

Summary
Ventricular fibrillation (VF) is the most important cause of sudden cardiac death. While traditionally thought to result from random activation of the ventricles by multiple independent wavelets, recent evidence suggests that VF may be determined by the sustained activation of a relatively small number of reentrant sources. In addition, recent experimental data in various species as well as computer simulations have provided important clues about its ionic and molecular mechanisms, particularly in regards to the role of potassium currents in such mechanisms. The results strongly argue that the inward rectifier current, Ik1, is an important current during functional reentry because it mediates the electrotonic interactions between the unexcited core and its immediate surroundings. In addition, IK1 is a stabilizer of reentry due to its ability to shorten action potential duration and reducing conduction velocity near the center of rotation. Increased I K1 prevents wavefront-wavetail interactions and thus averts rotor destabilization and breakup. Other studies have shown that while the slow component of the delayed rectifier potassium current, IKs, does not significantly modify rotor frequency or stability, it plays a major role in post-repolarization refractoriness and wavebreak formation. Therefore, the interplay between IK1 and the rapid sodium inward current (INa) is a major factor in the control of cardiac excitability and therefore the stability and frequency of reentry while IKs is an important determinant of fibrillatory conduction.
Introduction
…turbulent flows seem almost ‘alive’.
AT Winfree
Ventricular fibrillation (VF) is by far the most important immediate cause of sudden cardiac death (SCD). Every year, VF is responsible for an estimated 300,000 deaths annually in the United States of America alone1,2. Because of its highly complex electrocardiographic (ECG) appearance3, VF is commonly thought of as an exceptionally complex and disorganized cardiac activation, in which electrical waves propagate through the ventricles chaotically and unpredictably. In fact, during VF the ventricular activation sequence is profoundly abnormal; electrical wavefronts do not follow the usual paths. The heart rate accelerates to the extreme, and the electrical waves assume a complex vortex-like behavior that looks a lot like eddy formation and turbulence in water. Such turmoil renders the heart unable to pump blood. Thus, the blood pressure drops and immediate loss of consciousness follows. Unfortunately, despite many years of research and speculation, the mechanism underlying VF continues to be a matter of speculation and debate.
Over the last several years, evidence has accumulated supporting the idea that, while VF is indeed complex, it is in fact deterministic, quantifiable, and in theory, mechanistically understandable. In this regard, it seems reasonable to suggest that achieving a comprehensive knowledge of VF mechanisms will require a clear familiarity with the underlying bases of cardiac excitation and its frequency dependence, in which cardiac ion channels play important roles. More specifically, it is my contention that advances in VF understanding will require thorough quantitative knowledge of the ionic mechanisms of the extremely complex phenomena that underlie the initiation and maintenance of VF, including wavebreak and rotor formation, rotor stabilization, and spiral wave behavior. In this article, I briefly review the most salient aspects related to the dynamics of VF, as well as current knowledge, however incomplete, on the role played by inward rectifying potassium channels in the mechanisms of VF initiation and maintenance.
Functional reentry and spiral waves
Theoretical4,5 and experimental6 studies that began to appear in the relevant literature more than 60 years ago have established that the heart can sustain electrical activity that rotates about a functional obstacle. In this regard, the experimental work of Allessie and collaborators in the 1970’s that gave birth to the so-called “leading circle” concept was an essential initial step toward our current understanding of the phenomenon of functional reentry in cardiac tissue; that is, reentry without the involvement of an anatomical obstacle (Figure 1A–B).6 At about the same time, work conducted by Soviet scientists using the Belousov-Zhabotinski reaction and its numerical counterparts led to the idea that two-dimensional spiral autowaves (Figure 1C) could be a possible mechanism of cardiac arrhythmias.7 Subsequently, the notion of spiral autowaves was brilliantly expanded into the third dimension (Figure 1D) by the late Arthur T. Winfree,8 who in fact coined the term “rotor” to signify the actual vortex that generates the spiral (scroll) wave activity, and led to the virtual abandonment of the use of the term “leading circle”. Thereafter, much work has focused on rotors as the underlying mechanism of ventricular tachycardia and VF.
