

Abstract
MicroRNAs (miRNAs) are evolutionarily conserved, 18-25 nucleotide non-protein coding transcripts that play an important function in post-transcriptional regulation of gene expression during development (1-4). However, the significance of miRNAs in postmitotic cells, such as neurons in the mammalian CNS, is less well characterized. Here we investigate the role of miRNAs in the terminal differentiation, function, and survival of mammalian midbrain dopaminergic neurons (DNs). We identify a miRNA, miR-133b, that is specifically expressed in midbrain DNs and deficient in Parkinson's disease midbrain tissue that has lost midbrain DNs. MiR-133b regulates the maturation and function of midbrain DNs within a negative feedback circuit that includes the paired-like homeodomain transcription factor Pitx3.
Keywords: miRNA, miR-133b, midbrain dopamine neurons, Parkinson's disease, Dicer
miRNAs are derived from long primary transcripts through sequential processing by the Drosha ribonuclease (5) and the Dicer enzyme (1, 6). In the context of an RNA-induced silencing complex (RISC), miRNAs guide the cleavage of target mRNAs and/or inhibit their translation (2). miRNAs were first characterized in invertebrates, where they function to regulate developmental cell fate decisions in the nervous system (7, 8) and elsewhere (9).
Midbrain dopamine neurons (DNs) play a central role in complex behaviors such as reward and addiction, and these cells are lost in Parkinson's disease. Furthermore, a number of transcription factors have been identified that regulate midbrain DN development, function, and survival (16). However, the role of post-transcriptional mechanisms in these processes is uncharacterized. We sought to establish a role for miRNAs in mammalian dopamine neuron differentiation, function, and survival. To facilitate a kinetic analysis, we first used an in vitro model system: the differentiation of murine ES cells into DNs (17, 18). An ES cell line was obtained that expresses Dicer enzyme containing LoxP recombinase sites that flank both chromosomal copies of the Dicer gene (floxed Dicer alleles)(19). Introduction of Cre recombinase into these cells by lentiviral transduction leads to the deletion of Dicer in nearly 100% of cells (Supplementary Figure 1A). Floxed Dicer ES cultures were differentiated to a midbrain DN phenotype using the embryoid body protocol (EB; Supplementary Figure 1B)(18). Briefly, cells were initially grown in non-adherent conditions in the context of defined media, including growth factors, to generate neuronal precursors (stage 2); subsequently, neuronal precursors were expanded in the presence of basic fibroblast growth factor (bFGF; stages 3 and 4); and finally, the bFGF was withdrawn to obtain mature DNs (stage 5), which constitute 10-25% of the cells in these cultures (18).
Cre-mediated deletion of the floxed Dicer alleles at stage 4, when postmitotic dopamine neurons first arise, led to a nearly complete loss of dopamine neuron accumulation at stage 5, as quantified by the expression of markers including tyrosine hydroxylase (TH; Figure 1A). Other mature neuronal classes, including GABAergic neurons, were reduced in these cultures to a lesser extent (by approximately 50%), as were cells expressing TujI, an early general neuronal marker that first appears at the neural precursor stage of EB differentiation (stage 4). Cre lentivirus transduction had no affect on wild-type cells lacking the floxed alleles (Supplementary Figure 1C). The reduction in dopamine neuron accumulation appeared to be the result of both increased neuronal apoptosis, as demonstrated by the appearance of apoptotic markers including activated caspase-3 and TUNEL staining, as well as reduced neurogenesis, as quantified by BrdU incorporation (Figure 1B). The Dicer deletion phenotype is significantly rescued by transfection of low molecular weight RNA species (but not high molecular weight RNA species) derived from embryonic mouse midbrain, consistent with a model in which miRNAs play a role in midbrain dopamine neuron terminal differentiation and survival (Figure 1C and Supplementary Figure 1D).
Figure 1.
