

Abstract
P14 mice expressing a transgenic TCR specific for the lymphocytic choriomeningitis virus glycoprotein p33 epitope were used to study the induction of CTL effector activity by a variety of ligands. Surprisingly, p33 variants which are weaker agonists for the P14 TCR than the wild-type p33 peptide were able to induce more potent effectors with a broader range of cytolytic specificity. Similarly, low concentrations of p33 were more effective than higher concentrations. These results correlated with no or only moderate TCR down-regulation by variants of p33 and low p33 concentrations. This phenotype observed after 18 h of culture was transient as progressive restoration of reactivity was observed at 42 or 66 h in the cultures stimulated with high p33 concentrations and this correlated with recovery of TCR surface levels. TCR down-regulation was blocked by src family kinase inhibitors. These findings indicate that the specificity of a T cell can be fine-tuned by the nature of the primary stimulus correlating with surface TCR level and imply an important role for src family kinases in the differential regulation of surface TCR levels upon TCR engagement by different ligand/MHC complexes.
Keywords: Altered peptide ligand, TCR down-regulation, Serial triggering, LCMV, TCR transgenic mouse
1 Introduction
Single amino acid changes in antigenic T cell epitopes generate altered peptide ligands (APL) which can act as partial agonists, i.e. selectively activate only some of the various T cell effector functions or antagonize reactivity to agonistic epitopes [1-3]. Partial activation and TCR antagonism have been demonstrated for T cells of both CD4 and CD8 phenotype. It has been shown for a number of viruses, that such T cell epitope variants are generated in vivo due to selective pressure exerted by virus-specific CTL. It was shown that single amino acid exchanges in T cell epitopes can result in inhibition of responses to the wild-type epitope by non-productive TCR engagement [4] or TCR antagonism [5, 6]. Another possibility is impairment or complete loss of CTL recognition [7-14]. It was hypothesized that the generation of such variants could contribute to establishing a persistent virus infection. Pircher et al. [7] reported that infection of P14 mice expressing a transgenic TCR specific for the glycoprotein (GP) epitope GP p32-42 [15, 16] of lymphocytic choriomeningitis virus (LCMV) with a high dose of virus resulted in the development of immune escape variants. Viruses were isolated and single amino acid exchanges in this epitope identified. It was found that the variant epitopes were no longer detected by CTL from LCMV infected TCR transgenic mice. Using these P14 mice we compared the cognate peptide GP p33-41 (p33) [17] and four variants including the escape variants described by Pircher et al. [7] for their capacity to prime naive TCR transgenic T cells and to be recognized by CTL effectors primed with the different peptides as well as by a TCR transgenic CTL clone. We used TCR down-modulation as a means to define agonistic potential based on the serial triggering model developed by Valitutti et al. [18, 19]. These authors have shown that the usually small number of complexes of a given antigenic peptide bound to its restricting MHC molecule activate T cells by serially engaging a large number of TCR as measured by the down-regulation of triggered TCR from the cell surface. In addition, it has also been shown that different thresholds of TCR signaling exist for different T cell responses [20, 21] and that activation thresholds can be altered by co-stimulation [22-26].
Our experiments showed that in vitro stimulation of naive P14 T cells with peptide variants resulted in more potent CTL effectors with broader reactivity to multiple epitope variants compared to stimulation with p33. Similarly, stimulation with a low concentration of cognate peptide resulted in reactivity to weak agonists, including one previously characterized as an escape variant epitope in the P14 TCR transgenic mice [7]. When higher concentrations of peptide p33 were used to stimulate the P14 cells, reactivity to this peptide variant was completely absent and overall cytotoxic reactivity was decreased. Analysis of surface TCR levels revealed that the fine specificity of the stimulated transgenic P14 T cells correlated with the extent of TCR down-regulation. Desensitization of a transgenic T cell clone by pretreatment with cognate peptide or PMA also resulted in a significant decrease in reactivity to the weakest agonists and also correlated with TCR down-regulation.
Our data show that the fine specificity of naive T cells can be differentially “programmed” by the nature of the primary stimulus in that the strength of this stimulus determines T cell fine specificity most likely by modulation of surface TCR levels on the primed T cells. We also present evidence for a critical role of src family kinases in the regulation of surface TCR levels. Our findings have important implications for T cell responses in general and may shed more light on the complexity of immune escape mechanisms by CTL selection of epitope variants.
2 Results
2.1 Relative H-2Db binding affinity of p33 and its variants
The sequences of the p33 peptide and its variants are shown in Fig. 1. We performed standard RMA-S stabilization assays and destabilization assays to assess the relative binding affinities of the various peptides for H-2Db, which serves as the restriction element for p33. As shown in Fig. 1, p33 and the single amino acid variants used in this study have similar binding affinities. Peptide p33YG stabilizes the Db molecule more efficiently than p33 and p33YF and peptides p33VG and p33VL exhibit a slightly weaker binding affinity than p33 and p33YF (Fig. 1A). Similar dissociation kinetics for H-2Db were observed (Fig. 1B). An exception is peptide p33VG which exhibits a faster off rate than the other peptides.
Figure 1.
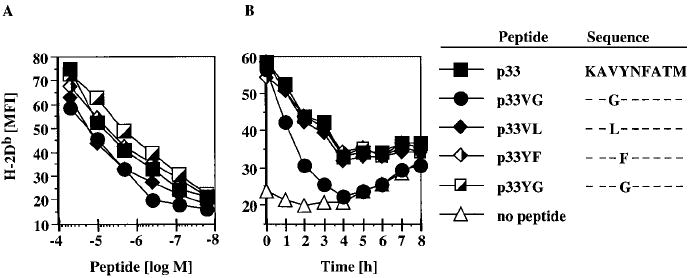
H-2Db stabilization by p33 and its variants. (A) TAP-deficient RMA-S cells were preincubated overnight at 31 °C and graded concentrations of the indicated peptides added for 1 h before cells were shifted to 37 °C for 4 h. (B) Following overnight incubation at 31 °C, peptides were added to RMA-S cells at 20 μM for 1 h. BFA was then added for 2 h, peptide was washed away and the cells shifted to 37 °C. Cells were then incubated in the continuous presence of BFA and aliquots stained every hour. Staining was done with an H-2Db-specific antibody and cells analyzed by flow cytometry.