Figure 1.
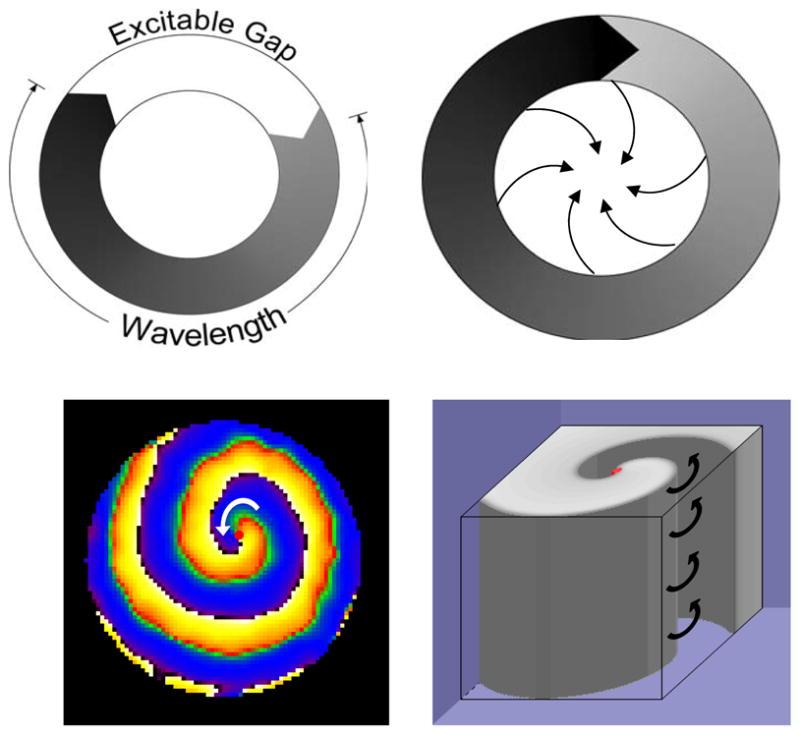
Four different concepts of reentry: A. Reentry around a ring-like anatomical obstacle. Successful reentry occurs when wavelength (black) is smaller than pathlength and allows for a fully excitable gap (white). B. Leading circle reentry around a functional obstacle. Partially excitable gap allows wave front to “bite” its wavetail of refractoriness. The hypothesis suggests that wavefronts invading the center result in refractoriness. C. Two-dimensional spiral wave rotates counter clockwise around an unexcited but excitable core (red dot) in a neonatal rat ventricular myocyte monolayer. D. Computer simulation of a three dimensional scroll wave rotating counter-clockwise.
Rotors and their breakup
Life threatening, complex cardiac tachyarrhythmias can be due to the activity of a reentrant electrical source, i.e., a rotor of characteristic size and angular velocity from which spiral waves radiate at a high frequency.8,9 The basic components of the rotor are a curved wave front, a curved wave tail and a core around which the wave front and tail rotate (Figure 1C). The rotor may drift and travel along complex paths or may be completely stationary with spiral waves emanating from it and propagating through the ventricles.3,10 The waves may undergo a variety of behaviors; for example, they may be highly stable and spiral periodically around their generating rotor as in Figure 2 to activate the ventricles at extremely high frequencies, or they may undergo breakup in the rotor’s periphery and result in fibrillatory conduction (see Figure 3), the net result being complex spatial and temporal patterns of ventricular activation. 11 In other words, the behavior of the rotor, and that of the waves generated by it, may be reflected on ECG as monomorphic or polymorphic ventricular tachycardias,12 or even VF.3
Figure 2.
