two of the studies contributed datasets to both the early and late meta-analyses, five datasets were used for the “early’ analysis and seven datasets were used for the “late” analysis (Supplemental Table T1). The two time points were analyzed entirely independently of each other throughout.
Of the ten studies selected for meta-analysis, raw microarray intensity data were unavailable for five, which necessitated the use of the normalized probeset expression values submitted to GEO by the investigator. Fortunately, nine of the ten studies provided normalized expression values calculated using one of two related methods: the RMA method or the closely related GCRMA method. In an effort to standardize the processing steps as much as possible between all of the studies in the meta-analysis, we did not attempt to recalculate normalized probeset expression values for the ten studies and used the RMA/GCRMA normalized expression values provided by the investigators. In this way, all but one study was processed using either RMA or GCRMA (Supplemental Table T1).
Principal component analysis
Prior to differential expression analysis between the 17βE2 treated arrays and their respective vehicle treated controls, principal component analysis (PCA) was done with both the early and late time point datasets (Figure 1 and Supplemental Figure S1 respectively). As seen in Figure 1 and Supplemental Figure S1 the largest source of variance between datasets can be attributed to the “laboratory” effect for both the early and late time points, a general phenomenon for microarrays identified by a recent large-scale study 18. Potential contributing factors to the observed laboratory effect may be the variety of 17βE2 doses used, variations in the MCF-7 cells themselves, and at least six distinct cell culture protocols (Supplemental Table T1). However, when each of the 10 datasets was analyzed independently, the 17βE2 treatment effect is the largest source of variance for the majority of the 10 studies in the meta-analysis (Supplemental Figure S2A and S2B). Rather than attempt to correct for the laboratory effects, we analyzed each study dataset separately for differential gene expression prior to meta-analysis. Given that incremental changes in expression of a gene repeated across multiple independent datasets might actually reflect a real biological effect, the filtering out of probe sets showing marginal variability in expression was omitted.
Figure 1. Principal component analysis of the five datasets included in the early analysis.
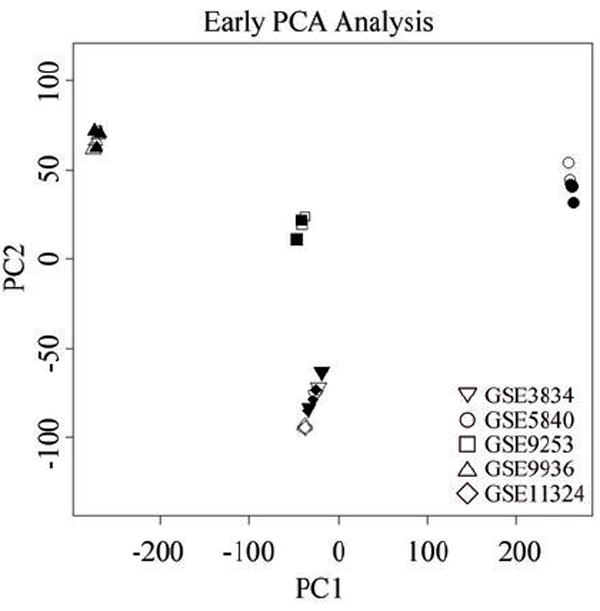
The graph depicts principal component one (PC1) graphed against principal component two (PC2). For each study the 17βE2-treated samples are shown in red and their corresponding vehicle treated controls are shown in black. The principal component analysis for the late time point is shown in Supplemental Figure S1.
Study relationship analysis
We set out first to determine the extent of overlap, in terms of regulated genes, between the independent datasets. Using a fold change cut off of 2 and a FDR-corrected p-value (referred to from here on as the q-value) < 0.05, both of which are commonly accepted parameters in microarray analysis, zero (0) differentially expressed genes were identified by all of the independent datasets at either time point. Moreover, relaxing the q-value cut -off to 0.2 resulted in only a modest increase in the number of genes in this intersection (data not shown here but available for download from the GEMS website). This initial result indicated that given the extent in variation across the datasets, traditional Venn analysis would be of limited use in arriving at a consensus gene expression response for 17βE2 in MCF-7 cells.