2.2 Agonist activity of p33 and its variants for TCR transgenic P14 thymocytes and CTL clones
A sensitive thymocyte deletion assay was used to determine whether the peptides interacted with the P14 transgenic TCR. When P14 transgenic thymocytes were incubated for 18 h with graded concentrations of the different peptides, we found differences in the capacity of the various peptides to induce deletion of DP thymocytes (Fig. 2A). These results allowed us to establish the following hierarchy of the agonisitic potential of the peptides: p33 > p33VG > p33VL > p33YF > p33YG. The same hierarchy was found when a TCR transgenic CTL clone, P14.G5, derived from P14 mice, was used and cytolytic activity with graded peptide concentrations assessed in a 51Cr-release assay (Fig. 2B). It is clear that the DP deletion assay is more sensitive than the effector CTL assay.
Figure 2.

Agonist activity of p33 and its variants. (A) TCR transgenic P14 thymocytes were incubated with EL-4 cells as APC and graded concentrations of the indicated peptides for 18 h and then stained with anti-CD4 and anti-CD8. Deletion of immature CD4hiCD8hi DP thymocytes is given as % of control without peptide. (B) Graded concentrations of the indicated peptides were preincubated with 51Cr-labeled EL-4 cells for 30 min. P14.G5 CTL were then added and a standard 4-h Cr-release assay performed. The effector/target ratio was 5/1.
2.3 CTL with different fine specificity patterns are induced by priming with p33 as compared to weaker agonists
In vitro priming of naive P14 spleen cells for 18 h with p33 or its variants resulted in different patterns of cytotoxic reactivity to the peptide variants (Fig. 3A). Priming with the wild-type peptide generated effectors which were able to lyse targets coated with peptides p33, p33VG and p33VL. Interestingly, priming with the variants p33VG and p33VL generated effectors able to recognize these ligands but, in these cases, significant reactivity to the weakest agonist, p33YF, was also detected. The activity against p33YF was completely absent when cells had been primed with the wild-type peptide. It is worth noting that the overall cytolytic activity was higher in cultures primed with the weaker agonists. Stimulation with p33YG, p33YF or the control peptide Ad E1A did not result in the generation of cytolytic effector cells.
Figure 3.
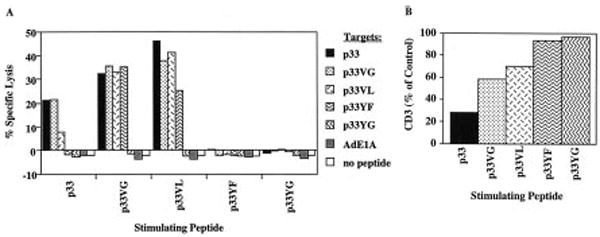
Different patterns of cytolysis are induced in P14 splenocytes by priming with wild-type or variant peptide. Correlation with surface CD3 level. TCR transgenic P14 spleen cells were stimulated for 18 h with EL-4 cells which had been prepulsed with 1 μM of the indicated peptides. (A) Cytotoxic activity was assayed after washing the cultures and addition of 51Cr-labeled, peptide-pulsed EL-4 target cells for 4 h at an E/T ratio of 100/1. (B) Corresponding CD3 levels on CD8+ cells of these cultures stimulated with the different peptides were determined by flow cytometry. Data are given as % MFI of control cells cultivated for 18 h with EL-4 cells without peptide.
When we analyzed the surface CD3 level of CD8+ cells in the different effector populations we found that stimulation of naive P14 spleen cells with the wild-type peptide induced a significant TCR down-regulation (Fig. 3B). Stimulation with p33VG or p33VL, in contrast caused a more modest TCR down-regulation, yet nevertheless, activated the T cells. The observed level of surface TCR may therefore be responsible for the differential sensitivity of the transgenic T cells as measured in 51Cr-release assays.
2.4 Type and density of TCR ligands determines T cell fine specificity and correlates with surface TCR levels
The above results suggested that weaker agonists stimulate the maturation of more potent effector cells. If this is related to the avidity of the naive T cell for the stimulating APC then lower concentrations of the original agonist might also induce more potent CTL. To address this question, we stimulated naive transgenic spleen cells for 18 h in vitro using EL-4 cells as APC. When the APC were coated with decreasing concentrations of p33, the overall cytotoxic reactivity to all peptides increased (Fig. 4A). Most interestingly, at low p33 concentration significant reactivity to p33YF became evident. When the CD3 levels on the CD8+ TCR transgenic cells were analyzed (Fig. 4B), we observed an inverse correlation with the concentration of p33 used for stimulation. In order to rule out the possibility that peptide-pulsed EL-4 APC carried over from the 18-h cultures into the Cr-release assay were acting as cold targets, we labeled EL-4 cells with a fluorescent dye prior to 18-h culture which allowed us to separate the T cells from the larger fluorescent tumor cells by FACS sorting before the Cr-release assay. This experiment (Fig. 4C) yielded the same result. We again observed a correlation between increased cytotoxic activity and surface CD3 levels on transgenic CD8+ T cells (Fig. 4D). Furthermore, when adherent MC57G fibrosarcoma cells were used as APC in order to exclude cold target inhibition, we again found similar results (data not shown).
Figure 4.

Different patterns of cytolysis are induced in P14 splenocytes by stimulation with different concentrations of the agonist peptide p33 or p33VG. Correlation with surface CD3 level. P14 splenocytes were cultured for 18 h with irradiated EL-4 cells that had been pulsed with titered levels of p33, washed and labeled with fluorescent dye, BCECF-AM. Cultures were harvested, washed, and assayed immediately for lysis of 51Cr-labeled EL-4 pulsed with various peptides at 5 μM (A), and stained for surface CD3 levels on CD8+ cells (B). Alternatively, TCR transgenic P14 splenocytes were sorted away from BCECF-labeled EL-4 cells before assaying for cytolysis (C) and determination of surface CD3 level (D). E/T was 100/1. (E, F) P14 spleen cells were stimulated for 18 h with EL-4 cells that had been prepulsed with titered levels of p33VG. Lysis assays and determination of CD3 levels were done as for (A, B). No lysis was detected for 18-h cultures set up in the absence of peptide (not shown).
The same type of experiment was performed using the weaker agonist p33VG for priming P14 spleen cells. Under these priming conditions we observed significant reactivity to the weakest agonists p33VL and p33YF over a much broader concentration range of stimulating peptide (Fig. 4E) as compared to p33 priming. This reactivity pattern correlated with higher surface TCR levels on the p33VG primed T cells (Fig. 4F).