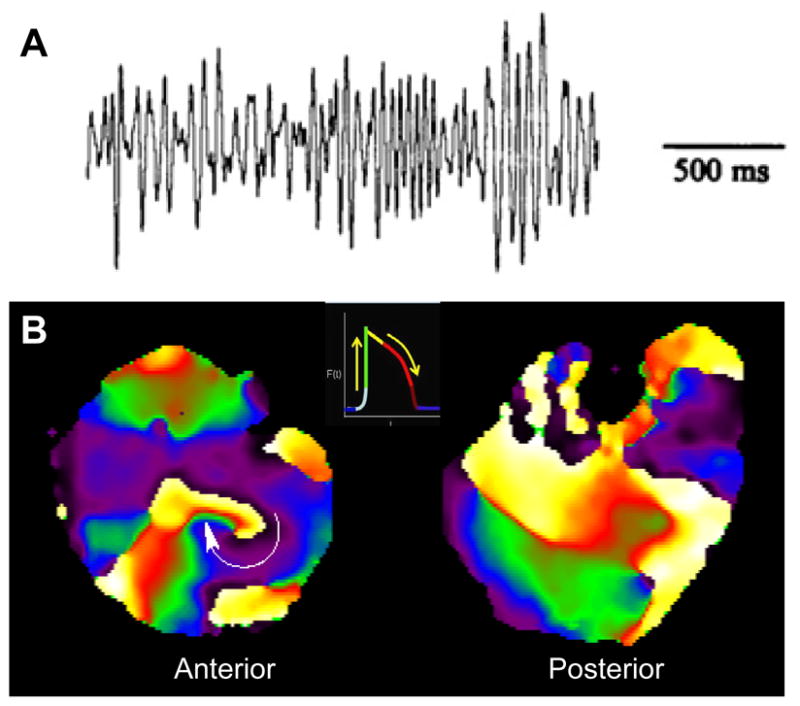
High-frequency stationary rotor results in fibrillation in an isolated, Langendorff-perfused guinea pig heart. A, ECG trace of VF. B, Snapshots of phase maps of the anterior (left) and posterior (right) epicardial surfaces of the ventricles during VF, which was maintained by a long-lasting rotor rotating clockwise on the anterior left ventricular wall. Colors indicate different phases of the action potential (inset) at each pixel location.
Figure 3.

A, IK1 and IKs have different roles in VF dynamics. A. Role of IK1 density in VF dynamics in a 2-D model (6×6 cm2) of the guinea pig ventricles consisting of >200,000 excitable elements (“cells”). The simulated left ventricle (LV, center) is surrounded by the right ventricle (RV, periphery). Top, phase map of VF maintained by a stable rotor in the LV, with fibrillatory conduction to RV. Red square shows the perimeter of the LV model (area, 2×2 cm2). As illustrated by the inset on the right, each color represents a different phase of the action potential in each cell. Bottom, current-voltage (IV) relations of IK1 used for LV (left) and RV (right). Note larger IK1 density in LV. Broken curve is IK1 with nominal density. For further explanation see text and ref13. B, IKs, Overexpression results in fibrillatory conduction even when IK1 is normal. Top, single rotor in wildtype monolayer. Bottom, multiple wavebreaks and fibrillatory conduction in monolayer overexpressing IKs. See ref24 for details.