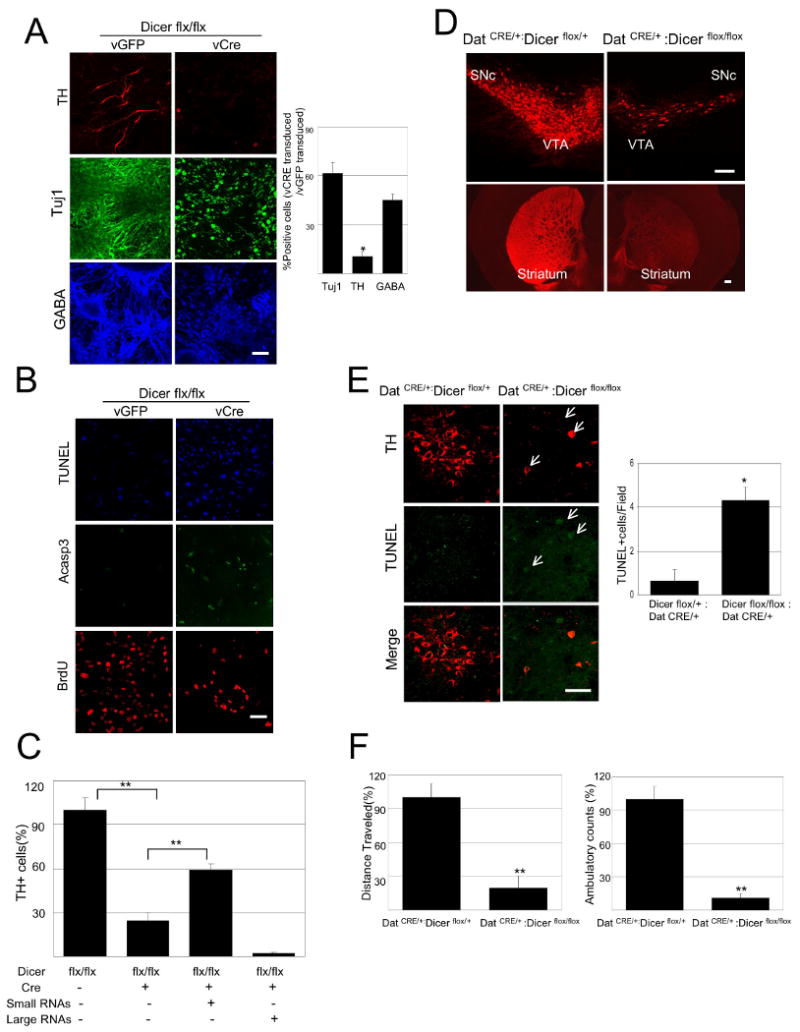
Dicer is essential for terminal differentiation of ES cells to a DN phenotype.
(A) Floxed Dicer conditional knockout cells were transduced with CRE lentivirus at EB stage 4 and analyzed by immunostaining at day 7 of stage 5 with antibodies specific for TH (Red), TujI (Green) and GABA (blue). Cultures transduced with a lentiviral Cre vector (vCre) are essentially devoid of TH+ neurons, whereas TujI+ and GABA+ cells are reduced by approximately 40%-60%. (Scale bars, 100μm). (n=3 independent samples per group; 2 additional independent sets of samples showed consistent results). Data represent means ± S.E.M. ANOVA-test, *p < 0.05.
(C) The Dicer deletion phenotype, as in (A) can be ‘rescued’ by transfection of midbrain-derived small RNAs (<200bp) not large RNAs (>200bp) (Two independent experiments of 3 sets each with 10 visual fields per set), Data represent means ± S.E.M. ANOVA-test, **p < 0.01.
(D) Immunostaining of brain sections from 8 week old DATCRE/+ :Dicerflox/flox mice for tyrosine hydroxylase (TH) demonstrates loss of approximately 90% of midbrain DNs in the substantia nigra (SN) and ventral tegmental area (VTA) and their axonal projections to the striatum relative to control littermates (DATCRE/+ : Dicerflox/+; N=3 for each genotype). (Scale bars,200μm)
(F) Locomotor activity of DATCRE/+ :Dicerflox/flox mice in the open field. The total distance traveled during the test was significantly decreased in DATCRE/+ :Dicerflox/flox mice. (N=4 for each genotype), Data represent means ± S.E.M. Student's t test, *p < 0.05, **p < 0.01.