2.5 Peptide pretreatment desensitizes a CTL clone and correlates with TCR down-regulation
We next wanted to determine whether preincubation of a TCR transgenic CTL clone, P14.G5, with graded concentrations of p33 would differentially affect its ability to react with target cells coated with the variant peptides. To test this, P14.G5 CTL were preincubated for 4 h with adherent MC57G cells that had been pulsed with graded concentrations of p33 and then assayed for lysis of peptide-pulsed target cells. We found that as the concentration of p33 used to coat the MC57G cells in the pretreatment was increased, there was a progressive loss of subsequent cytotoxic activity (Fig. 5A). The reactivity of the CTL correlated well with the surface TCR Vα2 levels (Fig. 5B). As with the naive TCR transgenic P14 spleen cells (Fig. 4), the reactivity to the weakest agonists was most sensitive to decreases in surface TCR levels induced by preretreatment with higher p33 concentrations. As a control for the carry-over of antigen from the first culture to the assay, when MC57 monolayers were treated in the absence of CTL in a manner identical to the plates which contained CTL and the supernatants of these plates were added to P14.G5 CTL which had not been preincubated with MC57G cells, there was no inhibition of CTL activity. It is worth noting that p33YF is recognized as an agonist by P14.G5 CTL carried in vitro with p33-coated spleen cell stimulators (see also Fig. 2B). This is in contrast to naive P14 spleen cells after 18-h culture on p33-coated APC (Fig. 3).
Figure 5.

Desensitization of a CTL clone for cytotoxic reactivity by agonist pretreatment. Adherent MC57G cells were pulsed with the indicated concentrations of p33 for 1 h and washed three times. P14.G5 cells were added for 4 h, harvested and washed repeatedly. A 4-h Cr-release assay was performed using peptide-pulsed, 51Cr-labeled EL-4 target cells (A). Corresponding TCR Vα2 levels on the CTL were determined by flow cytometry at the time of harvest from the MC57G monolayers (B).
2.6 PMA-induced TCR down-regulation affects reactivity to weak agonists
It has been shown previously that treatment of T cells with PMA induces TCR down-regulation [27]. We employed this method to analyze whether antigen-independent TCR down-regulation on transgenic P14 T cells would preferentially reduce T cell reactivity to weak agonists. Treatment of P14.G5 CTL with graded concentrations of PMA prior to addition of peptide pulsed EL-4 target cells resulted in a progressive loss of reactivity to the weak agonists p33VL and p33YF (Fig. 6A) as seen with p33 pretreatment. In contrast, the reactivity to the strongest agonists p33 and p33VG was not significantly affected, demonstrating that the cells are still functional after the PMA treatment. Analysis of surface TCR Vα2 levels (Fig. 6B) clearly showed that PMA also induced TCR down-regulation. At concentrations above 1 ng/ml PMA CD8 surface levels also drop significantly, which contributes to the observed effects (data not shown). Thus, PMA-induced TCR down-regulation correlated with a reactivity pattern that we had observed using graded concentrations of p33 for stimulation of naive transgenic P14 T cells and CTL clones.
Figure 6.
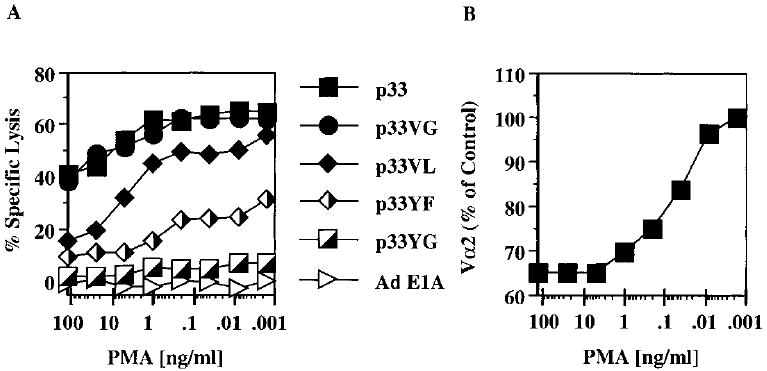
PMA-induced TCR down-regulation causes preferential loss of reactivity to weak agonists. P14.G5 cells were incubated for 1 h with the indicated concentrations of PMA. After three washes, peptide-pulsed EL-4 target cells were added and a 4-h 51Cr-release assay was performed (A). TCR Vα2 levels of CD8+ cells were determined by flow cytometry following the 1-h treatment with PMA (B).
2.7 TCR down-regulation requires src family kinase signaling
We performed TCR down-regulation experiments in the presence of the src family kinase specific inhibitor PP1 [28] and the more general protein tyrosine kinase inhibitors herbimycin A and genistein. P14 spleen cells (Fig. 7A, B) or P14.G5 CTL (Fig. 7C, D) were preincubated in the presence or absence of these inhibitors and peptide-pulsed APC were then added for 4 h. We found that both TCR down-regulation and CD69 up-regulation was efficiently blocked by all these inhibitors. Herbimycin A and genistein were less efficient as inhibitors of TCR down-regulation as compared to PP1. Our results are consistent with recent data by D’Oro et al. who found that these inhibitors block anti-CD3-induced TCR down-regulation in Jurkat T cells [29].
Figure 7.
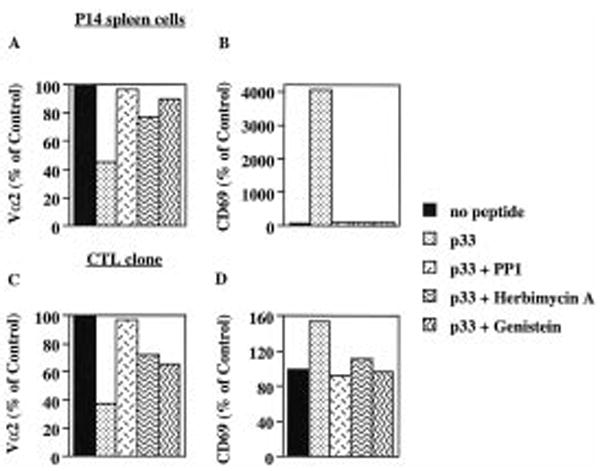
Inhibition of protein tyrosine kinase activity blocks TCR down-regulation. P14 TCR transgenic spleen cells (A, B) or P14.G5 CTL clone (C, D) were preincubated for 60 min in the presence of PP1 or genistein. Preincubation with herbimycin A was done overnight with P14 spleen cells or for 60 min with P14.G5 CTL. EL-4 cells pulsed with p33 (1 μM) were then added for 4 h and cells stained for flow cytometry with anti-CD8 and anti-Vα2 antibodies (A, C) or with anti-CD8 and anti-CD69 antibodies (B, D). Mean fluorescence intensities are expressed as % or MFI of control in the absence of peptide.
