

Abstract
Background & Aims
Chronic visceral hyperalgesia is considered an important pathophysiological symptom in irritable bowel syndrome (IBS); previous gastrointestinal inflammation is a potent etiological factor for developing IBS. Although there are several animal models of adult visceral hypersensitivity following neonatal perturbation or acute colonic inflammation, there is no suitable model of post-inflammatory chronic visceral hyperalgesia. The aim of this study was to establish a model of chronic visceral hyperalgesia following colonic inflammation in the rat.
Methods
Deoxycholic acid (DCA) was instilled into the rat colon daily for 3 days and animals were tested for up to 4 weeks.
Results
DCA induced mild, transient colonic inflammation within 3 days that resolved within 3 weeks. An exaggerated visceromotor response, referred pain to mechanical stimulation, increased spinal Fos expression, and colonic afferent and dorsal horn neuron activity were apparent by 1 week and persisted for at least 4 weeks, indicating chronic dorsal horn hyperexcitability and visceral hyperalgesia. There was no spontaneous pain, based on open field behavior. There was a significant increase in opioid receptor activity.
Conclusion
DCA induces mild, transient colitis, resulting in persistent visceral hyperalgesia and referred pain in rats, modeling some aspects of post-inflammatory IBS.
Keywords: visceral pain, hyperalgesia, deoxycholic acid (DCA), inflammation, colon, spinal cord, irritable bowel syndrome (IBS)
Introduction
Irritable bowel syndrome (IBS) is a functional, multifactorial disorder characterized by abdominal pain and altered bowel habits. Population-based surveys documented that IBS affects between 8 and 22% of the general population 1. In general, psychosocial factors, abnormal GI motility and secretion, and visceral hypersensitivity are thought to contribute to the symptoms of IBS. Visceral hypersensitivity is currently considered to be the most important pathophysiological factor in IBS and may partially result from the sensitization of primary afferent fibers innervating the gastrointestinal tract 2-4. However, central sensitization also contributes to the development of abdominal pain 2. As many as 26% of patients suffered from acute gastroenteritis that resolved prior to developing IBS 5,6. Therefore, establishing an appropriate chronic visceral hyperalgesia model that mimics characteristics of post-inflammatory IBS is of great clinical significance.
Several chemical irritants have been used to produce colonic inflammation resulting in visceral hyperalgesia. Acetic acid 7, mustard oil (MO) 8-12 and zymosan 13,14 evoke short-term hyperalgesia associated with transmural tissue damage/colonic inflammation. Dextran sulfate sodium induces colonic inflammation without visceral hypersensitivity 15-17. Intracolonic TNBS induces histological changes in the distal colon 24 hours after injection including severe colonic inflammation, necrosis, loss of myenteric neurons and a reduction of sensory terminals in the colon wall 18,19 and is considered a model for Crohn’s disease 20,21. This is accompanied by hyposensitivity to CRD within 2–3 days (unpublished observations) and hyperalgesia starting at 4–5 days, all symptoms diminishing by 14 days 22,23. However, hyperalgesia reappears at later time points in a subset of animals 8,24. Given the severe tissue damage and pattern of changes in visceral sensitivity, it cannot be ruled out that the delayed hypersensitivity is a form of neuropathic pain. Most recently butyrate enemas were reported to induce a sustained, concentration-dependent colonic hypersensitivity 25. However, there were no signs of macroscopic or histological tissue damage. Therefore, it can not be used as a model of post-inflammatory IBS.
Bile acids are derivatives of cholesterol synthesized in the hepatocytes. Deoxycholic acid (DCA), an unconjugated secondary bile acid, is produced in the colon from the salts of glycocholic and taurocholic acid converted by an action of anaerobic bacterial enzymes in the caecum/proximal colon. DCA acts as a detergent to solubilize fats for intestinal absorption and is cytotoxic to colonic epithelial cells inducing inflammatory changes within 12 to 48 hours after treatment at higher concentrations 21,26. Four mM DCA increases colonic mucosal permeability 27-29, causes colonic epithelial proliferation 30 and plays a role as colon cancer promoter 31,32. Bile salt malabsorption underlies some forms of post-infectious IBS 6. However, it has never been determined if colonic inflammation with DCA results in chronic visceral hyperalgesia.
The aim of this study was to establish an animal model of chronic visceral hyperalgesia post colonic inflammation. Our results suggest that DCA induces mild transient colitis and long term visceral hyperalgesia in rats, which may provide a suitable model for examining mechanisms underlying chronic visceral pain. Some of these data were presented in abstract form 33.
Methods and Materials
Animals
Adult male Sprague Dawley rats (240–320 g; Harlan Sprague Dawley, Indianapolis, IN) were used in this study. Animals were housed under a 12/12 hr light/dark cycle. Experiments were performed under approval of the University of Maryland Dental School Animal Care and Use Committee and conform to the ethical treatment of animals published by the International Association for the Study of Pain.
Induction of colonic inflammation
Rats were anesthetized with Nembutal (45 mg/kg i.p.). A gavage needle was inserted through the anus approximately 6 cm into the colon and 1 ml of 4 mM DCA in Kreb’s solution (in mmoles: NaCl, 122; KCl, 3.5; NaHCO3, 25; KH2PO4, 1.2; MgCl2, 1.2; pH 7.4) was injected while the needle was slowly withdrawn. Rats were left on a mound of bedding in a head down position to prevent leakage of DCA. Rats were injected once daily on 3 consecutive days, the first injection counting as Day 1. Rats in the control group received 1 ml 0.9 % saline instead of DCA.