Meta-analysis
Within each study, the differential expression analysis produced a set of t-stats for each probeset indicative of 17βE2 action. For the meta-analysis each study dataset was reduced to the set of probesets held in common across the Affymetrix U133 GeneChip® family. This resulted in paring each study dataset to a core of 22277 common array features, representing approximately 13000 genes. We next utilized an algorithm to construct a 17βE2/MCF-7 gene expression metasignature based on combining appropriately weighted evidence (t-stats) across all of the studies at the early and late time points. Using the weighted Z-method at a combined q-value < 0.05, our meta-analysis identified a 17βE2/MCF-7 metasignature of 2313 unique Entrez Gene IDs at 3-4h (“early”) and 4144 unique Entrez Gene IDs at 24h (“late”). The results are summarized in Table 2, where the early and late metasignature genes are binned according to a number of different individual dataset fold change criteria, from FC ≥ 2.0 in zero individual datasets, to FC ≥ 2 in all datasets. Full gene lists from Table 2 are provided in Supplemental Table T2. The lower the combined q-value for a given gene, the greater the weight of cumulative evidence across all the independent datasets for regulation in this system. As reference points, pS2/TFF1 and MYC are two genes whose robust 17βE2 regulation in MCF-7 cells has been indisputably confirmed using quantitative techniques such as Northern hybridization and quantitative real-time PCR. At the 24h (“late”) time point, these genes have combined q-values of 4.78 × 10-17 and 2.62 × 10-11 respectively.
Table 2. Genes with a combined q-value < 0.05 identified by the meta-analysis.
Genes are binned according to a number of different individual dataset fold change criteria, ranging from FC ≥ 2 in none of the individual datasets to FC ≥ 2 in all underlying datasets. Full gene lists at each level are provided in Supplemental Table T2.
| # of Independent Datasets with FC > 2.0 | Meta-analysis genes
|
|
|---|---|---|
| Early | Late | |
| --- | 2313 | 4144 |
| 1 | 526 | 1213 |
| 2 | 140 | 516 |
| 3 | 67 | 321 |
| 4 | 20 | 118 |
| 5 | 6 | 29 |
| 6 | NA | 5 |
| 7 | NA | 0 |
Gene Expression MetaSignatures (GEMS) web resource
In order to enhance the utility of our meta-analysis as a research resource we have designed a web interface, Gene Expression MetaSignatures (GEMS) that enables the user to rapidly and conveniently evaluate the 17βE2/MCF-7 gene expression metasignature on a gene-by-gene basis. The 17βE2/MCF-7 GEMS is freely accessible and located on the Nuclear Receptor Signaling Atlas (NURSA) website at www.nursa.org/gems.
The version to which this paper refers (v 1.0) provides for querying the 17βE2/MCF-7 metasignature for a specific gene. For the gene name or symbol entered, GEMS displays, by default, a simple view showing (i) the fold regulation range across independent datasets and (ii) the combined q-value, a measure of the collective evidence gleaned from the independent datasets pointing to that gene being differentially regulated. Both values should be given due consideration when evaluating the possibility of differential expression for that gene. For example, a gene can have negligible fold changes in the independent datasets and yet have a highly significant combined q-value resulting from low variability within the replicates of each dataset. Conversely, a gene can have high fold changes in the independent datasets but a high combined q-value resulting from high variability within the replicates of the independent datasets. In both these scenarios, evidence for differential expression is questionable. In contrast, for a historical, quantitatively validated 17βE2 target in MCF-7 cells, such as pS2/TFF1, which has both high fold changes in the independent datasets and a very low combined q-value, evidence for differential expression is much more reliable.
We next set out to evaluate the performance of GEMS in calling regulation of experimentally-identified 17βE2-responsive genes in MCF-7 cells. Since a statistically rigorous experimental validation was not feasible, we first carried out a MEDLINE literature survey to identify a set of genes which had been shown to be quantitatively regulated by 17β-estradiol in MCF-7 cells at either of the two time points in our meta-analysis. The list of genes is shown in Supplementary Table T3. We evaluated the fold change range and combined q-values assigned to these genes by GEMS. GEMS used the combined statistical power of the underlying datasets to call regulation (combined q-value <0.05) of these genes with nearly 85% accuracy. For comparison, Fisher’s method identified 70% correctly, and the overlap between the studies is 100%, ie all of the genes identified by Fisher are identified by the weighted-Z method (data not shown).
Discussion
As microarray studies have been published in increasing numbers it has become clear that the technique is particularly sensitive to variations in protocol, and independent laboratories continue to yield discrepant datasets, even in experimental systems as robust and well-characterized as that of 17βE2 in MCF-7 cells. The field until now has lacked a resource which allows the bench researcher unfamiliar with advanced microarray analysis to routinely avail of an objective weighting of these datasets to help determine the response of a specific gene or gene network to 17βE2 in these cells.
It is not our intent with this study to call into question any of the gene lists arising from the published datasets selected for meta-analysis, all of which are valid observations under their own prevailing experimental conditions. Rather, our study is an attempt to reconcile the disparate observations made by these studies and arrive at a statistical consensus for the system by leveraging the increased statistical power afforded by combining independent datasets in a single meta-analysis. By opening the results of our meta-analysis to query through a freely-accessible web resource, GEMS, we hope to provide the field with a convenient resource to fully exploit these datasets.