3 Discussion
The role of TCR down-regulation in T cell biology is not fully understood. It is not required nor sufficient for T cell activation [25, 30]. A T cell may use this mechanism for desensitization in order to avoid excessive stimulation resulting in anergy or activation-induced cell death. Thus, it has been recently shown that persistent TCR engagement can result in loss of TCR signaling and inactivation of the Lck signal [31]. It has also been demonstrated that TCR down-regulation can be employed as a mechanism for peripheral tolerance [32]. In thymic selection, maintenance of a low-to-intermediate TCR level on immature DP thymocytes may be essential to prevent negative selection due to the high sensitivity of thymocytes for apoptosis inducing stimuli at this stage of development. The importance of the regulation of surface TCR levels for thymic selection has been demonstrated by some recent reports [29, 33-36].
The results presented in our study show that regulation of TCR levels may be of importance for the fine tuning of T cell reactivity. We show that the nature and strength of the primary stimulus for a naive T cell can influence its fine specificity pattern. A strong correlation between T cell reactivity to variants of p33 and surface TCR levels using p33-specific TCR transgenic CD8+ T cells was observed. The APL used here differ in their affinity for the TCR as they have very similar Db binding affinities (Fig. 1). The hierarchy of the agonist potential of p33 and its variants was established based on their efficiency in deleting P14 TCR transgenic DP thymocytes (Fig. 2A) and triggering cytolysis by a TCR transgenic CTL clone (Fig. 2B). We found that the variant peptides p33VL and p33YF, which have been isolated as escape variants from LCMV growing in P14 mice, were the weakest agonists for peripheral T cells. Peptide p33YF was a true partial agonist, it triggered cytotoxicity in primed CTL (Fig. 2B, 3) but not proliferation of established or naive CTL (data not shown). In addition, we found that LCMV immune spleen cells lyse target cells coated with p33YF but do not proliferate when stimulated with this peptide (data not shown). p33VL triggered both effector functions although a 100-fold higher concentration was needed to obtain similar activity as with p33 (Fig. 2B). This is in contrast to the data by Pircher et al. [7] and is most likely due to their use of longer, non-optimal peptides. Peptide p33YG did not activate any effector function in mature peripheral P14 CTL (Fig. 2B, 3) but was able to delete DP thymocytes (Fig. 2A), demonstrating its capability to interact with the transgenic TCR. We have also shown that this APL has antagonist activity for P14 CTL (data not shown).
The differences in agonist potential of these ligands were further reflected in their differential ability to induce TCR down-regulation. Only the strongest agonist, p33, induced significant TCR down-regulation, whereas p33VG and p33VL induced only moderate reduction of surface TCR levels (Fig. 3B), and only at high levels of peptide. Nevertheless, all three of these peptides activated cytotoxicity (Fig. 3A) and proliferation (data not shown). The data clearly demonstrate that TCR down-regulation is not required for T cell activation in the presence of costimulation but rather reflects the strength of an agonist, a fact previously noted by others [25, 30].
The most interesting consequence of the different levels of surface TCR down-regulation following stimulation of naive TCR transgenic P14 spleen cells with either the APL (Fig. 3) or with low concentrations of p33 (Fig. 4) was the appearance of reactivity to p33YF. This activity was only detected when TCR levels were greater than 60–70 % of the initial level. A similar threshold was also seen for the P14-derived CTL clone P14.G5 (Figs. 5 and 6). It has been proposed by Valitutti et al. that, in order to become fully activated, a certain number of TCR have to be triggered by MHC-peptide complexes [18]. Accumulation of such triggering events over a certain threshold results in T cell activation. Different effector functions have been shown to have different activation thresholds [20, 21]. It has also been shown that MHC/APL complexes engage TCR with a more rapid dissociation rate due to their lower affinity for the TCR [21, 37, 38]. This shorter duration of TCR occupancy may result in engagement of TCR at a higher rate in the case of APL of lower TCR affinity as compared to the cognate peptide. The consequence may be transient signals that do not result in the full array of phosphorylation events, recruitment of components of the TCR activation complex or the generation of a critical level of second messengers. Thus, a weak signal may not allow the assembly of all the components which are required for efficient TCR internalization. In order to achieve the activation threshold for a given effector function, weaker agonists may therefore need to engage more TCR than stronger agonists to allow for the accumulation of a sufficient number of incomplete, partial signaling events. Consistent with this theory, an inverse correlation between the TCR affinity for its ligand and the number of TCR required for activation has been described [39]. Thus, below a critical surface TCR level, low-affinity ligands such as p33YF may not be able to trigger a sufficient number of TCR precluding the T cell from reaching the threshold for activation of cytotoxicity. As shown by Viola et al. [22], T cells respond when a threshold number of TCR has been triggered and this response is independent of the nature of the triggering ligand. Peptide p33YG, with an even lower TCR affinity, cannot activate any effector function and may engage very many TCR with a high dissociation rate that results in an unproductive signaling process. Such a consumption of TCR is likely to be the basis for its antagonistic activity and the fast kinetics explain the advantage of antagonists over agonists [40]. These kinetics can be visualized when agonists are used which induce TCR down-regulation as recently shown by Preckel et al. [41]. In contrast, APL such as p33VG and p33VL can still efficiently activate cytotoxicity at this low surface TCR level due to their higher affinity for the TCR resulting in longer TCR occupancy.
When such cultures were tested at 42 or 66 h, a progressive restoration with time of CTL reactivity to the weak agonists was observed upon p33 stimulation. This correlated with a recovery of the surface TCR levels on the transgenic T cells and is most likely due to a decay of antigen/MHC complexes and the lysis of APC by the CTL (data not shown). Interestingly, when cloned P14.G5 CTL were used early (day 2) after in vitro restimulation on p33-pulsed syngeneic spleen cells, we were unable to detect significant reactivity to the weak agonist p33YF. In contrast, this reactivity was readily detected on days 3–5 and also correlated with a recovery of surface TCR levels which are low early after restimulation (data not shown).