To extend these findings to the intact rodent CNS, mice were generated that are homozygous for the floxed Dicer allele and express Cre recombinase under the regulation of dopamine transporter regulatory sequences (DATCRE/+ :Dicerflox/flox; (19)), thus leading to the specific deletion of Dicer in postmitotic dopamine neurons (20). These mice display a progressive loss of midbrain DNs and of their nigrostriatal axonal processes, as quantified by TH and DAT immunostaining (Figure 1D). This loss is first apparent at 2 weeks of age and is nearly complete by 6 weeks of age (Supplementary Figures 1E and 1F). Further analysis of the substantia nigra of mutant mice revealed the induction of apoptosis, as determined by TUNEL staining and nuclear condensation (Figure 1E).
Behavioral studies of mice that harbor a midbrain dopamine neuron-specific deletion of Dicer revealed dramatically reduced locomotion in an open field assay (Figure 1F). Furthermore, this is mostly a consequence of long periods of immobility, which is reminiscent of the phenotype of human PD patients. Taken together, these results indicate that Dicer is essential for the terminal differentiation and maintenance of multiple neuron types, including midbrain DNs.
We next sought to identify specific miRNAs that function in midbrain DNs. A limitation to standard approaches for the purification of miRNAs from midbrain DNs is that these cells typically represent a minor population within tissue or primary culture preparations. To circumvent this, we took a subtractive approach and compared miRNA expression profiles of normal adult midbrain with the profiles of midbrain depleted of dopamine neurons. We used three independent models to this end: adult Aphakia mice deficient in the transcription factor Pitx3 (21-23); adult human Parkinson's disease (PD) patients; and mice treated with the dopamine neuron-specific toxin 6-hydroxydopamine (6-OHDA)(18). These models were validated by real-time quantitative PCR (qPCR) on cDNA preparations, demonstrating a dramatic reduction in midbrain DN marker expression (Supplementary Figure 2A).
Expression analyses were performed by quantitative real-time rtPCR (qPCR) for a panel of approximately 230 miRNAs precursors in midbrain, cerebellum, and cerebral cortex samples from PD patients and normal controls (Supplementary Figures 2C and 2D and Supplementary Table 1). Within this miRNA panel, 8 appeared to be enriched in midbrain relative to cerebral cortex or cerebellum. Expression of one of these precursor miRNA, miR-133b, was specifically deficient in the context of PD patient samples, as confirmed by RNase protection assays and Northern blotting for mature miR-133b (Figure 2A). Additional miRNAs within the panel were found to be specifically enriched in the midbrain, cerebellum or cerebral cortex, but were not altered in the PD patient samples.
Figure 2.
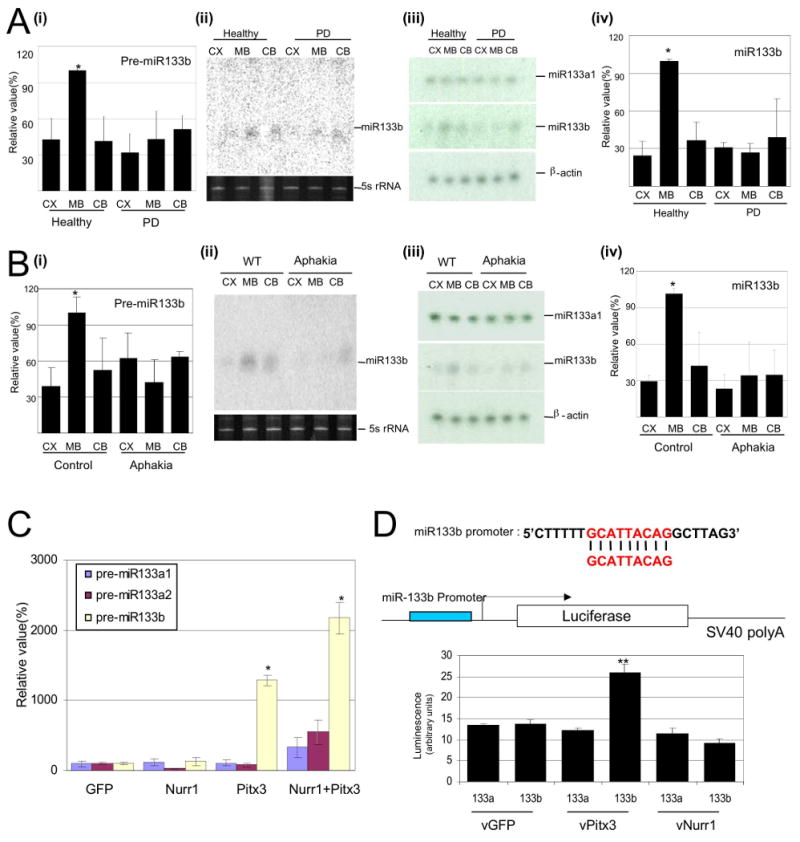
miR-133b is enriched in midbrain and is deficient in PD samples.
