

Abstract
A search for general regulators of cancer metastasis has yielded a set of microRNAs for which expression is specifically lost as human breast cancer cells develop metastatic potential. Here we show that restoring the expression of these microRNAs in malignant cells suppresses lung and bone metastasis by human cancer cells in vivo. Of these microRNAs, miR-126 restoration reduces overall tumour growth and proliferation, whereas miR-335 inhibits metastatic cell invasion. miR-335 regulates a set of genes whose collective expression in a large cohort of human tumours is associated with risk of distal metastasis. miR-335 suppresses metastasis and migration through targeting of the progenitor cell transcription factor SOX4 and extracellular matrix component tenascin C. Expression of miR-126 and miR-335 is lost in the majority of primary breast tumours from patients who relapse, and the loss of expression of either microRNA is associated with poor distal metastasis-free survival. miR-335 and miR-126 are thus identified as metastasis suppressor microRNAs in human breast cancer.
Although metastasis is the overwhelming cause of mortality in patients with solid tumours, our understanding of its molecular and cellular determinants is limited1-3. Transcriptional profiling has revealed sets of genes, or `signatures', for which expression in primary tumours correlates with metastatic relapse or poor survival4. Some of these genes endow cancer cells with a more invasive phenotype, enhanced angiogenic and intravasation activity, the ability to exit from the circulation, or an ability to modify the metastasis microenvironment5,6. Such gene sets are thus providing numerous candidate mediators of metastasis to be validated through functional and clinical studies. Much less insight, however, has been gained into the regulatory networks that establish such altered gene expression states7. MicroRNAs (miRNAs) are attractive candidates as upstream regulators of metastatic progression because miRNAs can post-transcriptionally regulate entire sets of genes8,9. Recent work has revealed important roles for miRNAs and miRNA processing in tumorigenesis10-12. Large sets of miRNAs are underexpressed in human tumours compared to normal tissues13, and interfering with miRNA processing enhances experimental tumorigenesis11. These findings suggest that this class of regulators contains suppressors of tumour progression and possibly metastasis. Therefore, we sought to identify human miRNAs that affect breast cancer metastasis to lung and bone—the two main sites of metastatic spread in this highly prevalent form of cancer.
Identification of miRNAs that suppress metastasis
We performed array-based miRNA profiling14 of MDA-MB-231 human breast cancer cell derivatives that are highly metastatic to bone (1833 or 2287; denoted BoM1 lines) or lung (4175, 4173, 4180 and 4142; LM2 lines), as well as the parental, unselected MDA-MB-231 cell population. Out of 453 human miRNAs assayed, 179 miRNAs were expressed above background levels in one or more of the highly metastatic cells (data not shown). Notably, hierarchical clustering based on the expression of these miRNAs correctly classified the MDA-MB-231 derivatives into three groups comprising the BoM1 lines, the LM2 lines and the parental lines, respectively.
Hierarchical clustering on the basis of the expression of the 20 miRNAs for which expression was most altered across various cell lines again correctly classified the MDA-MB-231 derivatives into these three groups (Fig. 1a). The most salient finding was a set of eight miRNAs for which expression was decreased across all metastatic sub-lines compared with their expression in the parental line (Fig. 1a). Through quantitative stem-loop polymerase chain reaction (qRT-PCR), we were able to validate the differential expression of seven of these miRNAs in MDA-MB-231 and representative LM2 and BoM1 sub-lines (Supplementary Fig. 1).
Figure 1. Systematic identification of miRNAs that suppress lung and bone metastasis in multiple human breast cancer cell derivatives.

a, Hierarchical clustering of normalized miRNA expression levels for the 20 miRNAs that displayed the highest coefficients of variation clusters the MDA-MB-231 derivatives into three groups comprised of the bone metastatic BM2 lines (2287 and 1833), the lung metastatic LM2 lines (4180, 4173, 4175, and 4142) and the parental MDA lines. The heatmap highlights a set of miRNAs whose expression decreases across all lung and bone metastatic derivatives (red bracket along the right). The scale bar across the bottom depicts standard deviation change from the mean. b, Bioluminescence imaging of lung metastasis by lung metastatic breast cancer cells with restored expression of specific miRNAs. 1 × 104 LM2 cells expressing individual miRNAs or the control hairpin, as well as the parental MDA-MB-231 cells, were inoculated intravenously into immunodeficient mice. Shown are representative mice corresponding to the LM2 set (top) and the MDA-MB-231 set (bottom) at day 50. Lung colonization was measured by bioluminescence and quantified. n = 5; error bars represent s.e.m.; asterisk, P < 0.05. c, Human-vimentin-stained images of representative lungs that emitted the median luciferase signal for each cohort. Lungs were extracted at 8 weeks after xenografting and representative images of sections were obtained at whole-field magnification. d,2 × 104 BM2 cells expressing individual miRNAs or the control hairpin were inoculated into the arterial circulation via intracardiac injection of athymic mice. Bone metastasis was measured by bioluminescence and quantified as the normalized hindlimb photon flux. Shown are representative mice corresponding to marked data points. n = 6-10; horizontal line represents median signal for each cohort; P-values based on a one-tailed rank-sum test. e, Representative whole-field magnification images of haematoxylin- and eosin-stained femurs extracted from representative mice 7 weeks after intraventricular injection of indicated cancer cells. f, 2 × 105 primary human cancer derivative CN34-LM1 cells expressing individual miRNAs or the control vector were inoculated intravenously into immunodeficient mice. Lung colonization was measured by bioluminescence, quantified and normalized. n = 8; error bars represent s.e.m.; P-values based on a one-sided rank-sum test. g, 2 × 105 primary human cancer derivative CN34-BoM1 cells expressing individual miRNAs or the control vector were inoculated into the arterial circulation via intracardiac injection of immunodeficient mice. Bone metastasis was measured by bioluminescence, quantified and normalized. n = 9-10; error bars represent s.e.m.; P-values based on a one-tailed rank-sum test.