Since p33YF is a partial agonist for P14 T cells which activates only cytotoxicity but not proliferation and it has been previously shown that such partial agonists induce only partial signals [42-45], it may be that as a consequence of a short duration of TCR occupancy the quality of the signals transduced by p33YF may be different from the ones transduced by the stronger agonists.
It is likely that the differences in fine specificity patterns measured in cytotoxicity assays are the result of the surface TCR levels observed in our experiments. Thus, although they involve different mechanisms [30, 46], our data show that ligand- or PMA-induced TCR down-regulation result in similar alterations of P14 CTL reactivity (Figs. 5, 6). At comparable TCR levels, we see a progressive decline of reactivity to p33VL and p33YF in both cases. Earlier studies have suggested that TCR signaling via protein tyrosine kinases is linked to TCR down-regulation [47, 48] and may involve association of tyrosine-phosphorylated CD3ζ chain with the cytoskeleton [49]. A recent report showed that activation of the src family kinase Lck targets cell surface TCR for lysosomal degradation in Jurkat T cells [29]. Furthermore, expression of a dominant negative form of Rab5, a GTPase involved in endocytosis, as a transgene in mice results in increased surface TCR levels and hyperresponsiveness of peripheral T cells [36]. Our own experiments show in TCR transgenic T cells that TCR internalization is blocked in the presence of general protein tyrosine kinase specific inhibitors or a src family kinase specific inhibitor (Fig. 7).
We have provided evidence that a T cell has a significant flexibility in its response to ligand by tightly regulating surface TCR levels. This process may be an important mechanism to fine-tune the sensitivity of a T cell in response to signals of different strength and allow the T cell to adapt to the quality and quantity of TCR ligands. We would like to speculate that the extent of TCR down-regulation reflects Lck activity. An interesting new aspect in this context is the recently discussed role of protein tyrosine kinase activity for contact zone formation between T cells and APC [50]. Our data show how the quality and quantity of a TCR ligand during the priming and the effector phase can influence the fine specificity and sensitivity of a T cell and are in agreement with the model of kinetic discrimination in T cell antigen recognition [51, 52].
4 Materials and methods
4.1 Animals
C57BL/6 mice were purchased from Taconic Farms (Germantown, NY). LCMV p33 specific TCR transgenic P14 mice have been described earlier [15, 16] and were purchased from Jackson Laboratories [Bar Harbor, ME, strain B6,D2-TgN(TcrLCMV0327Sdz)] and were bred in the animal facility of the University of Washington under specific pathogen-free conditions.
4.2 Peptides
Peptides were purchased from Research Genetics Inc. (Huntsville, AL) and were > 80 % pure. Peptide concentration was determined using the BCA assay (Pierce, Rockford, IL). Single amino acid variants of the LCMV p33 epitope and the H-2Db binding peptide from adenovirus E1A protein [53] have been described before [54].
4.3 Antibodies
Anti-CD3ε (500A2), anti-CD8α (53-6.7), anti-CD69 (H1.2F3), anti-TCR Vα2 and anti-H-2Db (KH95) were all purchased from Pharmingen (San Diego, CA) and were either FITC-, PE- or biotin conjugated. Tricolor®-conjugated streptavidin was from Caltag Laboratories (South San Francisco, CA).
4.4 Cell lines and clones
A CTL line was generated by cultivation of spleen cells from P14 TCR transgenic mice on p33-pulsed, irradiated (3000 rad) C57BL/6 spleen cells in RPMI with 10 % FCS/25 mM Hepes/2 mM l-glutamine/10μM 2-ME and antibiotics (RP-10) in 96-well U-bottom plates (Costar, Cambridge, MA). Clone P14.G5 was derived from this line by standard limiting dilution cloning. Cultures were restimulated every 7 days in the presence of 5 % Con A-induced rat spleen cell supernatant (RCAS) as a source of IL-2. Cells obtained were shown by flow cytometry to express CD8 and the transgenic TCR Vα2 and Vβ8.1.
Tumor cell lines (EL-4, RMA-S and MC57G) were grown in RP-5.
4.5 H-2Db stabilization and destabilization assays
RMA-S lymphoma cells were preincubated at 31 °C overnight. To measure H-2Db stabilization, graded concentrations of peptides were added for 1 h and the cells were then shifted to 37 °C for 4 h. For destabilization assays (adapted from Chen et al. [55], 2×106 cells/ml in RP-10 were incubated with 20 μM of peptide for 1 h at 31 °C. Brefeldin A (BFA, Sigma, St. Louis, MO) was then added to 10 μg/ml for 2 h, cells washed twice with RP-5/10 μg/ml BFA and resuspended in the same media and then shifted to 37 °C. Aliquots were stained every hour with a FITC-conjugated H-2Db-specific antibody and analyzed by flow cytometry.
4.6 In vitro thymocyte deletion assay
Peptides were titrated in 96-well U-bottom plates in RP-10. Irradiated (20 000 rad) EL-4 cells were added at 1 × 106/ml. A single-cell suspension of TCR transgenic P14 thymus was prepared and thymocytes added at 1×107/ml to the plates in the same medium. Cultures were incubated for 18 h at 37 °C/5 % CO2, then washed once with FACS buffer and stained with anti-CD4 and anti-CD8. Samples were analyzed using a total live gate and a tight CD4hiCD8hi (double-positive, DP) gate excluding EL-4 cells based on forward scatter characteristics. Deletion of DP thymocytes was calculated for 5×104 events and determined as: % deletion = [100× (number of CD4hiCD8hi thymocytes with peptide/number of CD4hiCD8hi thymocytes without peptide)].
4.7 Cytotoxicity assays
Standard 4-h chromium release assays were performed. EL-4 lymphoma cells were labeled with Na251CrO4 for 90 min at 37 °C in RPMI/10 % FCS/25 mM Hepes (Cr medium) and pulsed with 5 μM peptide during labeling. After three washes 2×103 cells were added to effector cells in 96-well U-bottom plates. For the 18-h spleen cell cultures effectors consisted of total viable trypan blue-excluding cells. P14 spleens typically contain about 30 % CD8+ transgenic T cells.