Rotors and VF in the human heart
During VF, there may be a wide spectrum of rotor behaviors.3,13,14 Under appropriate conditions, even in the structurally normal heart of small animals, rotors may be long lasting, and result in a high degree of spatial and temporal organization.13 Consequently, a question may be raised as to whether the same applies to the ventricles of larger animals such as dogs, pigs and of course humans. Recent studies,15–17 suggest that even in large hearts such as that of the pig, the dynamics of wave propagation during VF are not as complex as might occur if the mechanism were spiral breakup, or as random as would be expected from the multiple wavelet hypothesis.15–17 In fact, Rogers et al15 were unable to rule out the possibility that mother rotors located in unmapped regions in their swine heart experiments maintained the fibrillatory activity. Additionally, a study has proposed that the mother rotor and the multiple wavelets are both mechanisms of VF in the human heart.17–18 Yet another study, in which the epicardium of the human left ventricle was mapped concluded that there is significant organization of human VF.19
The results described in the previous paragraph are consistent with data of Thomas et al,20 who investigated the way in which activation is organized during VF induced in the presence of an old anterior left ventricular wall infarct. By using 20 needles carrying multiple monopolar electrodes to record the transmural activity of the left ventricle of the sheep heart, Thomas et al demonstrated that the regions with the highest activation frequency displayed less variable cycle lengths and were generally hidden within the ventricular myocardium. In some experiments, they were able to reveal regions deep in the myocardium whose activity was stable and extremely rapid but highly regular. In other words, while the characteristics of VF in the epicardium were constantly changing in space and time, the activity within the myocardium had a higher frequency and was also highly periodic and organized.20 Such findings are clearly compatible with the hypothesis that in that experimental model fibrillatory activity is maintained by a small number (one or two) of three-dimensional rotors.
Ionic Mechanisms of Wavebreak and Functional Reentry
Propagation of an action potential from a depolarized cell (the source) to a resting neighbor (the sink) are dependent on intercellular coupling (gap junctions) and on the complex, nonlinear interactions between different ionic currents, electrogenic pumps, and/or exchangers in each source and sink cell. It therefore seems intuitively obvious that the wavebreak that leads to the initiation of reentry is the direct result of a source-sink mismatch, which may occur when a propagating wavefront encounters a refractory tissue or when the source is too weak to elicit an active response in a fully polarized tissue. As such, wavebreak and reentry are expected to be a direct consequence of the nonlinear dynamic interplay between inward and outward currents underlying the excitation-recovery cycle.
Inward Currents and VF
Most studies on the role of the sodium current in VF have involved analyzing the effects of its antagonists. During VF, the administration of tetrodotoxin (TTX)—a potent, selective and reversible antagonist of sodium channels tripled the area of the core.21 Accordingly, TTX reduced the VF frequency, organized the electrical activity and reduced the number of wavelets. Lowering the excitability reduces the degree of curvature at which propagation is blocked near the spiral tip, and thus increases the perimeter of the core.22 As the core increases in size, the rotation frequency of the spiral that maintains the VF becomes lower. This facilitates 1:1 conduction, and reduces the degree of peripheral fragmentation.21
Blocking the L-type calcium current (ICa-L) with verapamil has been shown to stabilize reentry and convert VF to VT by reducing the appearance of singularity points, rotor frequency (due to an increased core radius and reduced APD) and wavefront fragmentation.23 In contrast, other authors have reported that verapamil transiently increases VF dominant frequency (DF) and reduces core size; these discrepancies have been attributed to methodological differences.23
Potassium channels and cardiac excitation
In the heart, potassium currents are diverse in that their individual properties depend not only on the membrane potential and their dissimilar activation and recovery kinetics, but also on the activation frequency. Diffusion of potassium ions through the cell membrane as the so-called inward rectifier potassium current (IK1) maintains the resting membrane potential in all cardiac cells. In most mammals, upon action potential (AP) depolarization (phase 0) the transient outward current (Ito), is rapidly activated to mediate the initial phase of repolarization (phase 1) followed by rapid inactivation. The more slowly activating delayed rectifier currents (IKs and IKr), contribute to the slow repolarization that characterizes the AP plateau (phase 2). During the terminal phase of repolarization (phase 3), IK1 again predominates, rapidly restoring the membrane potential to resting values (phase 4). During tachyarrhythmias, probably all potassium currents involved in cardiac repolarization participate to various degrees in helping to establish the dynamics of cardiac excitation and propagation. Here I will focus attention only on IK1.