We focused on the six miRNAs (miR-335, miR-126, miR-206, miR-122a, miR-199a*, miR-489) whose expression was most decreased in metastatic cells on the basis of the combination weight of fold change in hybridization and PCR-based detection methods. Restoring the expression of miR-335, miR-126 or miR-206 in LM2 cells through retroviral transduction10,15 (Supplementary Fig. 2) decreased the lung colonizing activity of these cells by more than fivefold (Fig. 1b). Restoration of miR-122a, miR-199a* or miR-489 expression decreased lung colonization at early time points but did not result in a significant decrease in lung colonization at the end point (Supplementary Fig. 3). Histological assessment of lungs revealed a marked decrease in the number of metastatic foci where bioluminescence imaging revealed a significant reduction (Fig. 1c and Supplementary Fig. 4). The decreased expression of these candidate miRNAs in both lung and bone metastatic cells suggested a role for these molecules in general metastatic activity of breast cancer cells. Indeed, expression of miR-335, miR-206 or miR-126 significantly decreased bone metastasis formation as assessed by bioluminescence imaging and histological analysis of hindlimbs subsequent to intracardiac injection of BoM1 cells into the arterial circulation (Fig. 1d, e).
Selective pressure for specific miRNA loss
Consistent with a selective pressure against these regulators during the metastatic process, quantitative PCR of rare metastatic foci revealed a reduction in expression of all three metastasis suppressor miRNAs in cells that had metastasized relative to the inoculated population (Supplementary Fig. 5a). Restoration of miR-335 or miR-206 expression in LM2 cells also significantly reduced their metastatic dissemination from the primary mammary tumour site (Supplementary Fig. 5b). miR-126 was not included in this assay because of its inhibitory effect on mammary tumour growth (see below).
To determine whether the expression of these miRNAs is lost in other human breast cancer cells with enhanced metastatic propensity, we inoculated mice with a purified population of malignant cells (CN34) obtained from the pleural fluid of a patient with metastatic breast cancer who was treated at our institution6. We then isolated human tumour cells from metastatic lesions that formed in the lungs (CN34-LM1 cells) and bones (CN34-BoM1 cells) of the mice. Both the CN34-LM1 and CN34-BoM1 derivatives displayed a loss of miR-335, miR-126 and miR-206 expression relative to the parental population from which they were isolated (Supplementary Fig. 5c). miR-335, miR-206 and miR-126 significantly reduced the ability of CN34-LM1 and CN34-BoM1 cells to metastasize to lung (Fig. 1f and Supplementary Fig. 5d) and bone (Fig. 1g), respectively. The expression of these miRNAs, therefore, is lost in multiple, independently derived, breast cancer samples and their restoration suppresses lung and bone metastasis in these distinct metastatic cell populations.
Distinct mechanisms of metastasis suppression
Cell proliferation, survival and migration are among the common functions required by tumour cells for metastatic progression in target microenvironments. Of the three miRNAs that suppressed metastasis, only miR-126 significantly suppressed overall tumour growth, as assessed by tumour volume (Fig. 2a and Supplementary Fig. 6). Immunohistochemistry on these mammary tumours revealed a decrease in proliferation (Supplementary Fig. 7a, b), but not apoptosis (Supplementary Fig. 7c). Consistent with this, restoration of miR-126 reduced the proliferation rate of LM2 cells as well as CN34-BoM1 cells in vitro (Fig. 2b). miR-126, therefore, suppresses tumorigenesis and metastasis, in part, through an inhibition of cancer cell proliferation.
Figure 2. miR-126 suppresses overall tumour growth and proliferation whereas miR-335 and miR-206 regulate migration and morphology.
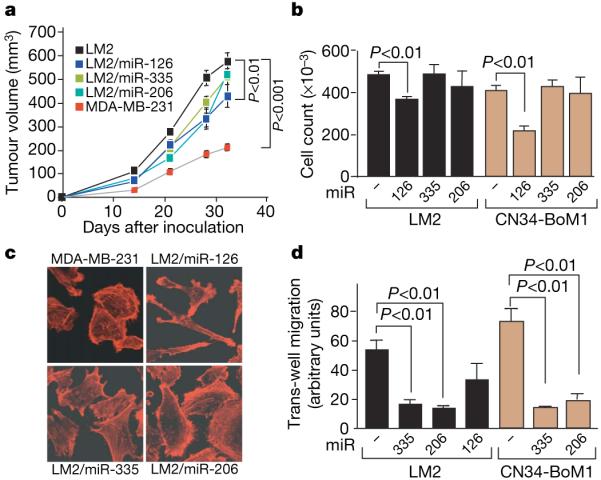
a, 5 × 105 LM2 cells expressing individual miRNAs or the control hairpin, as well as the parental MDA-MB-231 cells, were injected into the mammary fat pads of immunodeficient mice and tumour volumes were measured over time. n = 5 (MDA-MB-231, LM2/miR-126, LM2/miR-206) and n = 10 (LM2, LM2/miR-335); error bars indicate s.e.m.; P-values based on a one-sided Student's t-test at day 32. b, 5 × 104 LM2 or primary breast cancer line CN34-BoM1 expressing miR-126, miR-335 or miR-206 and control cells were seeded in triplicate and viable cells were counted at 5 days after seeding. n = 3; error bars represent s.e.m.; P-values obtained using a one-sided Student's t-test. c, Parental MDA-MB-231 cells and lung metastatic LM2 cells expressing miR-126, miR-335 or miR-206 were seeded onto glass slides. Cells were stained with the actin marker phalloidin and confocal images were obtained. d, 2.5 × 104 LM2 and CN34-BoM1 cells were transduced with the indicated miRNAs or a control hairpin, and trans-well migration was assessed. Images of cells that had migrated through trans-well inserts were obtained and analysed in automated fashion using Metamorph software. n = 3; error bars represent s.e.m.; P-values obtained using a one-sided Student's t-test.