Plasma extravasation
Plasma extravasation (PE) in the colon was determined with Evan’s blue dye. Rats injected with DCA or saline (1 and 4 weeks) were distended to 60 mmHg (VMR study). Subsequently, rats were anesthetized with Nembutal and injected with Evan’s blue (50 mg/kg, i.v.) for 10 min followed by perfusion with 150 ml saline. A 5 cm length of colon proximal to the level of the pubic symphysis was removed and placed in 5 ml dimethyl sulfoxide (DMSO) for 48 hours 34. The concentration of extracted Evan’s Blue was detected by spectrophotometer at 620 nm absorbance. Plasma extravasation was expressed as μg Evan’s Blue per gram of dry weight colon.
Myeloperoxidase assay (MPO)
Rats injected with DCA or saline (1,2,3,4 weeks) were distended (60 mmHg) and euthanized. Five cm of fresh colon tissue proximal to the pubic symphisis was removed. The distal half of each colon was rinsed briefly with saline, cut into smaller pieces, snap frozen and stored at −80°C until use. MPO activity was measured 48 hrs later. The proximal half was placed in 4% paraformaldehyde and used for histological examination (see below).
MPO assay
The tissue was weighed and placed in 0.5 ml of 0.5% hexadecyltrimethylammonium bromide (HTAB) in 50 mM phosphate buffer (PH 6.0). The tissue was minced with scissors for 20 sec on an ice-cold plate and then homogenized for 20 sec. Another 0.5 ml HTAB buffer was added to the homogenate, vortexed and centrifuged at 14000 rpm for 2 min. The supernatant (50 μl) was assayed for MPO activity by adding 1.45 ml O-dianisidine (0.167 mg/ml in 5mM phosphate buffer (pH 6.0), with 0.0005 % H2O2 added) and the absorbance was taken at 460 nm 4 times at 30 sec intervals. The MPO units (one unit of MPO is defined as 1 μmole H2O2 split in one minute which equals to 1.13 ×10−2 changes in absorbance) were normalized to the colon wet weight and expressed as MPO units/mg 35.
Histological examination of colonic samples
The proximal colon samples from above were fixed in 4% paraformaldehyde for 5 days and transferred to 30% sucrose. Sixteen micron transverse sections were mounted on glass slides, dehydrated in a graded ethanol series and stained with haematoxylin and eosin. A pathologist blinded to treatment counted three random fields at 400x and recorded the numbers of neutrophils located within the mucosa, submucosa and density within capillaries for each field.
Paraffin sections (6 μm) from the proximal half of the sample were cut and stained with toluidine blue. Mast cells were counted in 3 random sections at 200x.
Visceromotor response (vmr)
The visceromotor response is a pseudoaffective reflex contraction of the abdominal muscles in response to colorectal distention (CRD). Five days prior to starting the experiment, Teflon-coated 32 gauge stainless steel wire (Cooner Wire Company, Chatsworth, CA) made into EMG electrodes were implanted into the lateral abdominal wall. The electrode leads were exteriorized at the back of the neck 36. Rats were singly housed after surgery.
Rats were fasted overnight (with free access to water) in order to facilitate balloon placement. On the day of the experiment, rats were briefly sedated with isofluorane and a 5–6 cm balloon (made from the finger of a latex glove) attached to Tygon tubing was inserted through the anus into the rectum and descending colon, the distal end 1 cm proximal to the external anal sphincter. Rats were loosely restrained in Plexiglas tubes and allowed 30 min to recover. CRD was produced by inflating the balloon with air. Each trial consisted of five 60 mmHg distentions (20-s duration, 3-min interstimulus interval) or graded intensity stimulation trials (10, 20, 40, 60, 80 mmHg). Generally, 4 trials were run to achieve a stable response (less than 20% variability between the last two trials).
The EMG recordings were collected and analyzed with a CED Micro 1401 using Spike 2 for windows software (Cambridge Electronic Design, Cambridge, UK). The EMG was rectified, and the area under the curve (AUC) for the 20 sec before distention was subtracted from the AUC during the 20 sec distention to give the magnitude of the vmr.
Electrophysiology (spinal neurons and primary afferents)
Rats (1 or 4 weeks post DCA or saline) were anesthetized with Nembutal (50 mg/kg, i.p.) and maintained on 5–10 mg/kg/hr, i.v. Rats were respirated and paralyzed (pancuronium bromide, 0.2mg/kg/hr). The T12-S2 spinal cord was exposed by laminectomy and the response of primary afferent fibers or dorsal horn neurons to 80 mmHg CRD was recorded and analyzed in Spike 2.
Thoracolumbar (TL) and lumbosacral (LS) dorsal horn neurons were recorded 6–8 days and 4 weeks following injection of DCA or saline. Standard single unit recording techniques were used 34,37,38. Cells responding to 80 mmHg CRD were studied. The mean number of spikes per second (Hz) during a 20 sec distention was determined and compared between saline and DCA-treated rats.
Colonic afferent activity was recorded from fibers teased from the proximal end of the L6 or S1 dorsal roots cut close to the root entry zone in the spinal cord. Fibers were identified as colonic afferents if they responded to CRD and a single spike could be isolated with a window discriminator. Data were collected and analyzed as above.