(A) Expression analysis for precursor miR-133b in cerebral cortex (CX), midbrain (MB), or cerebellum (CB) of healthy human and Parkinson's disease (PD) patient brain. RNA protection assay for miR133a1 and miR133b expression from normal or PD human brain and quantification of RNA protection assay. n=3 independent experiments; data represent means ± S.E.M. ANOVA test, *p < 0.05.
(B) Expression analysis for precursor miR-133b in cerebral cortex (CX), midbrain (MB), or cerebellum (CB) of control mice and Aphakia mutant. RNA protection assay for miR133a1 and miR133b expression from control mice and Aphakia mutant mice brain and quantification of RNA protection assay. n=3 independent experiments; data represent means ± S.E.M. ANOVA test, *p < 0.05.
(C) qPCR analysis of murine ES cultures differentiated by EB and transduced at stage 4 with viral vectors for Nurr1, Pitx3, both, or GFP vector control. Pitx3 transduction (but not Nurr1 transduction) leads to the specific induction of miR-133b precursor expression assayed at stage 5; miR-133a1 and miR-133a2 precursors are not induced by Pitx3 overexpression. n=3 independent experiments; data represent means ± S.E.M. ANOVA test, *p < 0.05.
We investigated expression of miR-133b in 2 additional dopamine neuron deficiency models: Pitx3 mutant Aphakia mice and 6-OHDA-treated mice. miR-133b was specifically expressed in midbrain of normal mice, as in humans, and expression was dramatically reduced in both rodent dopamine deficiency models as demonstrated by qPCR, RNase protection assays, and Northern blotting (Figure 2B and Supplementary Figure 2B).
The relative deficiency of miR-133b expression in Aphakia midbrain was surprising, as adult Aphakia mice do maintain a population of midbrain dopamine neurons within the ventral tegmental area (VTA)(22); this suggested the possibility that miR-133b is a direct target of Pitx3 transcription activation (as Pitx3 is mutated in Aphakia mice). Consistent with this model, overexpression of Pitx3 in stage 4 murine ES cultures led to a dramatic up-regulation of miR-133b precursor expression (Figure 2C). Expression of a luciferase reporter gene that harbors 350 bp of proximal miR-133b promoter sequence is specifically induced by over-expression of Pitx3 in COS cells (Figure 2D). Taken together, these data suggest that Pitx3 regulates miR-133b expression in midbrain DNs.
Next, we sought to investigate the role of miR-133b in midbrain DN maturation, function, and survival. A lentiviral vector was generated that harbors miR-133b precursor sequences. Transduction of COS cells with this vector, but not a control vector (miR-18 or GFP), inhibits expression of a luciferase reporter that harbors a precise predicted target sequence for miR-133b within its 3′ untranslated sequences (Supplementary Figures 3A and 3B). We investigated the consequences of increased miR-133b expression in either ES cell-derived cultures or in primary embryonic day 14.5 (E14.5) midbrain cultures. miR-133b precursor overexpression induced a relative reduction in the transcription of the late midbrain DN maturation marker DAT, although transcription of early midbrain DN markers, such as Pitx3 and Nurr1, appeared unaltered or increased (Figure 3A). Transcription of TH, which is a relatively early marker of midbrain dopamine neurons (18), showed a trend towards reduction that did not reach statistical significance. Consistent with this, dopamine release in the context of potassium-induced depolarization was markedly reduced with miR-133b overexpression in either ES cell cultures or in primary midbrain cultures (Figure 3B and Supplementary Figures 3C and 3D). Overexpression of miR-133b at the neural precursor stage (stage 4) of EB-differentiated ES cultures led to a significant reduction in the number of TH-positive (but not TujI-positive) cells present at stage 5 (Figure 3C).