In contrast to miR-126, restoring the expression of miR-335 or miR-206 did not alter the proliferation or apoptotic rates of LM2 cells in vivo or in vitro but it induced an altered morphology. LM2 cells and CN34-BoM1 cells expressing miR-335 or miR-206 as a population contained a significantly lower fraction of elongated cells than did control cells (Fig. 2c and Supplementary Fig. 8). Despite these morphological changes, LM2 cells expressing these miRNAs continued to express the mesenchymal marker vimentin, in vivo, suggesting that the suppression of metastasis in these cells was not due to classical mesenchymal-epithelial transition (Supplementary Fig. 9). We postulated that such an alteration in shape may be associated with a decrease in cell motility, which would limit metastatic migration. Indeed, in trans-well migration assays, LM2 cells and CN34-BoM1 cells expressing miR-335 or miR-206 displayed a significant reduction in migration compared with controls (Fig. 2d). miR-335 and miR-206 also caused a significant reduction in invasive capacity, as assessed through matrigel invasion assays (Supplementary Fig. 10). These findings highlight cell-autonomous mechanisms of cell proliferation, migration and invasion through which specific miRNAs may suppress metastasis.
MicroRNA expression in clinical metastasis
To determine whether these miRNAs are associated with human metastasis, we determined by qRT-PCR the expression levels of miR-335 and miR-126 in 20 archived primary breast tumours. These tumours comprised large (>2.5 cm) and small (<2.5 cm) oestrogen receptor (ER)-positive and -negative subtypes that were surgically resected before administration of chemotherapy. This set consisted of primary tumours resected from 11 patients that ultimately relapsed to lung, bone, or brain as well as tumours resected from nine patients who did not suffer metastatic relapse (Supplementary Table 1). qRT-PCR (Supplementary Fig. 11) revealed that patients whose primary tumours displayed low expression of miR-335, miR-126 (Fig. 3a) or miR-206 (Supplementary Fig. 12) had a shorter median time to metastatic relapse. Notably, the low expression levels of miR-335 (P 0.0022; median survival of 1.84 yr; hazard ratio (ratio of the predicted hazard (metastasis) for a member of the control group compared to that for a member of the test group) of 8.95) or miR-126 (P 0.0156; median survival of 2.57 yr; hazard ratio of 5.08) were associated with very poor overall metastasis-free survival compared to the group whose tumours expressed a high level of these miRNAs. The median expression values of these miRNAs was more than eightfold lower in tumours from patients that ultimately relapsed compared to tumours from patients that did not relapse (Supplementary Fig. 11). The expression levels of these miRNAs were not significantly correlated with ER status or HER-2 (also called ERBB2) amplification status (Supplementary Table 1). The group of patients whose primary tumours had a low level of miR-122a or miR-199a expression, two miRNAs that do not suppress metastasis, showed no difference in metastasis-free survival compared to those with high expression (Fig. 3a and Supplementary Fig. 11). These findings uncover a significant association between the loss of metastasis suppressor miRNA expression in primary human breast tumours and the likelihood of future distal metastatic recurrence.
Figure 3. Clinical association of miR-335 and miR-126 with metastasis-free survival.

a, miR-335, miR-126, miR-122a and miR-199a expression was assessed in a set of 20 primary breast tumour samples through qRT-PCR. Kaplan-Meier curves depict metastasis-free survival of patients whose primary tumours contained low or high levels of the indicated miRNAs. P-values were obtained using a log-rank test. b, The parental MDA-MB-231 cells were transfected with antagomirs targeting endogenous miR-335, miR-199a* or a control antagomir. Four days after transfection, 1 × 104 LM2 cells from each cohort were inoculated intravenously into immunodeficient mice. Lung colonization was measured by bioluminescence at day 35 and quantified. n = 5; error bars represent s.e.m.; P-values based on a one-sided rank-sum test.
miR-335 regulates a set of metastasis genes
Given the strong association between the loss of miR-335 expression and clinical relapse, we wondered whether miR-335 loss could promote metastasis. To test this, we transfected the poorly metastatic MDA-MB-231 cells with an anti-miRNA antagomir16 targeting either miR-335, miR-199a* or a control sequence. Indeed, inhibition of miR-335 enhanced the lung-colonizing ability of MDA-MB-231 cells compared with control cells (Fig. 3b). To identify putative metastasis genes that miR-335 suppresses, we transcriptionally profiled LM2 cells with restored miR-335 expression and arrived at the set of 756 genes for which expression is decreased (using a permissive threshold; see Methods) compared with control LM2 cells. We separately identified genes for which expression levels were increased across both bone and lung metastatic cells. On the basis of available gene expression data sets17,18 we identified 116 genes for which expression was increased by at least twofold in both bone and lung metastatic MDA-MB-231 derivatives compared with the parental line (Supplementary Table 2). The overlap between these two lists yielded a set of six genes for which expression is high in metastatic cells and suppressed by miR-335 (Fig. 4a). This list of miR-335-regulated metastasis genes included genes previously implicated in extracellular matrix and cytoskeleton control, such as the type 1 collagen COL1A1 (ref. 19); in signal transduction, such as the receptor-type tyrosine protein phosphatase PTPRN2 (ref. 20), the c-Mer tyrosine kinase (MERTK)21 and the phospholipase PLCB1 (ref. 22); as well as in cell migration, such as tenascin C (TNC)23 and the SRY-box containing transcription factor SOX4 (refs 24, 25).