Fos immunocytochemistry
Seven days or 4 weeks following DCA or saline injection, rats were distended for 2 hrs (30 sec on/90 sec off), overdosed with Nembutal and perfused through the heart with saline followed by 4% paraformaldehyde. Control rats were not distended. The T13-L2 and L6-S2 spinal cord segments were removed, postfixed overnight and cryoprotected in 30% sucrose. Thirty micron transverse sections were cut on a cryostat. Every fourth section through the T13-L2 and L6-S2 spinal cord segments was collected for immunocytochemical labeling for c-Fos using a modified ABC method. Free-floating sections were incubated sequentially in primary antisera (rabbit anti-Fos; 1:50,000, Oncogene Science, Cambridge, MA), biotinylated goat anti-rabbit IgG (1:600, Jackson Immuno Research Laboratories, Inc. West Grove, PA), ABC (Vector Laboratories, Burlingame, CA) and reacted in 2.5% nickel sulfate, 0.02% diaminobenzidine tetrahydrochloride (DAB) and 0.003% H2O2 in 0.175 M sodium acetate buffer for 3–4 min. Sections were washed, mounted on gelatin-subbed slides, cleared in alcohol and coverslipped from xylenes. The number of Fos labeled cells in 5 random sections from the T13-L2 and L6-S2 segments were counted by a blinded observer.
Determination of referred pain
Animals were tested for referred hyperalgesia weekly for 4 weeks. The lower back was shaved, rats placed in individual plastic boxes and following acclimation a calibrated series of von Frey filaments (Stoelting Co., Wood Dale, IL) ranging from 15.1 g down to 1.2 g were applied perpendicularly to the lower back. A brisk escape was considered a positive response. Each filament was tested 5 times for 1–2 seconds. If the rat had a positive response (at least one escape response), the filament of next lower force was applied until two filaments were tested without a positive response. The lowest filament that evoked a 40% response rate was considered threshold.
Open field test
Open field behavior to assess ongoing discomfort or anxiety was measured using an automated device (Med Associates Inc. St. Albans, VT) to detect animal movement. Before and 7 days after DCA or saline adminstration, rats were placed individually into the center of the field and locomotor activity was quantified during a 5 minute observation period. The overall distance traveled and the number of zone entries was calculated by the system.
Systemic morphine and naloxone
One week following injection of DCA or saline, rats were sedated with isofluorane, a distention balloon was placed in the colon and a polyethylene (PE)-10 catheter was subcutaneously implanted in the back of the neck for morphine (0.1–10 mg/kg) administration using a cumulative dosing paradigm or naloxone (1 mg/kg). Following establishment of a baseline visceromotor response, morphine or naloxone was administered s.c. and the vmr recorded. The interval between each dose of morphine was 20 min 39.
Statistics
The vmr data are reported as area under the curve (AUC) or percent baseline response (% baseline) as appropriate. Data were analyzed using unpaired or paired Student’s t-test and one-way or two-way RM ANOVA followed by a multiple comparison test. All the data are expressed as mean ± SEM. p< 0.05 was considered significant.
Results
Measurement of colonic inflammation
Several measures were used to evaluate colonic inflammation. DCA alone increased PE 3 and 7 days following injection (t-test, p<0.05; Figure 1A). CRD 1 week following DCA further increased PE compared to saline which resolved by 4 weeks (ANOVA, p<0.05; Figure 1B).
Figure 1.

DCA induces mild, transient colonic inflammation. A: Plasma extravasation 3 and 7 days following intracolonic injection of DCA or saline, no CRD; * p<0.05 vs. saline, n= 4–8 rats/ group. B: Plasma extravasation 1 and 4 weeks following DCA or saline plus CRD; * p<0.05, n= 12/group. C: MPO activity measured weekly following DCA or saline injection; *p<0.05 vs. saline, n=4/group. D: The number of polymorphonuclear neutrophils per field following DCA or saline, n=8/group. E: The number of Mast cells per section of colon following DCA or saline; *p<0.05 vs. saline, n= 4–8/group. F: The number of fecal pellets following DCA or saline injection; *p<0.05 vs. saline, n=11/group.
MPO activity was increased for 3 weeks following DCA injection (two-way ANOVA, p<0.005; Figure 1C) before returning to the level of saline-treated rats by 4 weeks. The apparent increase in MPO at 1 week in saline-treated rats was likely due to minor irritation from the 3 enemas. However, this did not result in visceral hypersensitivity (see below).
Histological changes in colonic tissue were assessed macroscopically and microscopically from the same animals as the MPO assay. There was no gross change in appearance of the colon wall in DCA-treated rats compared to controls at any time point. Neutrophil number did not differ between DCA and saline-treated rats (p>0.05; Figure 1D) or change over time (p>0.05). There was an increase in mast cell number for 1 week that was back to control levels by 2 weeks (Figure 1E). Taken together these data suggest that 4 mM DCA produces a mild inflammation of the colon wall for 2–3 weeks that is fully resolved by 4 weeks.
Fecal pellet output and rat weight
Fecal pellet output was measured as an index of intestinal motility on Days 4–7 (n=11) and body weight was recorded on Days 4, 7 and 14. There were no signs of diarrhea or bloody stools from the rats injected with DCA. There was a transient increase in fecal pellet output in the DCA group on Day 4 compared to baseline (one-way RM ANOVA, p<0.001; Figure 1F). In contrast, saline had no effect on fecal pellet output (one way RM ANOVA, p=0.320).