Figure 3.
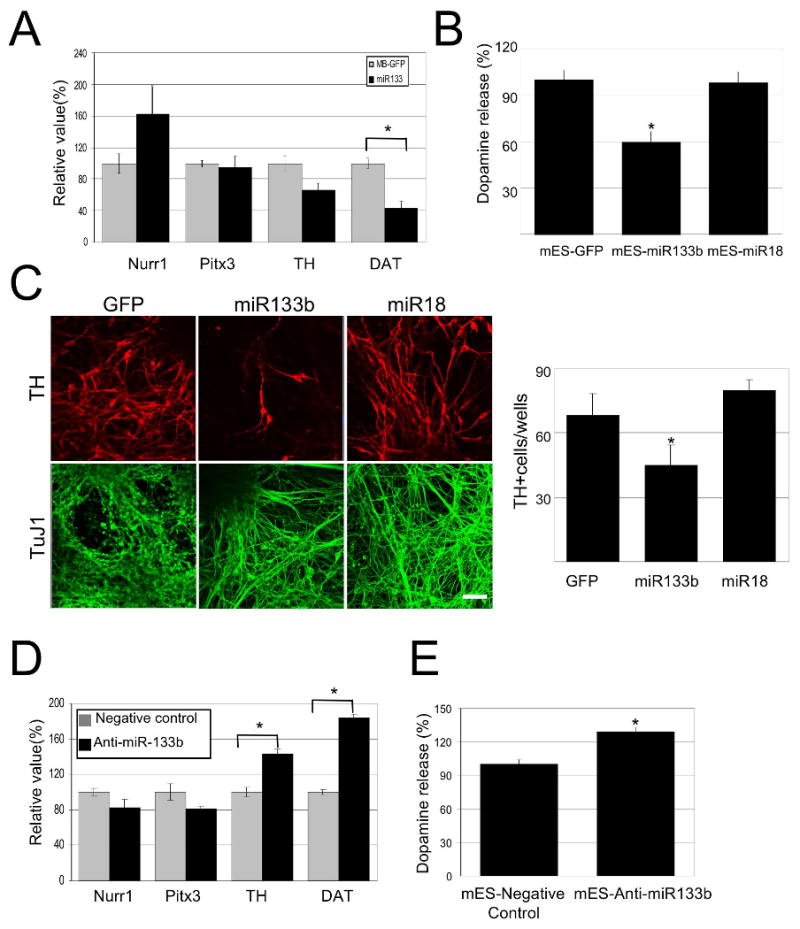
miR-133b suppresses dopamine neuron terminal differentiation and function.
(A) Overexpression of miR-133b precursor in primary embryonic rat midbrain cultures leads to decreased expression of dopamine neuron mRNAs including TH and DAT, whereas Nurr1 and Pitx3 mRNAs are not altered. Data represent means ± S.E.M. n=3 independent experiments; Student's t test, *p < 0.05.
(B) Depolarization-induced dopamine release was quantified in cultures transduced with miR-133b precursor vector or GFP control. miR-133b overexpression reduced dopamine release in murine ES cultures derived dopamine neurons. Data represent means ± S.E.M. n=5; ANOVA test, *p < 0.05.
(C) Overexpression of miR-133b during EB stage 4 of mouse ES cell differentiation resulted in a significant decrease in TH-positive cells. Data represent means ± S.E.M. n=3 independent experiments; ANOVA test, *p < 0.05.
(D) Knockdown of miR-133b by penetratin conjugated antisense miR133b 2OM oligo in primary embryonic rat midbrain cultures leads to increased expression of dopamine neuron mRNAs including TH and DAT, whereas Nurr1 and Pitx3 mRNAs are not significantly altered. Data represent means ± S.E.M. n=5 independent experiments; Student's t test, *p < 0.05.
(E) Depolarization-induced dopamine release was quantified in cultures with miR-133b knockdown or oligonucleotide control. miR-133b knockdown induced dopamine release in murine ES cultures. Data represent means ± S.E.M. n=3 independent experiments; Student's t test, *p < 0.05.