Figure 4. A miR-335-regulated gene set includes SOX4 as a miR-335 direct target.

a, The miR-335 metastasis signature consists of genes downregulated by miR-335 and are also overexpressed in bone as well as lung metastatic MDA-MB-231 derivatives. The heatmap depicts the variance-normalized expression values for each gene averaged across two MDA-MB-231 samples, four LM2 samples and two LM2/miR-335 samples. The scale bar depicts standard deviation change from the mean for the expression value of each gene. b, UTR reporter assays of miR-335 metastasis genes in LM2 and MDA-MB-231 cells. Reporter constructs consisting of the luciferase sequence fused to the 3′ UTRs of the miR-335 metastasis genes as well as the control gene UBE2F were transfected into the LM2 and parental MDA-MB-231 cell lines. Luciferase activity of cells was assayed at 32 h after transfection and the values were normalized to the LM2 cell line. n = 3; error bars represent s.e.m.; P-values were obtained using a one-sided Student's t-test. c, Luciferase activity of MDA-MB-231 cells co-transfected with 39 UTR reporter constructs with, or without, miR-335 antagomir was assayed at 32 h after transfection and normalized to control cells. n = 3; error bars represent s.e.m.; P-values obtained using a one-sided Student's t-test. d, Schematic diagram depicts seed sequence (red box) in SOX4 UTR. The 60 bp containing this sequence or the miR-335 seed target site mutant were subjected to UTR reporter assays in LM2 cells with restored miR-335 expression. Luciferase activity of cells was also assayed in the presence or absence of the miR-335 antagomir. n = 3; error bars represent s.e.m.; P-values obtained using a one-sided Student's t-test. e, SOX4 expression in LM2 and MDA-MB-231 cells was obtained through real time qRT-PCR in cells expressing miR-335 or transfected with the miR-335 antagomir. n = 3; error bars represent s.e.m.; P-values derived using a one-sided Student's t-test.
Bioinformatic analysis of the 3′ UTRs of these genes revealed them all to have at least six nucleotides of sequence complementarity to the miR-335 seed region (data not shown). To determine whether the altered expression of these genes in metastatic cells is, in part, mediated through their 3′ UTRs, we cloned the 3′ UTR of five of these genes downstream of a luciferase gene as a reporter, and assayed their expression in LM2 cells (low miR-335) and MDA-MB-231 cells (high miR-335). The expression from UTR reporters corresponding to SOX4, PTPRN2, TNC and MERTK, but not that of the control gene UBE2F lacking the miR-335 target sequence, was significantly lower in LM2 cells relative to MDA-MB-231 cells (Fig. 4b). Furthermore, inhibition of miR-335 in MDA-MB-231 cells by means of an antagomir was sufficient to enhance the expression of SOX4, PTPRN2 and MERTK, but not the expression of a control UBE2F reporter (Fig. 4c). These results suggest that SOX4, PTPRN2, MERTK and possibly TNC are direct targets of endogenous miR-335.
SOX transcription factors are known to regulate progenitor cell development and migration25. In reporter assays, mutation of the miR-335 seed sequence in the SOX4 UTR as well as miR-335 inhibition with an antagomir significantly increased reporter expression (Fig. 4d). Restoration of miR-335 expression in LM2 cells reduced endogenous SOX4 messenger RNA expression, whereas miR-335 inhibition increased SOX4 expression in MDA-MB-231 cells (Fig. 4e). The knockdown of SOX4 in LM2 cells with either of two unique short hairpin RNAs (shRNAs) (Supplementary Fig. 13a) reduced the overall fraction of elongated cells, similar to the phenotype observed with miR-335 restoration in these cells (Supplementary Fig. 14a, b). The morphological change resulting from SOX4 knockdown was also associated with a decrease in cell migration in a trans-well assay (Supplementary Fig. 14c). Another miR-335-regulated gene, TNC, also caught our attention, as its expression occurs in the invasive front of human carcinomas26. Knockdown of TNC in LM2 cells using either of two RNA interference molecules also reduced migration in a trans-well assay (Supplementary Fig. 14c). Notably, knockdown of SOX4 or TNC significantly diminished the invasive ability of LM2 cells (Fig. 5a). Moreover, short-hairpin inhibition of SOX4 or TNC significantly abolished metastasis by LM2 cells in lung colonization assays (Fig. 5b, c). Thus, miR-335 regulates metastasis through suppression of transcription factor SOX4 and extracellular matrix component tenascin C.
Figure 5. miR-335 regulates metastasis and invasion through suppression of SOX4 and TNC.