4.8 Flow cytometry
Cells were washed in PBS/0.02 NaN3/2 % FCS (FACS buffer). Biotin-conjugated antibodies were added for 20 min on ice and cells washed three times. FITC- and PE-conjugated antibodies were then added together with Tricolor®-conjugated streptavidin for 20 min on ice and the cells fixed in 1 % paraformaldehyde after three washes. Samples were analyzed using a FACScan instrument and CellQuest Software (Becton Dickinson, Mountain View, CA). Twenty thousand events were collected and live, CD8+ cells were analyzed using Reproman (TrueFacts Software Inc., Seattle, WA).
4.9 FACS sorting
Irradiated (20 000 rad) EL-4 cells were pulsed with graded concentrations of p33 for 60 min at 37 °C in RP-10, washed three times and resuspended in PBS containing 1 μM of BCECF-AM (Calbiochem, San Diego, CA). After 10 min at 37 °C, cells were washed three times in PBS, resuspended, and 6×105 cells added to 24-well plates (Costar, Cambridge, MA) in 1 ml RP-10. P14 spleen cells were added in 1 ml RP-10 at 5×106/well. The plates were then incubated for 18 h at 37 °C/5 % CO2. Cultures were harvested, washed twice with PBS/0.5 mM EDTA and resuspended in PBS/3 % FCS. Sorting was performed in a FACStar instrument (Becton Dickinson) based on forward scatter and fluorescence 1 (BCECF-AM) characteristics and the purity of the EL-4-depleted P14 spleen cells verified by flow cytometry. Cells were then washed with RP-10 and stained for flow cytometry or used in functional assays.
4.10 In vitro peptide stimulation
Peptides were titrated in RP-10 in 96-well U-bottom plates. EL-4 stimulator cells were irradiated (20 000 rad) and added at 6×104/well. Plates were incubated for 1 h and then washed three times with RP-10. P14 spleen cells (5×105) were added to the wells, incubated for 18 h at 37 °C/5 % CO2 and plates were then washed twice in PBS/0.5 mM EDTA to disrupt cell aggregates. Cells were then either used as effectors in a cytotoxicity assay or stained for flow cytometry.
4.11 T cell desensitization assay
Adherent MC57G fibrosarcoma cells were plated at 6×104/well in 96-well flat-bottom plates (Costar, Cambridge, MA) and allowed to adhere overnight. Graded concentrations or p33 were added for 1 h and plates washed three times with RP-10. P14.G5 CTL were washed repeatedly in RP-10 and added to the MC57G plates at 6×104/well for 4 h at 37 °C. The CTL were then gently washed off the MC57G monolayer and transferred to 96-well U-bottom plates. After repeated washes cells were stained for flow cytometry or peptide-pulsed, 51Cr labeled EL-4 target cells were added (2×103/ well) and a 4-h Cr-release assay performed.
4.12 PMA-induced TCR down-modulation
PMA (Sigma, St. Louis, MO) was titrated in RP-10 in 96-well U-bottom plates and P14.G5 CTL were added to the plates at 6×104/well in Cr medium. Plates were incubated for 1 h at 37 °C and then washed three times with Cr medium. Cells were stained for flow cytometry or peptide-pulsed 51Cr-labeled EL-4 cells were added to the wells and a standard 4-h Cr-release assay was performed.
4.13 Inhibition of src family kinase activity
P14.G5 CTL (6×104/well) or P14 spleen cells (5×105/well) were preincubated for 60 min with the src family kinase-specific inhibitor PP1 (10 μM and 50 μM, respectively) (Bio-mol, Plymouth Meeting, PA) [28]. Alternatively, cells were incubated with the protein tyrosine kinase-specific inhibitors herbimycin A (5 μM) or genistein (400 μM). For P14 spleen cells herbimycin A preincubation was done overnight. EL-4 cells (6×104/well) pulsed with 1 μM p33 were then added. After 4 h cells were washed twice in PBS/0.5 mM EDTA, once in FACS buffer and then stained for flow cytometry.
Acknowledgments
We would like to thank Ananda W. Goldrath and Dr. Eric A. Butz for critical reading of the manuscript, members of the Bevan lab for helpful discussions, Deborah Wilson for taking care of our animals and Kathy Allen for help with cell sorting.
This work was supported by National Institutes of Health grant A1-29802 and the Howard Hughes Medical Institute. S. Martin was supported in part by a research stipend of the Deutsche Forschungsgemeinschaft (DFG).
Abbreviations
- LCMV
Lymphocytic choriomeningitis virus
- APL
Altered peptide ligand
- GP
Glycoprotein
References
- 1.Sette A, Alexander J, Ruppert J, Snoke K, Franco A, Ishioka G, Grey HM. Antigen analogs/MHC complexes as specific T cell receptor antagonists. Annu Rev Immunol. 1994;12:413–431. doi: 10.1146/annurev.iy.12.040194.002213. [DOI] [PubMed] [Google Scholar]
- 2.Jameson SC, Bevan MJ. T cell receptor antagonists and partial agonists. Immunity. 1995;2:1–11. doi: 10.1016/1074-7613(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 3.Sloan-Lancaster J, Allen PM. Altered peptide ligand-induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu Rev Immunol. 1996;14:1–27. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Meier U-C, Klenermann P, Griffin P, James W, Köppe B, Larder B, McMichael A, Philipps R. Cytotoxic T lymphocyte lysis inhibited by viable HIV mutants. Science. 1995;270:1360–1362. doi: 10.1126/science.270.5240.1360. [DOI] [PubMed] [Google Scholar]
- 5.Bertoletti A, Sette A, Chisari FV, Penna A, Levrero M, De Carli M, Fiaccadori F, Ferrari C. Natural variants of cytotoxic epitopes are T-cell receptor antagonists for antiviral cytotoxic T cells. Nature. 1994;369:407–410. doi: 10.1038/369407a0. [DOI] [PubMed] [Google Scholar]
- 6.Klenerman P, Rowland-Jones S, McAdam S, Edwards J, Daenke S, Lalloo D, Koppe B, Rosenberg W, Boyd D, Edwards A, Giangrande P, Philipps RE, McMichael AJ. Cytotoxic T-cell activity antagonized by naturally occurring HIV-1 Gag variants. Nature. 1994;369:403–407. doi: 10.1038/369403a0. [DOI] [PubMed] [Google Scholar]