All rats gained weight at a comparable rate and there was no difference between DCA and saline-treated rats (ANOVA, p = 0.115).
Visceromotor response (vmr)
Colorectal sensitivity was tested by recording the vmr to noxious CRD (60 mmHg) before and 7 days after treatment with DCA or saline (Figure 2A). There was no change in the magnitude of the vmr in saline-treated rats, but a significant increase in the vmr in DCA-treated rats (two-way RM ANOVA, p<0.001). This represents a 76% increase in the magnitude of the vmr in DCA-treated rats compared with a 2% decrease in the saline-treated rats.
Figure 2.

DCA induces visceral hypersensitivity that persists beyond resolution of the inflammation. A: the magnitude of the vmr to 60 mmHg CRD before (baseline) and 1 week following saline or DCA injection, * p<0.001 vs. baseline, # p<0.01 vs. 1 week saline, n=12–16/group. B: Time course of visceral hypersensitivity to graded intensities of CRD in DCA-treated rats. * p<0.05 vs. all time points, n=10 rats.
The time course of the colorectal hyperalgesia was determined using graded intensities of CRD. Following a baseline measurement, the rats were injected with DCA and the vmr measured weekly for 4 weeks. DCA significantly increased colorectal sensitivity to noxious intensities of CRD (two-way RM ANOVA, p<0.001; Figure 2B), but not to innocuous intensities (10, 20 mmHg). There was a trend towards an increase in sensitivity to 40 mmHg which is approximately threshold for noxious distention.
Electrophysiology
Primary afferents
DCA increased the response of primary afferents in the LS spinal cord 1 and 4 weeks following injection compared to saline (two-way RM ANOVA, p<0.001, Figure 3A).
Figure 3.

DCA increases activity of colonic afferents and dorsal horn neurons. A: Effects of DCA on LS colonic afferents 1 and 4 weeks following injection, n=17–18 cells/group. * p<0.05, ** p<0.01, *** p<0.001 vs. saline; ++ p<0.01, +++ p<0.001 vs. 1 wk DCA. B: Effects of DCA on the response to CRD of TL and LS dorsal horn neurons 1 and 4 weeks following injection. * p<0.05 vs. saline, n= 8–14 cells/group.
Dorsal horn neurons with Abrupt responses to noxious CRD (80 mmHg) were compared between saline and DCA-treated rats 6–8 days and 4 weeks following injection. Similar to other models of acute colonic inflammation 37,40, DCA did not increase the response of Abrupt neurons in the LS spinal cord at either time tested. However, there was a significant increase in the response of Abrupt neurons in the TL spinal cord at one week that persisted at least 4 weeks (ANOVA, p<0.05; Figure 3B).
Fos expression in the spinal cord
TL and LS spinal cord sections from DCA or saline-treated rats with or without distention were labeled for Fos expression. DCA ± CRD significantly increased Fos expression in the TL and LS spinal segments by one week (ANOVA, p<0.001 for TL, p<0.001 for LS; Figure 4). In both the TL and LS spinal segments, DCA alone significantly increased Fos expression in the dorsal horn, suggesting that a mild inflammation produced by DCA is sufficient to induce Fos expression, comparable to CRD, in visceroceptive dorsal horn neurons. CRD in addition to colonic inflammation further increased Fos expression in the LS, but not TL, spinal cord. However, by 4 weeks the number of Fos labeled neurons returned to the level of CRD alone (saline at 1 week).
Figure 4.
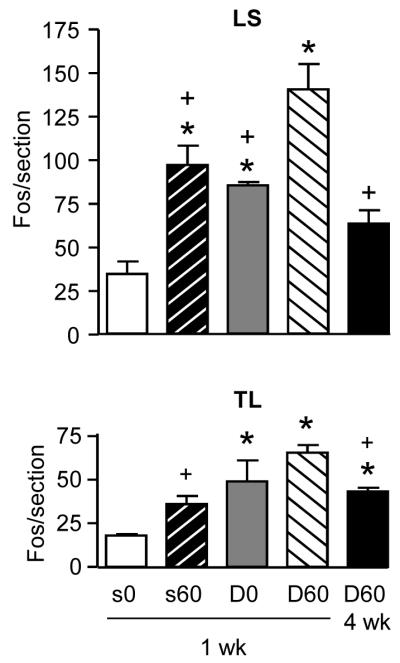
DCA increases Fos expression in the LS and TL spinal segments. Effect of DCA and CRD on Fos expression in the LS (top) and TL (bottom) spinal segments 1 and 4 weeks after injection. S0/D0: saline or DCA, no CRD; S60/D60: saline or DCA + 60 mmHg CRD. * p<0.05 vs. saline (s0). + p<0.05 vs. DCA + CRD at 1 week (D60), n=4–8/group.
Referred Pain
Probing the back over the hindlimbs proximal to the tail with von Frey filaments in normal rats evoked an escape response to 15.1 g (the greatest force tested) 35% of the time (Figure 5). In contrast, a 35% escape frequency was achieved with 6 g or less at all time points following DCA injection. This resulted in a significant decrease in the median threshold to evoke an escape response from 15.1 g at baseline to 3.6 g at 4 weeks post DCA (Friedman’s RM ANOVA on ranks, p<0.005).
Figure 5.