a, 5.0 × 104 LM2 cells were transduced with short hairpin control vector, either of two shRNAs targeting SOX4, a hairpin targeting TNC, or an siRNA targeting TNC, and invasion of cells through a trans-well matrigel-coated membrane insert was quantified. n = 6; error bars represent s.e.m.; P-values based on a one-sided Student's t-test. b, 2 × 105 LM2 cells transduced with either a control short hairpin, a short hairpin targeting SOX4 or one targeting TNC were inoculated intravenously into immunodeficient mice. Lung colonization was measured by bioluminescence and quantified. n = 7; error bars represent s.e.m.; P-values based on a one-tailed rank-sum test. c, Haematoxylin-and-eosin-stained images of representative lungs that were extracted at 4 weeks after cell inoculation. Representative images were obtained at 10× magnification. d, Kaplan-Meier curves for the combined Memorial Sloan Kettering and Erasmus Medical Center breast tumour cohorts (368 tumours) depicting metastasis-free survival of patients whose primary tumours expressed the miR-335 six-gene signature (positive) and those that did not (negative). n = 368; P-value based on the Mantel-Cox log-rank test.
To determine whether the expression of the genes that miR-335 regulates is associated with human breast cancer metastasis, we examined primary breast tumour gene expression data sets with corresponding disease outcome annotation18,27. Tumours in these cohorts for which aggregate six-gene expression scores exceeded one standard deviation from the mean were considered miR-335-signature-positive (equivalent to loss of miR-335 function). Patients whose primary breast tumours were positive for the miR-335 signature had a significantly worse metastasis-free survival both in the combined cohort of 368 patients (Fig. 5d) as well as in each cohort separately classified (Supplementary Fig. 15a). Consistent with the results of our functional studies, the miR-335 signature performed better as a predictor of overall metastasis rather than lung- or bone-specific metastasis (Supplementary Fig. 15b).
MicroRNAs as suppressors of metastasis
Multiple lines of evidence provided here argue for the involvement of specific miRNAs in suppressing breast cancer metastasis. miR-335, miR-206 and miR-126 are selectively downregulated across a number of highly metastatic human cell lines compared to the general tumour cell population and have demonstrated abilities to suppress metastasis of breast cancer cells to different organ sites. The expression of miR-335 and miR-126 in human mammary tumours is inversely associated with metastatic relapse of these tumours to distant organs, and the expression of miR-206 also shows a trend in the same direction. The expression of a set of genes regulated by one of these miRNAs, miR-335, is directly associated with relapse. We also establish two of these genes, SOX4 and TNC, as in vivo mediators of metastasis. The role of SOX4 in haematopoietic progenitor development28 suggests that its transcriptional programme may be re-used not only for cancer cell invasion in cooperation with tenascin C but also for tumour initiation in the metastatic niche. miR-335 and miR-126 are expressed in normal human breast tissue13. Our findings on their roles in the pathogenesis of human breast cancer argue for an important function for these regulators in maintaining normal tissue integrity. Recently, miR-10b was identified as a miRNA whose over-expression in breast cancer cells promoted tumour growth and lung micrometastasis12. Our work expands on this by identifying miRNAs as clinically meaningful suppressors of metastasis. MicroRNAs are thus uncovered as another class of molecules, along with metastasis suppressor genes29, that negatively regulate tumour progression.
The recent implication of miRNAs and miRNA processing in tumorigenesis11,13 has raised interest in identifying the specific miRNAs for which loss of expression enhances tumorigenesis, the mechanisms by which they act, and the phenotypic advantages afforded to cells that lose miRNA expression. Our findings suggest that within a tumour, the loss of specific miRNAs provides a selective advantage for cells destined for metastatic colonization. A global downregulation of miRNAs in cancer may serve metastasis by reducing the threshold for loss of specific miRNAs in a subset of cells that will ultimately metastasize. That one such miRNA (miR-335) regulates a set of putative, and a subset of validated, metastasis genes argues that the multi-gene regulatory capacity of a miRNA can function as a barrier to tumour progression in humans. The strong association of the loss of miR-335 and miR-126 expression with metastatic relapse suggests the potential for the use of these molecules in prognostic stratification of breast cancer patients in addition to conventional clinical and pathological staging markers.
METHODS SUMMARY
The MDA-MB-231 cell line and its metastatic derivatives18,17,30 as well as the pleural effusion-derived CN34 cancer cells have been described previously31. All metastatic derivatives were obtained through in vivo selection in immunodeficient mice17,18. MicroRNA microarray profiling was performed using LC Sciences technology (LC Sciences, LLC). Mature miRNA expression was assayed using Taqman MicroRNA assays as previously described32. MicroRNA expression was achieved through retroviral transduction of metastatic cells using miR-Vec technology15. The miRNA vectors were provided by R. Agami10. In brief, they consist of approximately 500-bp fragments that span a given miRNA genomic region under the control of a CMV promoter10,15. All animal work was done in accordance with the MSKCC Institutional Animal Care and Use Committee. Statistical analyses were performed using the Graphpad Prism 5 software package. See Supplementary Methods for additional information regarding retroviral infections, transfections, shRNA and siRNA constructs, miRNA inhibition, analysis of miRNA and mRNA expression, miRNA expression profiling and hierarchical clustering, transcriptional profiling, tumour miRNA and gene expression analysis, survival analysis, trans-well migration and invasion assays, bioluminescence imaging, animal injections and inoculations, metastatic cell extraction, tumour and lung immunohistochemical and histological analyses, and image quantification.
METHODS
Cell culture
The MDA-MB-231 line and its metastatic derivatives have been described previously18,30. All cell lines were cultured in DMEM supplemented with 10% FBS, glutamine, pyruvate, penicillin, streptomycin and fungizone. CN34 cancer cells were isolated from the malignant pleural effusion of a patient with metastatic breast cancer treated at our institution on obtaining written consent following IRB regulations as previously described31. The GPG29 retroviral packaging cell line was maintained in DMEM containing 10% FBS supplemented with G418, puromycin, penicillin, streptomycin, doxycycline and fungizone, as previously described18. All transfections were done using Lipofectamine 2000 (Invitrogen). Subsequent to transfection, GPG29 cells were maintained in DMEM supplemented with 10% FBS and pyruvate.