The activity of miR-133b can be inhibited using a 2′OMe-modified RNA oligonucleotide homologous to the miR-133b sequence and linked to a short peptide derived from the Drosophila Antennapedia protein that mediates cell transduction (15, 24)(Supplementary Figure 3A). Suppression of miR-133b in ES cell EB differentiation cultures induced expression of dopamine neuron markers including DAT and TH (quantified by qPCR analyses; Figure 3D). In ES-derived cultures, transduction of the miR-133b inhibitory oligonucleotide potentiated potassium-stimulated dopamine release (Figure 3E). Finally, inhibition of miR-133b at stage 4 of EB differentiation led to a trend with apparent increase in the number of TH+ cells, but this did not reach statistical significance (Supplementary Figure 3E). Taken together, these data implicate miR-133b in the regulation of midbrain dopamine neuron maturation and function.
Individual microRNAs appear to regulate the expression of numerous targets post-translationally (4). To identify potential physiological targets for miR-133b activity, we used available miRNA target prediction programs based on 3′ untranslated sequence homology to miR-133b (25, 26). Interestingly, the Pitx3 3′-untranslated region (UTR) was identified as a potential target of miR-133b activity (Supplementary Figure 4A). To test whether Pitx3 3′ UTR sequences are subject to regulation by miR-133b activity in vivo, we placed these sequences downstream of a luciferase reporter gene. miR-133b overexpression inhibited luciferase accumulation in the context of the Pitx3 3′-UTR sequences, whereas this inhibition was abrogated in the context of Pitx3 3′UTR sequences mutated in the miR-133b target sequence (Figure 4A).
Figure 4.
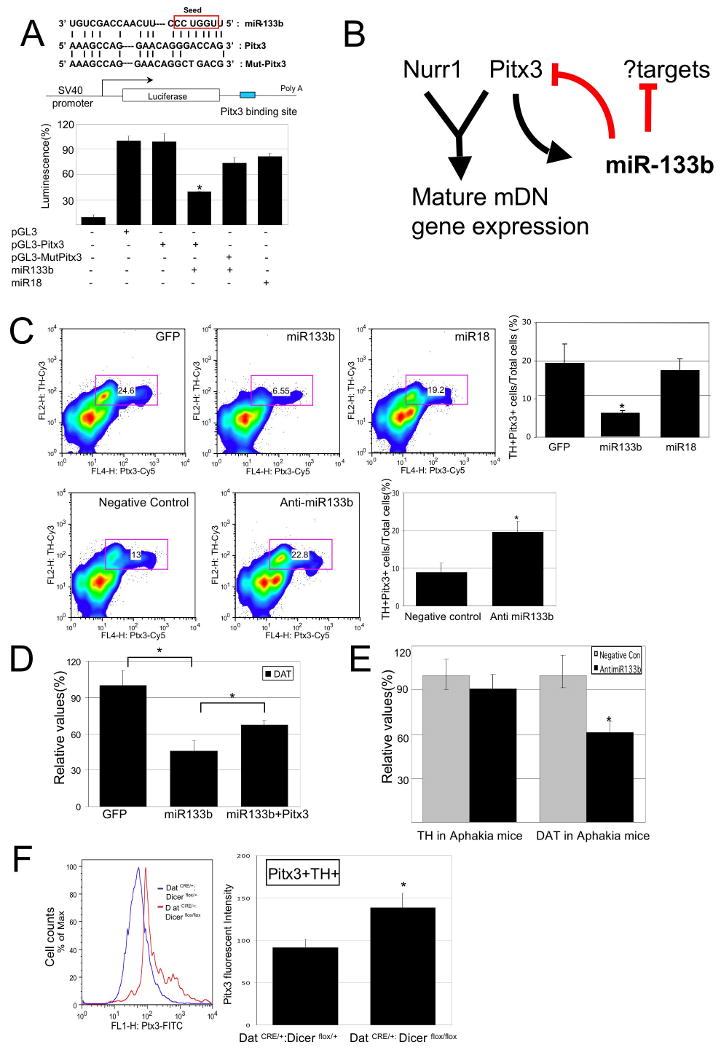
Pitx3 is a target of miR-133b activity.