DCA induces referred pain. The % withdrawals to von Frey filaments applied to the caudal back following DCA, n=8 rats.
Open field test
To determine whether this model of chronic hyperalgesia is associated with anxiety-like behavior, we assessed the response to open-field exposure at baseline and 1 week following DCA administration. DCA had no effect on the exploratory behavior of the rats. There were no changes in total distance traveled (1644.35 ± 217.4 cm vs.1293.6 ± 629.5 cm, p = 0.303) or number of zone entries (213.7 ± 72.1 vs. 201.0 ± 113.5, p = 0.827) between DCA and saline-treated rats.
Morphine
Morphine dose-dependently attenuated the magnitude of the visceromotor response 1 week following injection of DCA or saline. There was a small, but significant difference in the dose-response curve to morphine between DCA and saline-treated rats (two-way ANOVA, p<0.05; Figure 6A).
Figure 6.

DCA increases opioid analgesia. A, Left: The magnitude of the visceromotor response (bar graph) before (b) and 1 week following injection of saline or DCA; * p<0.05. Right: Dose-response curves for morphine in saline and DCA-treated rats as the % maximum possible effect (%MPE); * p<0.05 vs. saline, n=8/group. B: Naloxone facilitated the vmr 1 week following DCA, but not saline, administration; * paired t-test, p<0.05 vs. baseline, n=4–6/group.
Naloxone (1mg/kg) had no effect on the visceromotor response of saline-treated rats. However, 1 week following DCA administration, naloxone significantly increased the magnitude of the vmr (paired t-test, p<0.05; Figure 6B).
Discussion
Deoxycholic acic (DCA) is an “unconjugated secondary bile acid” that induces colonic inflammation 21,26-29. Colonic lesions, when present, are confined to the surface epithelium, without causing disruption of the epithelial lining, thus avoiding severe colonic mucosal damage including penetrating ulcers, fissures, sinuses and fistulas which are characteristic morphological changes in inflammatory bowel diseases 27,41-43.
The present study shows repetitive administration of 4 mM DCA induces mild colonic inflammation and persistent visceral hyperalgesia. Inflammation as assessed by PE was present as early as 3 days after the start of DCA injections. During the first 3 weeks plasma extravasation and MPO activity increased. There was no increase in polymorphonuclear leukocyte infiltration and a transient increase in the number of mast cells. There was no evidence of ulceration or epithelial damage, nor was there diarrhea or rectal bleeding. The concentration of 3α-hydroxy bile acids in the cecum of normal individuals is 0.4±0.2 mM 44. This increases 10-fold in patients with bile malabsorption 45, a concentration similar to the present study. Patients present with diarrhea, but the short time DCA was present in this model only led to colonic afferent hypersensitivity and central sensitization. Taken together, these data suggest repetitive administration of 4 mM DCA induces a mild, short-lasting inflammatory response in the descending colon.
Although the inflammation was transient, DCA induced persistent visceral hyperalgesia. Hyperalgesia to 60 and 80 mmHg CRD was present within 1 week and persisted at least 4 weeks. There was however, no hypersensitivity to innocuous distention pressures (10 and 20 mmHg) and no change in exploratory behavior unlike other models of acute visceral inflammation and hyperalgesia 46,47. These data suggest that although there was visceral hyperalgesia to noxious colorectal distention, the animals were not experiencing inordinate spontaneous pain or discomfort.
The electrophysiology and Fos data suggest both peripheral and central changes contribute to the persistent visceral hyperalgesia. The persistent increase in colonic afferent activity at 4 weeks is consistent with primary afferent sensitization in patients with IBS 4. The discrepancy between the Fos data and neuronal activity of LS Abrupt visceroceptive neurons suggests colonic inflammation increases the number of visceroceptive neurons, perhaps by activating mechanically insensitive afferents, but does not necessarily increase the magnitude of response of Abrupt neurons, consistent with acute inflammatory models 37,40. However, LS Sustained neurons which were not studied have increased responses in acute inflammation models. In contrast, the inflammation-induced increase in TL dorsal horn Abrupt neuron activity that is maintained after resolution of the inflammation is consistent with the expanded role of the TL spinal cord in visceral hyperalgesia 12,14,38.
Spinal plasticity in this model is reinforced by the persistent referred mechanical hyperalgesia. The decreased threshold to evoke an escape response was maintained through 4 weeks, showing a trend towards greater sensitivity in the latter weeks. Referred hyperalgesia is qualitatively similar to secondary hyperalgesia; resulting from an increase in the response of dorsal horn neurons to mechanical stimulation of noninjured tissue. Referred pain involves viscerosomatic convergence indicating plasticity is at least at the level of the spinal cord, suggesting DCA-induced hyperalgesia involves central sensitization.
The molecular mechanisms underlying the effect of DCA on the visceral hypersensitivity are unknown. Colonic inflammation and subsequent abnormal neuroimmune interactions might contribute. Previous studies have shown that DCA elicited a proinflammatory phenotype through activation of kinase pathways, including protein kinase C 48, ERK 49, and c-jun NH2-terminal kinase 50. DCA dose-dependently increased the transcription factor early growth response factor-1 (Egr-1) protein in hepatocytes 51 and promoted endothelial activation through the stimulation of NF-κB and p38 MAPK signaling pathways 52. Therefore, it is conceivable that DCA induced visceral hyperalgesia through the alteration of neuroimmune interaction. The present data suggest that inflammation elicited by DCA may promote the release of inflammatory mediators, which sensitizes primary afferents, recruits silent nociceptors and triggers the release of algesic mediators. The sensitized primary afferents in turn induce central sensitization. Further study related to the molecular mechanisms remains to be carried out.