Analysis of miRNA and mRNA expression
Total RNA from MDA-MB-231 cells, its derivatives, or CN34 cells and its derivatives was extracted and purified using the MiRvana kit (Ambion). Tissues from primary breast cancers were obtained from therapeutic procedures performed as part of routine clinical management at our institution. Methods for primary tumour sample processing were described previously18. In brief, RNA from these samples was extracted from frozen tissues by homogenization in TRIzol reagent (Gibco/BRL), as described previously18. Taqman microRNA assays (Applied Biosystems) were used to quantify mature miRNA expression as previously described32. RNU6 (Applied Biosystems) was used as endogenous control for miRNA expression studies. For cell line miRNA expression quantification, each reverse-transcriptase (RT) reaction consisted of 50 ng of purified total RNA, 1× RT buffer, dNTPs (each at 0.375 mM), 5 U μ ml-1 MultiScribe reverse transcriptase, 50 nM stem-loop RT primer and 0.38 U μ ml-1 RNase inhibitor (Applied Biosystems). RT reactions were incubated at 16 °C for 30 min, 42 °C for 30 min and 85 °C for 5 min. Real-time PCR reactions for miRNAs from cell lines were performed in quadruplicate in 10 μl volumes. The Real-time reaction mix consisted of 0.67 μl of RT product, 0.5 μl of 20× TaqMan microRNA assay mix, 5 μl TaqMan 2× universal PCR Master Mix. Quantitative miRNA expression data were acquired and analysed using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems).
For primary tumour miRNA expression quantification, each RT reaction contained 10 ng of purified total RNA. RT-PCR reactions from archived tumour RNA were performed in replicates of six. For mRNA expression quantification, 400 ng of total purified RNA was reverse transcribed using the SuperScript III first-strand synthesis system (Invitrogen). cDNA corresponding to approximately 5 ng of initial RNA was used for each quadruplicate quantitative PCR reaction. Human SOX4 and β-glucuronidase (as endogenous control) were amplified using commercially designed Taqman gene expression assays (Applied Biosystems) and the Taqman Universal PCR master mix (Applied Biosystems). Quantitative mRNA expression data were acquired and analysed in 384-well-plate format using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems).
MicroRNA microarray hybridization
Twenty micrograms of total RNA obtained from MDA-MB-231 and its derivatives was labelled and hybridized on miRNA microarrays by LC sciences. The arrays were designed to detect miRNA transcripts corresponding to 453 miRNAs contained in the Sanger miRBase Release 8.2. Of all the probes assayed, those corresponding to 179 miRNAs revealed a signal above a threshold above background in at least one of the MDA-MB-231 cell lines. Raw signal intensities representing hybridization to probes were mean-normalized across cell lines. Hierarchical clustering of variance-normalized expression values was performed using Matlab software. Hierarchical clustering was performed using euclidean distance as a metric and using average linkage. Clustering of cell types along organotropic and metastatic phenotypes was achieved using both the expression of 179 miRNAs as well as the 20 miRNAs that displayed the highest coefficients of variation across cell lines.
RNA microarray hybridization and miR-335 signature
The gene expression data sets from MDA-MB-231 cell derivatives as well as the primary breast tumour gene expression data sets from the Memorial Sloan Kettering (MSK) and Erasmus Medical Center (EMC) cohorts have been published previously18,27. Transcriptional profiling of LM2 cells expressing miR-335 was performed as previously described18 through the use of the Affymetrix HG-U133 plus 2.0 array by the genomics core laboratory at MSKCC. A total of 874 probes corresponding to 756 genes displayed a greater than 0.2 log10 (ratio) fold decrease in expression relative to LM2 cells expressing a short hairpin control. This low permissive threshold was chosen so as to include genes for which fold reduction at the transcript level as detected by hybridization may be much less than their potential reduction at the protein level. The number of transcripts downregulated at the permissive threshold as well as the number of transcripts downregulated at the less-permissive threshold of twofold (458 probes) is comparable to profiling analyses with other miRNAs33. The miR-335-regulated six-gene signature comprises the set of genes corresponding to the 756 miR-335 downregulated genes that (1) were also upregulated by more than twofold in both lung and bone metastatic MDA-MB-231 derivatives relative to the parental line (available gene expression data set18) and (2) were expressed by multiple MDA-MB-231 cell lines.