(A) Upper Panel: Schematic of a luciferase assay vector, pGL3-Pitx3 and pGL3-Mut-Pitx3 that harbors 300bp Pitx3 3′-UTR sequences predicted to be subject to miR-133b regulation.
Lower panel: Overexpression of miR-133b (but not miR-18) in 293 cells decreases luciferase expression from pGL3-Pitx3 (but not control vector and pGL3-mut Pitx3). Data represent means ± S.E.M. n=3 independent experiments; Student's t test, *p < 0.05.
(B) miR-133b and Pitx3 define a negative feedback loop in midbrain DN function and differentiation. It is likely that miR-133b has additional significant targets.
(C) Upper panels: FACS analysis of primary midbrain cultures transduced with miR-133b or control vectors using antibodies for Pitx3 and TH reveals that miR-133b overexpression decrease in both Pitx3 and TH protein expression. Pitx3 is reduced in TH-positive cells transduced with miR-133b relative to control vector or miR-18. Data represent means ± S.E.M. n=3 independent experiments; ANOVA test, *p < 0.05.
Lower panels: Knockdown of miR-133b using modified oligonucleotides leads to a significant induction in Pitx3 and TH protein levels relative to control oligonucleotides. Data represent means ± S.E.M. n=3 independent experiments; Student's t test, *p < 0.05.
(E) miR-133b inhibition by modified oligonucleotides in Pitx3-deficient Aphakia primary neuron cultures fails to induce TH or DAT transcription. Data represent means ± S.E.M. n=3 independent experiments; Student's t test, *p < 0.05.
A hypothetical model for the observed phenotypes associated with altered miR-133b expression is that miR-133b functions within a negative feedback circuit that normally suppresses Pitx3 expression post-transcriptionally (Figure 4B), and, in turn, Pitx3 activates midbrain DN gene expression (18, 27) and induces transcription of miR-133b. This model is supported by the finding that downstream targets of Pitx3 activation, including TH and DAT, are modified in their transcription in the context of miR-133b mis-expression (Figures 3A and 3D). Fluorescence activated cell sorter (FACS) analysis of permeablized primary rat midbrain cells with an antibody that recognizes Pitx3 revealed that miR-133b overexpression induced a reduction in Pitx3 protein expression in TH+ cells, whereas miR-133b knockdown led to an increase in Pitx3 protein in TH+ cells (Figure 4C). Of note, Pitx3 mRNA transcript levels do not appear altered by miR-133b misexpression (Figures 3A and 3D), consistent with post-transcriptional regulation.
If Pitx3 is a significant direct target of miR-133b, one prediction is that overexpression of a Pitx3 transgene lacking 3′-UTR regulatory elements would ‘rescue’ the reduced midbrain DN marker expression phenotype associated with miR-133b overexpression. Consistent with this, miR-133b induced reduction in DAT transcription can be partially suppressed by overexpression of Pitx3 lacking the 3′UTR miR-133b target sequences (Figure 4D). Also consistent with this model, miR-133b inhibition by modified oligonucleotide transduction fails to induce TH and DAT expression in Pitx3-deficient Aphakia primary neuron cultures (Figure 4E). Finally, FACS analysis on acutely dissociated midbrain DNs from young Dicer mutant mice (10-day old, derived from DATCRE/+ :Dicerflox/flox mice) revealed that Pitx3 protein expression is upregulated in miRNA deficient TH-positive neurons (relative to control DATCRE/+ : Dicerflox/+ cells; Figures 4F and Supplementary Figure 4B), further indicating a role for miRNA in Pitx3 regulation.
In summary, our data supports a model in which miR-133b functions within a feedback loop, as Pitx3 specifically induces transcription of miR-133b, and Pitx3 activity is downregulated by miR-133b post-transcriptionally (Figure 4B). Such a feedback loop has recently been described in the context of cell cycle regulation (28). Midbrain dopamine neuron function is dynamic, and consistent with this, expression of TH, DAT, and other enzymes is tightly regulated and highly inducible (29, 30). Negative feedback circuitry has been shown to increase the robustness and to speed response time and stability in the context of dynamic changes (31).