In conclusion, repetitive colorectal instillation of DCA leads to transient colonic inflammation, persistent visceral and referred mechanical hypersensitivity. The specific character of this model is that visceral hypersensitivity maintains at a stable and continuous level, which will be beneficial for the study of the mechanisms of chronic visceral pain. As this model mimics clinical manifestations of post inflammatory IBS it is of great importance in exploring the pathogenesis of IBS and for assessment of novel treatments.
Acknowledgements
This study was supported by National Institutes of Health Grants P01 NS 41384.
Supported by NIH P01 NS 41384.
Abbreviations
- DCA
Deoxycholic acid
- IBS
irritable bowel syndrome
Footnotes
No conflicts of interest exist.
Reference List
- 1.Rhodes DY, Wallace M. Post-infectious irritable bowel syndrome. Curr Gastroenterol Rep. 2006;8:327–332. doi: 10.1007/s11894-006-0054-0. [DOI] [PubMed] [Google Scholar]
- 2.Price DD, Zhou Q, Moshiree B, Robinson ME, Verne GN. Peripheral and central contributions to hyperalgesia in irritable bowel syndrome. J Pain. 2006;7:529–535. doi: 10.1016/j.jpain.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein CN, Niazi N, Robert M, Mertz H, Kodner A, Munakata J, Naliboff B, Mayer EA. Rectal afferent function in patients with inflammatory and functional intestinal disorders. Pain. 1996;66:151–161. doi: 10.1016/0304-3959(96)03062-x. [DOI] [PubMed] [Google Scholar]
- 4.Lembo T, Munakata J, Mertz H, Niazi N, Kodner A, Nikas V, Mayer EA. Evidence for the hypersensitivity of lumbar splanchnic afferents in irritable bowel syndrome. Gastroenterology. 1994;107:1686–1696. doi: 10.1016/0016-5085(94)90809-5. [DOI] [PubMed] [Google Scholar]
- 5.Parry S, Forgacs I. Intestinal infection and irritable bowel syndrome. Eur J Gastroenterol Hepatol. 2005;17:5–9. doi: 10.1097/00042737-200501000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Spiller R. Postinfectious irritable bowel syndrome. Gastroenterology. 2003;124:1662–1671. doi: 10.1016/s0016-5085(03)00324-x. [DOI] [PubMed] [Google Scholar]
- 7.Burton MB, Gebhart GF. Effects of intracolonic acetic acid on responses to colorectal distention in the rat. Brain Res. 1995;672:77–82. doi: 10.1016/0006-8993(94)01382-r. [DOI] [PubMed] [Google Scholar]
- 8.Adam B, Liebregts T, Gschossmann JM, Krippner C, Scholl F, Ruwe M, Holtmann G. Severity of mucosal inflammation as a predictor for alterations of visceral sensory function in a rat model. Pain. 2006;123:179–186. doi: 10.1016/j.pain.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 9.Kimball ES, Palmer JM, D’Andrea MR, Hornby PJ, Wade PR. Acute colitis induction by oil of mustard results in later development of an IBS-like accelerated upper GI transit in mice. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1266–G1273. doi: 10.1152/ajpgi.00444.2004. [DOI] [PubMed] [Google Scholar]
- 10.Palecek J, Willis WD. The dorsal column pathway facilitates visceromotor responses to colorectal distention after colon inflammation in rats. Pain. 2003;104:501–507. doi: 10.1016/S0304-3959(03)00075-7. [DOI] [PubMed] [Google Scholar]
- 11.Laird JM, Souslova V, Wood JN, Cervero F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J Neurosci. 2002;22:8352–8356. doi: 10.1523/JNEUROSCI.22-19-08352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Traub RJ. Evidence for thoracolumbar spinal cord processing of inflammatory, but not acute colonic pain. NeuroReport. 2000;11:2113–2116. doi: 10.1097/00001756-200007140-00011. [DOI] [PubMed] [Google Scholar]
- 13.Coutinho SV, Meller ST, Gebhart GF. Intracolonic zymosan produces visceral hyperalgesia in the rat that is mediated by spinal NMDA and nonNMDA receptors. Brain Res. 1996;736:7–15. doi: 10.1016/0006-8993(96)00661-0. [DOI] [PubMed] [Google Scholar]
- 14.Traub RJ, Murphy AZ. Colonic inflammation induces Fos expression in the thoracolumbar spinal cord increasing activity in the spinoparabrachial pathway. Pain. 2002;95:93–102. doi: 10.1016/s0304-3959(01)00381-5. [DOI] [PubMed] [Google Scholar]
- 15.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 16.Larsson MH, Rapp L, Lindstrom E. Effect of DSS-induced colitis on visceral sensitivity to colorectal distension in mice. Neurogastroenterol Motil. 2006;18:144–152. doi: 10.1111/j.1365-2982.2005.00736.x. [DOI] [PubMed] [Google Scholar]
- 17.Gaudio E, Taddei G, Vetuschi A, Sferra R, Frieri G, Ricciardi G, Caprilli R. Dextran sulfate sodium (DSS) colitis in rats: clinical, structural, and ultrastructural aspects. Dig Dis Sci. 1999;44:1458–1475. doi: 10.1023/a:1026620322859. [DOI] [PubMed] [Google Scholar]