The inwardly rectifying potassium current (IK1) controls VF frequency
IK1 flows through membrane channels formed by members of the strong inward rectifier (Kir2.x) sub-family of proteins. Inward rectifier potassium channels are part of a large family of membrane spanning proteins that have a conserved GYG sequence. Each protein spans the membrane twice, with both N and C termini being intracellular.24,25 Four homomeric or heteromeric subunits may form a channel.26 These channels are termed inward rectifiers because their permeability to potassium is greater in the inward than in the outward directions. Rectification is due to a voltage dependent blockade of the channel by penetration of positively charged molecules such as polyamines (PA) and Mg2+ from the intracellular space into the pore.27,28 Spermine and spermidine are the most potent PA blockers of IK1.29 At voltages positive to the resting membrane potential, PA’s and Mg2+ are drawn to the pore. They block it, reducing the outflow of potassium ions and resulting in a smaller outward current relative to the inward component.27
In the heart, the strong inward rectifier channels are formed by tetramers of Kir2.x proteins. Of the Kir2.x subfamily members, the Kir2.1 subunit is a key carrier of IK125,26 in the ventricles. The highly nonlinear current/voltage relationship of IK1 allows it to act as a stabilizer of the resting membrane potential.25 The role of IK1 is also apparent during depolarization and the late phase of repolarization of the AP. Increasing the density of IK1 causes APD abbreviation. Decreasing its density has the reciprocal effect.25
Rotor Stability and Fibrillatory Conduction
The role of IK1 in rotor behavior has been investigated in both computer simulations and experiments. In the guinea pig heart, VF is manifested by a stable high-frequency rotor that is sustained and anchored in the left ventricle (LV), as illustrated in Figure 2.13 This causes the frequency of activation of the LV (Panel B, 32 Hz) to be higher than the right ventricle (RV, 14 Hz). The anchoring of the rotor in the LV and the subsequent regional difference in frequency between the LV and RV have been attributed to differences in IK1 density between the 2 chambers.13 As shown in Figure 3A, incorporation of the characteristics demonstrated for IK1 in the LV and RV into a computer-based ionic model of the cardiac ventricular myocytes13 reproduced a stable rotor with a rotation frequency of 35 Hz in the region with larger IK1. Fibrillatory conduction characterized the region of the model with right ventricular IK1 in which the outward component of the current is relatively low.
It should be noted at this point that recent experiments in neonatal ventricular cardiomyocyte monolayers have demonstrated a significant role of the slow component of the delayed rectifier current, IKs, in the mechanism of fibrillatory conduction.27 As illustrated in Figure 3B, fibrillatory conduction is manifest when the IKs density in the monolayer is increased in the presence of nominal IK1, an effect which may be attributed to post-repolarization refractoriness and spatially distributed wavebreaks.27
Further experiments used Ba2+ at relatively low concentrations (1–50 μM) to selectively block IK1 in the isolated, Langendorff-perfused guinea pig heart.28 The major finding was that Ba 2+ perfusion resulted in a dose dependent decrease in the frequency of reentry. At 50 μM, Ba2+ perfusion terminated VF.28 It was shown as well that Ba2+ caused a proportional decrease in the density of IK1 in isolated myocytes. Those experiments indicated that IK1 affected the stability and duration of reentry.
From the molecule to the cell and to the organ
A more recent study provided the first demonstration at the molecular level of the role played by IK1 in the control of the stability and frequency of rotors and of VF.29 Cardiac specific upregulation of IK1 in a transgenic mouse heart accelerated the final phase of AP repolarization, which significantly shortened the APD and the QT interval.30 During reentry, this translates into a shorter wavelength and relative membrane hyperpolarization, both of which contribute to greater Na channel availability during the excitable gap and thus to increased excitability ahead of the rotating wavefront. In addition, IK1 overexpression augments the voltage gradient established between resting cells in the core and the active cells in its immediate surroundings. These effects help to enhance the electrotonic currents that flow continuously between resting and active cells, which further contributes to hasten the repolarization of the active cells.29,31 The end result is a steeper rise in the local CV as a function of the distance from the core and a faster, more stable rotor in transgenic, compared to the wildtype hearts. Further, during reentry in the IK1 overexpressing hearts, the unexcited cells at the very center of the core provide a larger than normal outward conductance which decreases the likelihood of being excited by the depolarizing influence of their immediate, actively depolarized neighbors (sink-to-source mismatch), helping to reduce core size and meandering and to stabilize the rotor.