GEMS is not intended as a static entity and will be improved by the incorporation of future suitable expression microarray datasets, and we welcome all contributions from the community. Moreover, the anticipated development of platforms with increasingly comprehensive genome coverage, and improved sensitivity in data capture and analysis, will also serve to hone the accuracy of GEMS. That said, it should be noted that certain conditions for individual studies may result in the identification of bona fide targets specific to those conditions that would not be identified by the meta-analysis.
Traditional models of scientific publication do not readily afford the bench researcher convenient access for mining of high content datasets. As a result these datasets in many cases fail to realize their full potential as research resources. The volume of gene regulation data in the field is reaching a point where there are reasonable grounds for organizing independent datasets as a whole, and applying statistical approaches to identify biological trends that are sufficiently robust to efface the experimental variables characteristic of each individual dataset. Backed by the collective contributions of researchers in this field, we anticipate GEMS evolving into a unique resource for analyzing tissue-specific regulation of gene expression by NRs, their ligands and coregulators, for the entire community.
Supplementary Material
Table 1.
Studies selected for meta-analysis.
| Dataset | Early | Late | n/group |
|---|---|---|---|
| GSE352913 | ● | 2 | |
| GSE38348 | ● | ● | 2 early/2 late |
| GSE40067 | ● | 2 | |
| GSE402510 | ● | 2.5 | |
| GSE58409 | ● | 4 | |
| GSE680011 | ● | 2 | |
| GSE85974 | ● | 4 | |
| GSE925312 | ● | 2 | |
| GSE99366 | ● | ● | 3 early/5.5 late* |
| GSE113245 | ● | 3 |
n/group = the average number of arrays per treatment group.
GSE9936 at the late time point used 6 control arrays and 5 17βE2 treated arrays for an average of 5.5 arrays per group.
Acknowledgments
We thank the principal investigators who made their datasets publicly available. This work was supported by NIDDK NURSA U19 DK62434.
References
- 1.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Molecular endocrinology (Baltimore, Md) 2005;19:833–42. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 2.Bourdeau V, Deschenes J, Laperriere D, Aid M, White JH, Mader S. Mechanisms of primary and secondary estrogen target gene regulation in breast cancer cells. Nucleic acids research. 2008;36:76–93. doi: 10.1093/nar/gkm945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll JS, Meyer CA, Song J, et al. Genome-wide analysis of estrogen receptor binding sites. Nature genetics. 2006;38:1289–97. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 4.Chang EC, Charn TH, Park SH, et al. Estrogen Receptors Alpha and Beta as Determinants of Gene Expression: Influence of Ligand, Dose, and Chromatin Binding. Molecular endocrinology (Baltimore, Md) 2008 doi: 10.1210/me.2007-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–42. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 6.Creighton CJ, Cordero KE, Larios JM, et al. Genes regulated by estrogen in breast tumor cells in vitro are similarly regulated in vivo in tumor xenografts and human breast tumors. Genome biology. 2006;7:R28. doi: 10.1186/gb-2006-7-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan M, Yan PS, Hartman-Frey C, et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer research. 2006;66:11954–66. doi: 10.1158/0008-5472.CAN-06-1666. [DOI] [PubMed] [Google Scholar]
- 8.Frasor J, Chang EC, Komm B, et al. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer research. 2006;66:7334–40. doi: 10.1158/0008-5472.CAN-05-4269. [DOI] [PubMed] [Google Scholar]
- 9.Gaube F, Wolfl S, Pusch L, Kroll TC, Hamburger M. Gene expression profiling reveals effects of Cimicifuga racemosa (L.) NUTT. (black cohosh) on the estrogen receptor positive human breast cancer cell line MCF-7. BMC pharmacology. 2007;7:11. doi: 10.1186/1471-2210-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kininis M, Chen BS, Diehl AG, et al. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Molecular and cellular biology. 2007;27:5090–104. doi: 10.1128/MCB.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rae JM, Johnson MD, Scheys JO, Cordero KE, Larios JM, Lippman ME. GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast cancer research and treatment. 2005;92:141–9. doi: 10.1007/s10549-005-1483-4. [DOI] [PubMed] [Google Scholar]
- 12.Kininis M, Kraus WL. A global view of transcriptional regulation by nuclear receptors: gene expression, factor localization, and DNA sequence analysis. Nuclear receptor signaling. 2008;6:e005. doi: 10.1621/nrs.06005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 14.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3:Article 3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 15.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;57:289–300. [Google Scholar]
- 16.Mosteller F, Bush R. Selected quantitative techniques. In: Lindzey G, editor. Handbook of Social Psychology. Cambridge, MA: Addison-Wesley; 1954. pp. 289–334. [Google Scholar]
- 17.Liptak T. On the combination of independent tests. Magyar Tud Akad Mat Kutato Int Kozl. 1958;3:171–97. [Google Scholar]
- 18.Shi L, Reid LH, Jones WD, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nature biotechnology. 2006;24:1151–61. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.