- 18.Linden DR, Couvrette JM, Ciolino A, McQuoid C, Blaszyk H, Sharkey KA, Mawe GM. Indiscriminate loss of myenteric neurones in the TNBS-inflamed guinea-pig distal colon. Neurogastroenterol Motil. 2005;17:751–760. doi: 10.1111/j.1365-2982.2005.00703.x. [DOI] [PubMed] [Google Scholar]
- 19.Miampamba M, Sharkey KA. Distribution of calcitonin gene-related peptide, somatostatin, substance P and vasoactive intestinal polypeptide in experimental colitis in rats. Neurogastroenterol Motil. 1998;10:315–329. doi: 10.1046/j.1365-2982.1998.00111.x. [DOI] [PubMed] [Google Scholar]
- 20.Kruschewski M, Foitzik T, Perez-Canto A, Hubotter A, Buhr HJ. Changes of colonic mucosal microcirculation and histology in two colitis models: an experimental study using intravital microscopy and a new histological scoring system. Dig Dis Sci. 2001;46:2336–2343. doi: 10.1023/a:1012334727509. [DOI] [PubMed] [Google Scholar]
- 21.Wargovich MJ, Eng VW, Newmark HL, Bruce WR. Calcium ameliorates the toxic effect of deoxycholic acid on colonic epithelium. Carcinogenesis. 1983;4:1205–1207. doi: 10.1093/carcin/4.9.1205. [DOI] [PubMed] [Google Scholar]
- 22.Lamb K, Zhong F, Gebhart GF, Bielefeldt K. Experimental colitis in mice and sensitization of converging visceral and somatic afferent pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G451–G457. doi: 10.1152/ajpgi.00353.2005. [DOI] [PubMed] [Google Scholar]
- 23.Gschossmann JM, Adam B, Liebregts T, Buenger L, Ruwe M, Gerken G, Mayer EA, Holtmann G. Effect of transient chemically induced colitis on the visceromotor response to mechanical colorectal distension. Eur J Gastroenterol Hepatol. 2002;14:1067–1072. doi: 10.1097/00042737-200210000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Q, Price DD, Caudle RM, Verne GN. Visceral and somatic hypersensitivity in a subset of rats following TNBS-induced colitis. Pain. 2008;134:9–15. doi: 10.1016/j.pain.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourdu S, Dapoigny M, Chapuy E, Artigue F, Vasson MP, Dechelotte P, Bommelaer G, Eschalier A, Ardid D. Rectal Instillation of Butyrate Provides a Novel Clinically Relevant Model of Noninflammatory Colonic Hypersensitivity in Rats. Gastroenterology. 2005;128:1996–2008. doi: 10.1053/j.gastro.2005.03.082. [DOI] [PubMed] [Google Scholar]
- 26.Lapre JA, Termont DS, Groen AK, Van der MR. Lytic effects of mixed micelles of fatty acids and bile acids. Am J Physiol. 1992;263:G333–G337. doi: 10.1152/ajpgi.1992.263.3.G333. [DOI] [PubMed] [Google Scholar]
- 27.Chadwick VS, Gaginella TS, Carlson GL, Debongnie JC, Phillips SF, Hofmann AF. Effect of molecular structure on bile acid-induced alterations in absorptive function, permeability, and morphology in the perfused rabbit colon. J Lab Clin Med. 1979;94:661–674. [PubMed] [Google Scholar]
- 28.Sun Y, Fihn BM, Sjovall H, Jodal M. Enteric neurones modulate the colonic permeability response to luminal bile acids in rat colon in vivo. Gut. 2004;53:362–367. doi: 10.1136/gut.2003.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Y, Fihn BM, Jodal M, Sjovall H. Inhibition of nitric oxide synthesis potentiates the colonic permeability increase triggered by luminal bile acids. Acta Physiol Scand. 2004;180:167–175. doi: 10.1046/j.0001-6772.2003.01226.x. [DOI] [PubMed] [Google Scholar]
- 30.Morotomi M, Guillem JG, LoGerfo P, Weinstein IB. Production of diacylglycerol, an activator of protein kinase C, by human intestinal microflora. Cancer Res. 1990;50:3595–3599. [PubMed] [Google Scholar]
- 31.Nair PP, Davis KE, Shami S, Lagerholm S. The induction of SOS function in Escherichia coli K-12/PQ37 by 4-nitroquinoline oxide (4-NQO) and fecapentaenes-12 and -14 is bile salt sensitive: implications for colon carcinogenesis. Mutat Res. 2000;447:179–185. doi: 10.1016/s0027-5107(99)00205-5. [DOI] [PubMed] [Google Scholar]
- 32.Martinez JD, Stratagoules ED, LaRue JM, Powell AA, Gause PR, Craven MT, Payne CM, Powell MB, Gerner EW, Earnest DL. Different bile acids exhibit distinct biological effects: the tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferation. Nutr Cancer. 1998;31:111–118. doi: 10.1080/01635589809514689. [DOI] [PubMed] [Google Scholar]
- 33.Sun Y, Traub RJ. A model of chronic inflammatory visceral pain induced by deoxycholic acid; Neuroscience Meeting Planner; Atlanta, GA: Society for Neuroscience. 2006; Online 2006;Program No. 346.6. [Google Scholar]