Recently, we used a combination of computer simulations and patch-clamp experiments in guinea pig myocytes and HEK cells to provide a more stringent test for the relationship between the IK1-induced changes in single cell properties and the Ba2+ induced changes in VF frequency in the whole heart.32 In that study, we defined a new electrophysiological parameter, the single cell frequency shift (Δfcell ), which was derived as the horizontal distance between individual plots of the phase difference between transmembrane current and transmembrane voltage in a single cell (i.e., the admittance phase) versus the VF activation frequency at 0, 10 and 50 μM Ba2+. The results showed that Δfcell responded to IK1 blockade in the same fashion as the effect of IK1 blockade on VF frequency. Biophysical analysis of those results demonstrated that although the rotation frequency does change as a result of IK1 blockade, the phase difference between transmembrane current and transmembrane voltage remains constant, which enabled us to quantitatively predict the change of VF frequency that results from IK1 blockade, based on single-cell measurement.32
Taken together, experimental data in isolated guinea pig and transgenic mouse hearts as well as computer simulations and single-cell to whole-heart correlations strongly argue that Ik1 is a stabilizer of reentry because of its ability to shorten APD and reducing CV near the center of rotation.10,13,29,31,32 Increased IK1 should prevent wavefront-wavetail interactions and thus prevent rotor destabilization and breakup. 10,13,30 In this regard, Ik1 is an important current during functional reentry because it mediates the electrotonic interactions between the unexcited core and its immediate surroundings.10,13,31,32 In addition, the interplay between IK1 and INa is a major factor in the control of cardiac excitability and therefore the stability and frequency of reentry29 These data open a new avenue for a search for, and careful selection of, new therapeutic agents capable of sufficiently reducing the outward component of IK1 to effectively prevent, or destabilize and terminate VF. On the other hand, as discussed above, IKs plays a role in wavebreak formation27, and is an important factor in fibrillatory conduction, but it does not significantly affect rotor frequency or stability.27 As such, IKs blockade would be expected to reduce wavebreak and VF complexity, and possibly convert VF to ventricular tachycardia, but arrhythmia termination would be less likely.
Acknowledgments
Supported by NHLBI Grants P01-HL039707, P01-HL087226; and R01-HL080159.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Myerburg RJ, Spooner PM. Opportunities for sudden death prevention: directions for new clinical and basic research. Cardiovasc Res. 2001;50:177–85. doi: 10.1016/s0008-6363(01)00253-x. [DOI] [PubMed] [Google Scholar]
- 2.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 3.Jalife J. Ventricular fibrillation: mechanisms of initiation and maintenance. Annu Rev Physiol. 2000;62:25–50. doi: 10.1146/annurev.physiol.62.1.25. [DOI] [PubMed] [Google Scholar]
- 4.Wiener NRA. The mathematical formulation of the problem of conduction of impulses in a network of connected excitable elements, specifically in cardiac muscle. Arch Inst Cardiol Mex. 1946;16:1–61. [PubMed] [Google Scholar]