- 7.Pircher H, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel RM. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature. 1990;346:629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 8.Aebischer T, Moskophidis D, Hoffmann Rohrer U, Zinkernagel RM, Hengartner H. In vitro selection of lymphocytic choriomeningitis virus escape mutants by cytotoxic T lymphocytes. Proc Natl Acad Sci USA. 1991;88:11047–11051. doi: 10.1073/pnas.88.24.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewicki H, Tishon A, Borrow P, Evans CF, Gairin JE, Hahn KM, Jewell DA, Wilson IA, Oldstone MBA. CTL escape variants I. Generation and molecular characterization. Virology. 1995;210:29–40. doi: 10.1006/viro.1995.1314. [DOI] [PubMed] [Google Scholar]
- 10.Philipps RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, Ogunlesi AO, Elvin JG, Rothbard J, Bangham CRM, Rizza CR, McMichael AJ. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature. 1991;354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 11.Price DA, Goulder PJR, Klenerman P, Sewell AK, Easterbrook PJ, Troop M, Bangham CR, Philipps RE. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc Natl Acad Sci USA. 1997;94:1890–1895. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couillin I, Culmann-Penciolelli B, Gomard E, Choppin J, Levy J-P, Guillet J-G, Saragosti S. Impaired cytotoxic T lymphocyte recognition due to genetic variations in the main immunogenic region of the human immunodeficiency virus 1 NEF protein. J Exp Med. 1994;180:1129–1134. doi: 10.1084/jem.180.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertoletti A, Costanzo A, Chisari FV, Levrero M, Artini M, Sette A, Penna A, Giuberti T, Fiaccadori F, Ferrari C. Cytotoxic T lymphocyte response to a wild type hepatitis B virus epitope in patients chronically infected by variant viruses carrying substitutions within the epitope. J Exp Med. 1994;180:933–943. doi: 10.1084/jem.180.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pewe L, Wu GF, Barnett EM, Castro RF, Perlman S. Cytotoxic T cell-resistant variants are selected in a virus-induced demyelinating disease. Immunity. 1996;5:253–262. doi: 10.1016/s1074-7613(00)80320-9. [DOI] [PubMed] [Google Scholar]
- 15.Pircher H, Buerki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 16.Pircher H, Mak TW, Lang R, Ballhausen W, Ruedi E, Hengartner H, Zinkernagel RM, Buerki K. T cell tolerance to Mlsa encoded antigens in T cell receptor Vβ8.1 chain transgenic mice. EMBO J. 1989;8:719–727. doi: 10.1002/j.1460-2075.1989.tb03431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kyburz D, Aichele P, Speiser DE, Hengartner H, Zinkernagel R, Pircher H. T cell immunity after a viral infection versus tolerance induced by soluble viral peptides. Eur J Immunol. 1993;23:1956–1962. doi: 10.1002/eji.1830230834. [DOI] [PubMed] [Google Scholar]
- 18.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 19.Valitutti S, Lanzavecchia A. Serial triggering of TCRs: a basis for the sensitivity and specificity of antigen recognition. Immunol Today. 1997;18:299–304. [PubMed] [Google Scholar]
- 20.Valitutti S, Müller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med. 1996;183:1917–1921. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itoh Y, Germain RN. Single cell analysis reveals regulated hierarchical T cell antigen receptor signaling thresholds and intraclonal heterogeneity for individual cytokine responses of CD4+ T cells. J Exp Med. 1997;186:757–766. doi: 10.1084/jem.186.5.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 23.Kündig TM, Shahinian A, Kawai K, Mittrücker H-W, Sebzda E, Bachmann MF, Mak TW, Ohashi PS. Duration of TCR stimulation determines costimulatory requirements of T cells. Immunity. 1996;5:41–52. doi: 10.1016/s1074-7613(00)80308-8. [DOI] [PubMed] [Google Scholar]
- 24.Bachmann MF, Sebzda E, Kündig TM, Shahinian A, Speiser D, Mak TW, Ohashi PS. T cell responses are governed by avidity and costimulatory thresholds. Eur J Immunol. 1996;26:2017–2022. doi: 10.1002/eji.1830260908. [DOI] [PubMed] [Google Scholar]
- 25.Cai Z, Kishimoto H, Brunmark A, Jackson MR, Peterson PA, Sprent J. Requirements for peptide-induced T cell receptor downregulation on naive CD8+ T cells. J Exp Med. 1997;185:641–651. doi: 10.1084/jem.185.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bachmann MF, McKall-Faienza K, Schmits R, Bouchard D, Bach J, Speiser DE, Mak TW, Ohashi PS. Distinct roles for LFA-1 and CD28 during activation of naive T cells: adhesion versus costimulation. Immunity. 1997;7:549–557. doi: 10.1016/s1074-7613(00)80376-3. [DOI] [PubMed] [Google Scholar]
- 27.Cantrell DA, Davies AA, Crumpton MJ. Activators of proteinkinase C down-regulate and phosphorylate the T3/T cell antigen receptor complex of human T lymphocytes. Proc Natl Acad Sci. 1985;82:8158–8162. doi: 10.1073/pnas.82.23.8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollock BA, Connelly PA. Discovery of a novel, potent, and Src-family selective tyrosine kinase inhibitor. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 29.D’Oro U, Vacchio MS, Weissman AM, Ashwell JD. Activation of the Lck tyrosine kinase targets cell surface T cell antigen receptors for lysosomal degradation. Immunity. 1997;7:619–628. doi: 10.1016/s1074-7613(00)80383-0. [DOI] [PubMed] [Google Scholar]
- 30.Salio M, Valitutti S, Lanzavecchia A. Agonist-induced T cell receptor down-regulation: molecular requirements and dissociation from T cell activation. Eur J Immunol. 1997;27:1769–1773. doi: 10.1002/eji.1830270726. [DOI] [PubMed] [Google Scholar]
- 31.Lee JE, Cossoy MB, Chau LA, Singh B, Madrenas J. Inactivation of lck and loss of TCR-mediated signaling upon persistent engagement with complexes of peptide: MHC molecules. J Immunol. 1997;159:61–69. [PubMed] [Google Scholar]
- 32.Schönrich G, Kalinke U, Momburg F, Malissen M, Schmitt-Verhulst A-M, Malissen B, Hämmerling GJ, Arnold B. Down-regulation of T cell receptors on self-reactive T cells as a novel mechanism for extra-thymic tolerance induction. Cell. 1991;65:293–304. doi: 10.1016/0092-8674(91)90163-s. [DOI] [PubMed] [Google Scholar]