- 34.Ji Y, Tang B, Traub RJ. Estrogen increases and progesterone decreases behavioral and neuronal responses to colorectal distention following colonic inflammation in the rat. Pain. 2005;117:433–442. doi: 10.1016/j.pain.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 35.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 36.Traub RJ, Zhai QZ, Ji Y, Kovalenko M. NMDA receptor antagonists attenuate noxious and nonnoxious colorectal distention-induced Fos expression and the visceromotor reflex. Neurosci. 2002;113:205–211. doi: 10.1016/s0306-4522(02)00170-7. [DOI] [PubMed] [Google Scholar]
- 37.Wang G, Tang B, Traub RJ. Differential processing of noxious colonic input by thoracolumbar and lumbosacral dorsal horn neurons in the rat. J Neurophysiol. 2005;94:3788–3794. doi: 10.1152/jn.00230.2005. [DOI] [PubMed] [Google Scholar]
- 38.Wang G, Tang B, Traub RJ. Pelvic nerve input mediates descending modulation of homovisceral processing in the thoracolumbar spinal cord of the rat. Gastroenterology. 2007;133:1544–1553. doi: 10.1053/j.gastro.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji Y, Murphy AZ, Traub RJ. Sex differences in morphine induced analgesia of visceral pain are supraspinally and peripherally mediated. Am J Physiol Regul Integr Comp Physiol. 2006;291:R307–R314. doi: 10.1152/ajpregu.00824.2005. [DOI] [PubMed] [Google Scholar]
- 40.Ness TJ, Gebhart GF. Acute inflammation differentially alters the activity of two classes of rat spinal visceral nociceptive neurons. Neurosci Lett. 2000;281:131–134. doi: 10.1016/s0304-3940(00)00832-6. [DOI] [PubMed] [Google Scholar]
- 41.Saunders DR, Hedges JR, Sillery J, Esther L, Matsumura K, Rubin CE. Morphological and functional effects of bile salts on rat colon. Gastroenterology. 1975;68:1236–1245. [PubMed] [Google Scholar]
- 42.Henrikson CK, Argenzio RA, Liacos JA, Khosla J. Morphologic and functional effects of bile salt on the porcine colon during injury and repair. Lab Invest. 1989;60:72–87. [PubMed] [Google Scholar]
- 43.Breuer NF, Rampton DS, Tammar A, Murphy GM, Dowling RH. Effect of colonic perfusion with sulfated and nonsulfated bile acids on mucosal structure and function in the rat. Gastroenterology. 1983;84:969–977. [PubMed] [Google Scholar]
- 44.Hamilton JP, Xie G, Raufman JP, Hogan S, Griffin TL, Packard CA, Chatfield DA, Hagey LR, Steinbach JH, Hofmann AF. Human cecal bile acids: concentration and spectrum. Am J Physiol Gastrointest Liver Physiol. 2007;293:G256–G263. doi: 10.1152/ajpgi.00027.2007. [DOI] [PubMed] [Google Scholar]
- 45.McJunkin B, Fromm H, Sarva RP, Amin P. Factors in the mechanism of diarrhea in bile acid malabsorption: fecal pH--a key determinant. Gastroenterology. 1981;80:1454–1464. [PubMed] [Google Scholar]
- 46.Palecek J, Paleckova V, Willis WD. The roles of pathways in the spinal cord lateral and dorsal funiculi in signaling nociceptive somatic and visceral stimuli in rats. Pain. 2002;96:297–307. doi: 10.1016/S0304-3959(01)00459-6. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Zhang X, Westlund KN. Restoration of spontaneous exploratory behaviors with an intrathecal NMDA receptor antagonist or a PKC inhibitor in rats with acute pancreatitis. Pharmacol Biochem Behav. 2004;77:145–153. doi: 10.1016/j.pbb.2003.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stravitz RT, Rao YP, Vlahcevic ZR, Gurley EC, Jarvis WD, Hylemon PB. Hepatocellular protein kinase C activation by bile acids: implications for regulation of cholesterol 7 alpha-hydroxylase. Am J Physiol. 1996;271:G293–G303. doi: 10.1152/ajpgi.1996.271.2.G293. [DOI] [PubMed] [Google Scholar]
- 49.Qiao D, Chen W, Stratagoules ED, Martinez JD. Bile acid-induced activation of activator protein-1 requires both extracellular signal-regulated kinase and protein kinase C signaling. J Biol Chem. 2000;275:15090–15098. doi: 10.1074/jbc.M908890199. [DOI] [PubMed] [Google Scholar]
- 50.Gupta S, Stravitz RT, Dent P, Hylemon PB. Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J Biol Chem. 2001;276:15816–15822. doi: 10.1074/jbc.M010878200. [DOI] [PubMed] [Google Scholar]
- 51.Kim ND, Moon JO, Slitt AL, Copple BL. Early growth response factor-1 is critical for cholestatic liver injury. Toxicol Sci. 2006;90:586–595. doi: 10.1093/toxsci/kfj111. [DOI] [PubMed] [Google Scholar]
- 52.Qin P, Tang X, Elloso MM, Harnish DC. Bile acids induce adhesion molecule expression in endothelial cells through activation of reactive oxygen species, NF-kappaB, and p38. Am J Physiol Heart Circ Physiol. 2006;291:H741–H747. doi: 10.1152/ajpheart.01182.2005. [DOI] [PubMed] [Google Scholar]