- 5.Selfridge O. Studies on flutter and fibrillation. V. Some notes on the theory of flutter. Arch Inst Cardiol Mex. 1948;18:177–187. [PubMed] [Google Scholar]
- 6.Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res. 1977;41:9–18. doi: 10.1161/01.res.41.1.9. [DOI] [PubMed] [Google Scholar]
- 7.Krinsky VI, Biktashev VN, Pertsov AM. Autowave approaches to cessation of reentrant arrhythmias. Ann N Y Acad Sci. 1990;591:232–246. doi: 10.1111/j.1749-6632.1990.tb15092.x. [DOI] [PubMed] [Google Scholar]
- 8.Winfree AT. Electrical instability in cardiac muscle: phase singularities and rotors. J Theor Biol. 1989;138:353–405. doi: 10.1016/s0022-5193(89)80200-0. [DOI] [PubMed] [Google Scholar]
- 9.Jalife J, Berenfeld O. Molecular mechanisms and global dynamics of fibrillation: an integrative approach to the underlying basis of vortex-like reentry. J Theor Biol. 2004;230:475–487. doi: 10.1016/j.jtbi.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 10.Beaumont J, Jalife J. Rotors and spiral waves in two dimensions. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology from Cell to Bedside. Saunders; 2000. pp. 327–335. [Google Scholar]
- 11.Chen J, Mandapati R, Berenfeld O, Skanes AC, Gray RA, Jalife J. Dynamics of wavelets and their role in atrial fibrillation in the isolated sheep heart. Cardiovasc Res. 2000;48:220–232. doi: 10.1016/s0008-6363(00)00177-2. [DOI] [PubMed] [Google Scholar]
- 12.Garfinkel A, Qu Z. Nonlinear dynamics, spirals, and heart rhythms. In: Zipes D, Jalife J, editors. Cardiac Electrophysiology. From cell to bedside. 3. W.B. Saunders Company; 2000. pp. 315–327. [Google Scholar]
- 13.Samie FH, Berenfeld O, Anumonwo J, Mironov SF, Udassi S, Beaumont J, Taffet S, Pertsov AM, Jalife J. Rectification of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ Res. 2001;89:1216–1223. doi: 10.1161/hh2401.100818. [DOI] [PubMed] [Google Scholar]
- 14.Davidenko JM, Pertsov AV, Salomonsz R, Baxter W, Jalife J. Stationary and drifting spiral waves of excitation in isolated cardiac muscle. Nature. 1992;355:349–351. doi: 10.1038/355349a0. [DOI] [PubMed] [Google Scholar]
- 15.Rogers JM, Huang J, Melnick SB, Ideker RE. Sustained reentry in the left ventricle of fibrillating pig hearts. Circ Res. 2003;92:539–545. doi: 10.1161/01.RES.0000061569.23879.20. [DOI] [PubMed] [Google Scholar]
- 16.Huang J, Rogers JM, Killingsworth CR, Singh KP, Smith WM, Ideker RE. Evolution of activation patterns during long-duration ventricular fibrillation in dogs. Am J Physiol Heart Circ Physiol. 2004;286:H1193–H1200. doi: 10.1152/ajpheart.00773.2003. [DOI] [PubMed] [Google Scholar]
- 17.Nanthakumar K, Jalife J, Masse S, Downar E, Pop M, Asta J, Ross H, Rao V, Mironov SF, Sevaptsidis E, Rogers JM, Wright G, Dhopeshwarkar R. Optical Mapping of Langendorff Perfused Human Hearts: Establishing a Model for the Study of Ventricular Fibrillation in Humans. Am J Physiol Heart Circ Physiol. 2007;2931:H875–H880. doi: 10.1152/ajpheart.01415.2006. [DOI] [PubMed] [Google Scholar]
- 18.Nash MP, Mourad A, Clayton RH, Sutton PM, Bradley CP, Hayward M, Paterson DJ, Taggart P. Evidence for multiple mechanisms in human ventricular fibrillation. Circulation. 2006;114:536–542. doi: 10.1161/CIRCULATIONAHA.105.602870. [DOI] [PubMed] [Google Scholar]