Generation of retrovirus, miRNA expressing cells and knockdown cells
The plasmid vectors encoding the miRNAs were provided by R. Agami10, and have been previously described. To produce retrovirus for restoration of miRNA expression in cells, these PMSCV-based vectors were individually transfected into the GPG29 amphotropic packaging cell line. Virus particles were collected at 48 and 72 h after transfection, filtered and concentrated through ultracentrifugation, as previously described6. These concentrated retroviral particles were then used to transduce cells in the presence of 8 μg ml-1 polybrene. Vector-transduced cells were selected with blasticidin. MicroRNA expression was confirmed in all cases using quantitative RT-PCR for the mature forms of individual miRNAs, suggesting that the precursor transcripts were endogenously processed to the mature forms. Notably, miRNA expression did not interfere with endogenous processing of miRNAs as revealed by qRT-PCR for the expression of other miRNAs in these cell lines. For in vitro miRNA inhibition studies, MDAMB-231 or LM2 cells were transfected with antagomirs16 (25 nM; Ambion) based on the manufacturer's recommendations. For in vivo miRNA-inhibition metastasis studies, MDA-MB-231 cells were transiently transfected with the control anti-miRNA locked nucleic acid (LNA)-159, LNA-335, or LNA-199a* (20 nM; Exiqon) based on the manufacturer's recommendations. Transfected cells were inoculated into immunodeficient mice via tail-vein injection 4 days after transfection. SOX4 knockdown cells were generated through infection with Mission TRC lentiviral particles (Sigma-Aldrich) containing short hairpins targeting the human SOX4 gene. The TRC numbers and the shRNA sequences that yielded the greatest knockdown were TRCN0000018216, 5′-CCGGTGGGCACATCAAGCGACCCATCTCGAGATGGGTCGCTTGATGTGCCCATTTTT-3′ and TRCN0000018217, 5′-CCGGGAAGAAGGTGAAGCGCGTCTACTCGAGTAGACGCGCTTCACCTTCTTCTTTTT-3′. TNC knockdown cells were generated either by infection with a pRetrosuper vector expressing the following forward sequence, 5′-GATCCCCGAGGCTCACAATCTCACGGTTCAAGAGACCGTGAGATTGTGAGCCTCTTTTTGGAAA-3′, or by transfecting cells with the following ON-TARGET siRNA with sense sequence 5′-GGUACACCCUCCAUGGUAAUU-3′ (Dharmacon).
UTR reporter assays
The full-length 3′ UTRs of the miR-335-regulated human genes COL1A1, SOX4, PTPRN2, TNC and MERTK, as well as the control gene UBE2F, were amplified from human genomic DNA (Novagen) and individually cloned into the Psicheck 2 dual luciferase reporter vector (Promega). MDA-MB-231 or LM2 cells were then transfected with each reporter construct and MDA-MB-231 cells were also transfected with a miR-335 antagomir (25 nM) targeting endogenous miR-335 (Ambion). Cells were lysed at 30 h after transfection and the ratio of Renilla to firefly luciferase was measured with the dual luciferase assay (Promega). Normalized Renilla to firefly ratios were determined in the presence or absence of miR-335 inhibition. The segment (base pairs 449-509) of the SOX43′ UTR containing the wild-type or mutated miR-335 target sequence at base pair 483 (TCTTGG to TGAGGG) was also cloned into the Psicheck 2 dual luciferase vector.
Animal studies
All animal work was done in accordance with an approved protocol by the MSKCC Institutional Animal Care and Use Committee. NOD/SCID female mice (NCI) age-matched between 6-8 weeks were used for lung colonization, tumour growth and orthotopic metastasis xenograft assays. Athymic female mice (NCI) age-matched between 6-8 weeks were used for bone metastasis xenograft assays. For primary tumour growth assays and orthotopic metastasis assays, 5 × 105 viable cells were re-suspended in a 1:1 mixture of PBS and growth-factor-reduced Matrigel (BD Biosciences) and injected orthotopically into the fourth mammary gland in a total volume of 25 μl. Primary tumour growth rates were analysed by measuring tumour length (L) and width (W), and calculating volume through the use of the formula πLW2/6, as previously described18. Orthotopic lung metastasis was quantified using non-invasive bioluminescence at 32 days after xenografting. For experimental metastasis assays, 1 × 104 viable cells were re-suspended in 0.1 ml PBS and injected into the lateral tail vein. Lung metastatic colonization was monitored and quantified using non-invasive bioluminescence as previously described18. For bone metastasis assays, 2 × 104 viable cells were re-suspended in 0.1 ml PBS and introduced into the arterial circulation via intra-cardiac injection, as previously described17. Bone metastatic colonization was monitored and quantified using non-invasive bioluminescence of normalized hindlimb photon flux at day 35.
Trans-well migration and invasion assays
Cancer cells were conditioned overnight in 0.2% FBS ECM media without growth factors. The next day, cells were pulsed with 5 μM cell tracker green (Invitrogen) for 30 min, trypsinized and re-suspended in 0.2% FBS ECM media without growth factors. Tumour cells were then seeded at 25,000 cells per well into trans-well inserts (3μ pore size, BD Falcon) for migration assays or at 50,000 cells per well into matrigel-coated, growth-factor-reduced, invasion chambers (8m pore size, BD Biosciences). The wells were washed with PBS and fixed with 4% paraformaldehyde after 6 h for migration assays or after 22 h for invasion assays. The cells on the apical side of each insert were then scraped off. The number of cells that had migrated to the basal side of the membrane was visualized with a Zeiss Axiovert 200M microscope at ×10 magnification. Pictures of three random fields from three or six replicate wells were obtained and the number of cells that had migrated quantified in automated fashion using Metamorph analysis software.
Lung and tumour immunostaining
Mice were killed and perfused with PBS followed by 4% paraformaldehyde. Tumours and lungs were extracted and fixed in 4% paraformaldehyde in PBS. Paraffin-embedding, sectioning and haematoxylin and eosin staining of tumours and lungs were performed by Histoserv technologies. Immunohistochemical staining for cleaved caspase-3 (Cell Signaling) and phospho-histone 3 (Upstate) was performed on paraffin-embedded sections by the MSKCC Molecular Cytology Core Facility. Images were obtained at ×10 magnification using the Axioplan2 microscopy system (Zeiss), and apoptosis (cleaved caspase 3) and cell proliferation (phosphohistone 3) were quantified as number of cells per high power field averaged across 18 images from 3 tumours per cell line. Immunohistochemical staining for human vimentin (Novocastra) was performed on paraffin-embedded sections by the MSKCC Molecular Cytology Core Facility.