Methods
See Supplementary Material
Acknowledgments
We thank Oliver Hobert, William Dauer, and Ron Liem for comments and critical reading of the manuscript. This work was funded by the NIH, NINDS, Spitzer, and Kenner Funds.
References
- 1.He L, Hannon GJ. Nat Rev Genet. 2004 Jul;5:522. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 2.Meister G, Tuschl T. Nature. 2004 Sep 16;431:343. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 3.Ambros V. Nature. 2004 Sep 16;431:350. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. Cell. 2004 Jan 23;116:281. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y, et al. Nature. 2003 Sep 25;425:415. [Google Scholar]
- 6.Grishok A, et al. Cell. 2001 Jul 13;106:23. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 7.Chang S, Johnston RJ, Jr, Frokjaer-Jensen C, Lockery S, Hobert O. Nature. 2004 Aug 12;430:785. doi: 10.1038/nature02752. [DOI] [PubMed] [Google Scholar]
- 8.Johnston RJ, Hobert O. Nature. 2003 Dec 18;426:845. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- 9.Ambros V. Cell. 2003 Jun 13;113:673. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 10.Giraldez AJ, et al. Science. 2005 May 6;308:833. doi: 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- 11.Giraldez AJ, et al. Science. 2006 Apr 7;312:75. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- 12.Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS. Rna. 2003 Oct;9:1274. doi: 10.1261/rna.5980303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kosik KS, Krichevsky AM. Neuron. 2005 Sep 15;47:779. doi: 10.1016/j.neuron.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 14.Krichevsky AM, Sonntag KC, Isacson O, Kosik KS. Stem Cells. 2006 Apr;24:857. doi: 10.1634/stemcells.2005-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schratt GM, et al. Nature. 2006 Jan 19;439:283. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- 16.Wallen A, Perlmann T. Ann N Y Acad Sci. 2003 Jun;991:48. doi: 10.1111/j.1749-6632.2003.tb07462.x. [DOI] [PubMed] [Google Scholar]
- 17.Kim JH, et al. Nature. 2002 Jul 4;418:50. [Google Scholar]
- 18.Martinat C, et al. Proc Natl Acad Sci U S A. 2006 Feb 21;103:2874. doi: 10.1073/pnas.0511153103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ. Proc Natl Acad Sci U S A. 2005 Aug 23;102:12135. doi: 10.1073/pnas.0505479102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chuhma N, et al. J Neurosci. 2004 Jan 28;24:972. doi: 10.1523/JNEUROSCI.4317-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang DY, Ardayfio P, Kang UJ, Semina EV, Kim KS. Brain Res Mol Brain Res. 2003 Jun 10;114:123. doi: 10.1016/s0169-328x(03)00162-1. [DOI] [PubMed] [Google Scholar]
- 22.Nunes I, Tovmasian LT, Silva RM, Burke RE, Goff SP. Proc Natl Acad Sci U S A. 2003 Apr 1;100:4245. doi: 10.1073/pnas.0230529100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van den Munckhof P, et al. Development. 2003 Jun;130:2535. doi: 10.1242/dev.00464. [DOI] [PubMed] [Google Scholar]
- 24.Davidson TJ, et al. J Neurosci. 2004 Nov 10;24:10040. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.John B, et al. PLoS Biol. 2004 Nov;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Cell. 2003 Dec 26;115:787. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 27.Smidt MP, Smits SM, Burbach JP. Cell Tissue Res. 2004 Oct;318:35. doi: 10.1007/s00441-004-0943-1. [DOI] [PubMed] [Google Scholar]
- 28.Sylvestre Y, et al. J Biol Chem. 2007 Jan 26;282:2135. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 29.Kumer SC, Vrana KE. J Neurochem. 1996 Aug;67:443. doi: 10.1046/j.1471-4159.1996.67020443.x. [DOI] [PubMed] [Google Scholar]
- 30.Lebel M, Gauthier Y, Moreau A, Drouin J. J Neurochem. 2001 Apr;77:558. doi: 10.1046/j.1471-4159.2001.00257.x. [DOI] [PubMed] [Google Scholar]
- 31.Becskei A, Serrano L. Nature. 2000 Jun 1;405:590. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- 32.Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. Nat Neurosci. 2006 Mar;9:443. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]