- 19.Nanthakumar K, Walcott GP, Melnick S, Rogers JM, Kay MW, Smith WM, Ideker RE, Holman W. Epicardial organization of human ventricular fibrillation. Heart Rhythm. 2004;1:14–23. doi: 10.1016/j.hrthm.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Thomas SP, Thiagalingam A, Wallace E, Kovoor P, Ross DL. Organization of myocardial activation during ventricular fibrillation after myocardial infarction: evidence for sustained high-frequency sources. Circulation. 2005;112:157–163. doi: 10.1161/CIRCULATIONAHA.104.503631. [DOI] [PubMed] [Google Scholar]
- 21.Mandapati R, Asano Y, Baxter WT, Gray R, Davidenko J, Jalife J. Quantification of effects of global ischemia on dynamics of ventricular fibrillation in isolated rabbit heart. Circulation. 1998;98:1688–1696. doi: 10.1161/01.cir.98.16.1688. [DOI] [PubMed] [Google Scholar]
- 22.Cabo C, Pertsov AM, Baxter WT, Davidenko JM, Gray RA, Jalife J. Wave-front curvature as a cause of slow conduction and block in isolated cardiac muscle. Circ Res. 1994;75:1014–1028. doi: 10.1161/01.res.75.6.1014. [DOI] [PubMed] [Google Scholar]
- 23.Samie FH, Jalife J. Mechanisms underlying ventricular tachycardia and its transition to ventricular fibrillation in the structurally normal heart. Cardiovasc Res. 2001;50:242–250. doi: 10.1016/s0008-6363(00)00289-3. [DOI] [PubMed] [Google Scholar]
- 24.Kubo Y, Reuveny E, Slesinger PA, Jan YN, Jan LY. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature. 1993;364:802–806. doi: 10.1038/364802a0. [DOI] [PubMed] [Google Scholar]
- 25.Lopatin AN, Nichols CG. Inward rectifiers in the heart: an update on I(K1) J Mol Cell Cardiol. 2001;33:625–38. doi: 10.1006/jmcc.2001.1344. [DOI] [PubMed] [Google Scholar]
- 26.Dhamoon AS, Pandit SV, Sarmast F, Parisian KR, Guha P, Li Y, Bagwe S, Taffet SM, Anumonwo JM. Unique Kir2.x properties determine regional and species differences in the cardiac inward rectifier K+ current. Circ Res. 2004;94:1332–1339. doi: 10.1161/01.RES.0000128408.66946.67. [DOI] [PubMed] [Google Scholar]
- 27.Muñoz V, Grzeda KR, Desplantez T, Pandit SV, Mironov S, Taffet SM, Rohr S, Kléber AG, Jalife J. Adenoviral expression of IKs contributes to wavebreak and fibrillatory conduction in neonatal rat ventricular cardiomyocyte monolayers. Circ Res. 2007;101:475–483. doi: 10.1161/CIRCRESAHA.107.149617. [DOI] [PubMed] [Google Scholar]
- 28.Warren M, Guha PK, Berenfeld O, Zaitsev A, Anumonwo JM, Dhamoon AS, Bagwe S, Taffet SM, Jalife J. Blockade of the inward rectifying potassium current terminates ventricular fibrillation in the guinea pig heart. J Cardiovasc Electrophysiol. 2003;14:621–631. doi: 10.1046/j.1540-8167.2003.03006.x. [DOI] [PubMed] [Google Scholar]
- 29.Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN, Jalife J. Up-regulation of the inward rectifier K+ current (IK1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578:315–326. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, McLerie M, Lopatin AN. Transgenic Up-Regulation of IK1 in The Mouse Heart Leads to Multiple Abnormalities of Cardiac Excitability. Am J Physiol Heart Circ Physiol. 2004:H2790–H2802. doi: 10.1152/ajpheart.00114.2004. [DOI] [PubMed] [Google Scholar]
- 31.Beaumont J, Davidenko N, Davidenko JM, Jalife J. Spiral waves in two-dimensional models of ventricular muscle: formation of a stationary core. Biophys J. 1998;75:1–14. doi: 10.1016/S0006-3495(98)77490-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grzeda KR, Anumonwo JM, O’Connell R, Jalife J. A single-cell model of phase-driven control of ventricular fibrillation frequency. Biophys J. 2009;96:2961–2976. doi: 10.1016/j.bpj.2008.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]