- 33.Wiest DL, Yuan L, Jefferson J, Benveniste P, Tsokos M, Klausner RD, Glimcher LH, Samelson LE, Singer A. Regulation of T cell receptor expression in immature CD4+ CD8+ thymocytes by p56lck tyrosine kinase: basis for differential signaling by CD4 and CD8 in immature thymocytes expressing both coreceptor molecules. J Exp Med. 1993;178:1701–1712. doi: 10.1084/jem.178.5.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ericsson P-O, Teh H-S. The protein tyrosine kinase p56lck regulates TCR expression and T cell selection. Int Immunol. 1995;7:617–624. doi: 10.1093/intimm/7.4.617. [DOI] [PubMed] [Google Scholar]
- 35.Yamazaki T, Arase H, Ono S, Ohno H, Watanabe H, Saito T. A shift from negative to positive selection of autoreactive T cells by the reduced level of TCR signal in TCR-transgenic CD3 zeta-deficient mice. J Immunol. 1997;158:1634–1640. [PubMed] [Google Scholar]
- 36.Andre P, Boretto A, Hueber A-O, Regnier-Vigoroux A, Gorvel J-P, Ferrier P, Chavrier P. A dominant-negative mutant of the Rab5 GTPase enhances T cell signaling by interfering with TCR down-modulation in transgenic mice. J Immunol. 1997;159:5253–5263. [PubMed] [Google Scholar]
- 37.Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NRJ. T-cell-receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 38.Lyons D, Liebermann SA, Hampl J, Boniface JJ, Chien Y-H, Berg LJ, Davis MM. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 39.Schodin BA, Tsomides TJ, Kranz DM. Correlation between the number of T cell receptors required for T cell activation and TCR-ligand affinity. Immunity. 1996;5:137–146. doi: 10.1016/s1074-7613(00)80490-2. [DOI] [PubMed] [Google Scholar]
- 40.Lanzavecchia A. Understanding the mechanisms of sustained signaling and T cell activation. J Exp Med. 1997;185:1717–1719. doi: 10.1084/jem.185.10.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preckel T, Grimm R, Martin S, Weltzien HU. Altered hapten ligands antagonize trinitrophenyl-specific cytotoxic T cells and block internalization of hapten-specific receptors. J Exp Med. 1997;185:1803–1813. doi: 10.1084/jem.185.10.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sloan-Lancaster J, Shaw AS, Rothbard JB, Allen PM. Partial T cell signaling: altred phospho-and lack of ZAP-70 recruitment in APL-induced T cell anergy. Cell. 1994;79:913–922. doi: 10.1016/0092-8674(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 43.Madrenas J, Wange RL, Wang JL, Isakov N, Samelson LE, Germain RN. ζ-phosphorylation without ZAP-70 activation induced by TCR antagonists or partial agonists. Science. 1995;267:515–518. doi: 10.1126/science.7824949. [DOI] [PubMed] [Google Scholar]
- 44.Rabinowitz JD, Beeson C, Wülfing C, Tate K, Allen PM, Davis MM, McConnell HM. Altered T cell receptor ligands trigger a subset of early T cell signals. Immunity. 1996;5:125–135. doi: 10.1016/s1074-7613(00)80489-6. [DOI] [PubMed] [Google Scholar]
- 45.Reis e Sousa C, Levine EH, Germain RN. Partial signaling by CD8+ T cells in response to antagonist ligands. J Exp Med. 1996;184:149–157. doi: 10.1084/jem.184.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bäckström BT, Rubin B, Peter A, Tiefenthaler G, Palmer E. T cell receptor α-chain tail is required for protein kinase C-mediated down-regulation, but not for signaling. Eur J Immunol. 1997;27:1433–1441. doi: 10.1002/eji.1830270621. [DOI] [PubMed] [Google Scholar]
- 47.Boyer C, Auphan N, Gabert J, Blanc D, Malissen B, Schmitt-Verhulst A-M. Comparison of phosphorylation and internalization of the antigen receptor/ CD3 complex, CD8, and class I MHC-encoded proteins. Role of the intracytoplasmic domains analyzed with hybrid CD8/class I molecules. J Immunol. 1989;143:1905–1914. published erratum appears in J. Immunol. 190 Jan 1; 144 (1): 408. [PubMed] [Google Scholar]
- 48.Luton F, Buferne M, Davoust J, Schmitt-Verhulst AM, Boyer C. Evidence for protein tyrosine kinase involvement in ligand-induced TCR/CD3 internalization and surface redistribution. J Immunol. 1994;153:63–72. [PubMed] [Google Scholar]
- 49.Rodzial MM, Malissen B, Finkel TH. Tyrosine-phosphorylated T cell receptor ζ chain associates with the actin cytoskeleton upon activation of mature T lymphocytes. Immunity. 1995;3:623–633. doi: 10.1016/1074-7613(95)90133-7. [DOI] [PubMed] [Google Scholar]
- 50.Delon J, Berovici N, Liblau R, Trautmann A. Imaging antigen recognition by naive CD4+ T cells: compulsory cytoskeletal alterations for the triggering of an intracellular calcium response. Eur J Immunol. 1998;28:716–729. doi: 10.1002/(SICI)1521-4141(199802)28:02<716::AID-IMMU716>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 51.McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci rUSA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rabinowitz JD, Beeson C, Lyons DS, Davis MM, McConnell HM. Kinetic discrimination in T-cell activation. Proc Natl Acad Sci USA. 1996;93:1401–1405. doi: 10.1073/pnas.93.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kast WM, Melief CJM. Fine peptide specificity of cytotoxic T lymphocytes directed against adenovirus-induced tumours and peptide-MHC binding. Int J Cancer (Suppl) 1991;6:90–94. doi: 10.1002/ijc.2910470718. [DOI] [PubMed] [Google Scholar]
- 54.Martin S, Kohler H, Weltzien HU, Leipner C. Selective activation of CD8 T cell effector functions by epitope variants of LCMV glycoprotein. J Immunol. 1996;157:2358–2365. [PubMed] [Google Scholar]
- 55.Chen W, Khilko S, Fecondo J, Margulies DH, McCluskey J. Determinant selection of major histocompatibility complex class I-restricted antigenic peptides is explained by class I-peptide affinity and is strongly influenced by nondominant anchor residues. J Exp Med. 1994;180:1471–1483. doi: 10.1084/jem.180.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]