Statistical analysis and clinical validation of miR-335 signature
The Kaplan-Meier method was used for survival curve analysis, and the log-rank (Mantel-Cox) test was used to determine the statistical significance of difference between survival curves using Graphpad Prism 5 software. To determine the association of miRNA expression with metastatic relapse, the raw miRNA expression values for each tumour sample were normalized to the median expression value across the 20 tumour samples. Tumours whose expression values for each miRNA fell in the lowest third (7 out of 20) of the entire set of tumours were deemed the low expression group for each miRNA. The Mantel-Cox log-rank test was then used to determine the statistical significance for differences between survival curves of patients from whom the tumours were resected. The Mann-Whitney test was used to determine the significance of difference between the expression levels for miRNAs in tumours that ultimately relapsed relative to those that did not. The correlation of miR-335 or miR-126 expression status with ER histological or HER-2/NEU amplification status was determined by calculating the significance of the Spearman correlation coefficients in a two-tailed test using Graphpad Prism 5 software. Primary tumour gene expression data from the MSK18 and EMC27 cohorts were used to classify each tumour as miR-335-signature-positive if the sum of Z-scores for the expression values of the six miR-335-regulated genes was greater than one standard deviation from the mean. Previously published metastasis-free survival data corresponding to these tumours were then plotted in Kaplan-Meier form for patients whose tumours were miR-335-signature-positive versus -negative.
Supplementary Material
Acknowledgements
We thank S. Tavazoie, M. Tavazoie, D. Nguyen, S. Kurdistani and X. Zhang for discussions and technical suggestions. We are grateful to R. Agami, C. Le Sage and R. Nagel for providing the miR-Vec constructs. We thank J. Baez, E. Montalvo, E. Suh, Z. Lazar, Y. Romin, A. Barlas, K. Manova-Todorova and members of the Molecular Cytology Core Facility. We thank X. Zhou of LC Sciences for miRNA profiling services as well as the MSKCC core facility for transcriptional profiling. J.M. was funded by a National Institutes of Health grant, and by grants of the Hearst Foundation and the Kleberg Foundation. S.F.T. is supported by the Olson Foundation grant and a Clinical Scholars Award. J.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
References
- 1.Fidler IJ. The pathogenesis of cancer metastasis: the `seed and soil' hypothesis revisited. Nature Rev. Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 2.Weigelt B, Peterse JL, van't Veer LJ. Breast cancer metastasis: markers and models. Nature Rev. Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 3.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Nevins JR, Potti A. Mining gene expression profiles: expression signatures as cancer phenotypes. Nature Rev. Genet. 2007;8:601–609. doi: 10.1038/nrg2137. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nature Rev. Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 6.Gupta GP, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446:765–770. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- 7.Seligson DB, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 8.Lim LP, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 9.Chan CS, Elemento O, Tavazoie S. Revealing posttranscriptional regulatory elements through network-level conservation. PLoS Comput. Biol. 2005;1:e69. doi: 10.1371/journal.pcbi.0010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voorhoeve PM, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 11.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nature Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 12.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 13.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 14.Calin GA, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl Acad. Sci. USA. 2004;101:11755–11760. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 16.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang Y, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 18.Minn AJ, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egeblad M, et al. Type I collagen is a genetic modifier of matrix metalloproteinase 2 in murine skeletal development. Dev. Dyn. 2007;236:1683–1693. doi: 10.1002/dvdy.21159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varadi A, Tsuboi T, Rutter GA. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 beta-cells. Mol. Biol. Cell. 2005;16:2670–2680. doi: 10.1091/mbc.E04-11-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graham DK, et al. Cloning and developmental expression analysis of the murine c-mer tyrosine kinase. Oncogene. 1995;10:2349–2359. [PubMed] [Google Scholar]
- 22.Lyu MS, Park DJ, Rhee SG, Kozak CA. Genetic mapping of the human and mouse phospholipase C genes. Mamm. Genome. 1996;7:501–504. doi: 10.1007/s003359900151. [DOI] [PubMed] [Google Scholar]
- 23.Ilunga K, et al. Co-stimulation of human breast cancer cells with transforming growth factor-β and tenascin-C enhances matrix metalloproteinase-9 expression and cancer cell invasion. Int. J. Exp. Pathol. 2004;85:373–379. doi: 10.1111/j.0959-9673.2004.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van de Wetering M, Oosterwegel M, van Norren K, Clevers H. Sox-4, an Sry-like HMG box protein, is a transcriptional activator in lymphocytes. EMBO J. 1993;12:3847–3854. doi: 10.1002/j.1460-2075.1993.tb06063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoser M, et al. Prolonged glial expression of Sox4 in the CNS leads to architectural cerebellar defects and ataxia. J. Neurosci. 2007;27:5495–5505. doi: 10.1523/JNEUROSCI.1384-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orend G, Chiquet-Ehrismann R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006;244:143–163. doi: 10.1016/j.canlet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 28.Schilham MW, Moerer P, Cumano A, Clevers HC. Sox-4 facilitates thymocyte differentiation. Eur. J. Immunol. 1997;27:1292–1295. doi: 10.1002/eji.1830270534. [DOI] [PubMed] [Google Scholar]
- 29.Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nature Rev. Cancer. 2003;3:55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 30.Minn AJ, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Invest. 2005;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomis RR, Alarcon C, Nadal C, Van Poznak C, Massague J. C/EBPβ at the core of the TGFβ cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell. 2006;10:203–214. doi: 10.1016/j.ccr.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 32.Chen C, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He L, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.