

Abstract
Insulin-like Growth Factor Binding Protein-3 (IGFBP3) is a high affinity binding protein shown to regulate cell growth, differentiation, and apoptosis in a variety of cellular systems. The primary aim of this study was to characterize IGFBP3 expression in the human corneal epithelium and in a corneal epithelial cell line and to establish a potential role for IGFBP3 mediated apoptotic signaling in corneal epithelial cells.
Using a telomerase-immortalized human corneal epithelial (hTCEpi) cell line cultured in serum-free media and fresh human eye bank donor tissue, expression and localization of IGFBP3 were established in situ and in vitro by indirect immunofluorescence and western blotting. Real time PCR was used to measure IGFBP3 mRNA levels following Trichostatin-A (TSA) treatment and as a function of confluence. IGFBP3 protein levels were assessed in resting human tears and in conditioned media by western blotting; as was the ability of recombinant human IGFBP3 protein to associate with the cell surface. Apoptotic signaling was assessed in vitro using TSA and recombinant human (rh)IGFBP3. Apoptosis was measured by Viability/Cytotoxicity, Annexin V, and TUNEL assays.
IGFBP3 localized to the plasma membrane of human corneal epithelial cells in situ and was upregulated in surface cells in the central cornea. IGFBP3 was secreted in conditioned media of growing cells, with a robust upregulation following confluence (P<0.001) and differentiation. IGFBP3 was undetectable in human tears. Addition of TSA to the culture media resulted in an upregulation of IGFBP3 mRNA (P<0.001) and protein. In addition, TSA treatment led to a significant increase in Annexin V positive cells at 18 and 24 hours (P<0.001) and TUNEL positive cells at 24 and 48 hours (P<0.001). The addition of rhIGFBP3 to the cell culture media appeared to induce occasional membrane blebbing, but cells failed to become positive with Annexin V or TUNEL.
Taken together, these results demonstrate that cell membrane-associated IGFBP3 is produced by corneal epithelial cells and associates with the plasma membrane of superficial cells in situ and in cultured cells, but is not present in human tears. The differential localization and effect(s) on apoptosis suggest that the effects of IGFBP3 are likely tissue compartment and receptor specific and may be regulated by glycosylation.
Introduction
Insulin-like Growth Factor Binding Protein-3 (IGFBP3) is an N-linked glycosylated, phosphorylated, secretory protein with known growth inhibitory and apoptotic roles (Butt and Williams, 2001). IGFPB3, one of six highly conserved IGF-binding proteins, modulates proliferation through extracellular interactions with Insulin-like Growth Factor-I (IGF-I). IGF-I, and the homologue IGF–II, both function as mitogens in many epithelial and fibroblast cell systems; and may have additional roles in regulating protein synthesis, cell cycling, and differentiation.
IGFBP3 is produced locally in many tissues, including the skin, where it has been reported as the most abundant IGF-binding protein secreted by keratinocytes and functions by inhibiting IGF-I binding to IGF-IR, thus regulating IGF-I bioavailability and subsequent growth response (Edmondson et al., 1999). IGFBP3 has also been shown to be present in serum, where it forms a large, 150 kDa ternary complex with IGF-I or IGF-II and the serum glycoprotein acid-labile subunit, ALS (Baxter and Martin, 1989). In this latter form, IGF-I or –II are systemically distributed to tissues throughout the body. In ocular tissue, IGFBP3 has postulated roles in the human cornea in regulating myofibroblast proliferation and differentiation (Izumi et al., 2006); however, the localization and expression of IGFBP3 in the corneal epithelium has not been characterized. By contrast, IGF-1R has been localized to human corneal and conjunctival cells and IGF-1 has been shown to play an important role in corneal epithelial wound healing, stimulating both proliferation of corneal epithelial cells and migration (Lee et al., 2006; Rocha et al., 2001). Clinically, the topical administration of IGF-1 in conjunction with Substance P has been shown to stimulate epithelial repair in patients with persistent corneal epithelial defects (Nagama et al., 2003; Nishida, 2005).
An additional role for IGFBP3 involves the regulation of apoptosis independently of the IGF axis. Specifically, IGFBP3 has been shown to induce apoptosis through the phosphorylation and inactivation of BCL2 (Rajah et al., 2002). BCL2, an anti-apoptotic protein, has been previously localized to the nuclei of corneal epithelial cells and loss of nuclear expression of BCL2 has been shown to precede apoptosis in surface epithelial cells (Yamamoto et al., 2001a; Yamamoto et al., 2001b). IGFBP3 has also been shown to upregulate the pro-apoptotic protein BAX, thus functioning as a regulator of the BCL2:BAX rheostat in cell death (Butt et al., 2002). The ability of secreted IGFBP3 to induce apoptosis through extracellular cell surface-mediated binding linked to intracellular vesicular trafficking to the nucleus (autocrine, paracrine loop) is currently unclear and appears to be cell type and tissue specific.
In the present study, we characterized IGFBP3 expression and localization in the human corneal epithelium in situ and in hTCEpi cells in vitro during proliferation and calcium induced multi-layer differentiation. We further assessed the ability of recombinant human IGFPB3 and endogenously upregulated IGFBP3 to mediate apoptosis in corneal epithelial cells.
Methods
RT PCR
RNA was extracted from hTCEpi cells using RNA STAT 60 (Tel-TEST, Friendswood, TX). cDNA was generated from 2 μg of total RNA using Superscript First Round Synthesis with random primers (Invitrogen, Carlsbad, CA) according to manufacturer instructions. A 50 μl PCR reaction was performed as follows: 5 μmol/l each primer, 0.20 mM dNTPs, 5 μl of 10× PCR buffer (Sigma, St. Louis, MO), 7.5 mM MgCl2 (Sigma, St. Louis, MO), 1.0 μl Taq DNA Polymerase (5U/μl, Sigma, St. Louis, MO), and 2 μl cDNA. Gene specific primers were used to amplify the full length IGFBP3 coding sequence: forward 5′ ATGCAGCGGGCGCGAC 3′ and reverse 5′ CTACTTGCTCTGCATGCTGTAGCA 3′ with melting temperatures of 62.9°C and 59.2°C, respectively. The reaction conditions for PCR were an initial denaturation for one cycle at 94°C for 3 minutes, followed by 25 cycles at 94.0°C for 1 minute, 60°C for 45 seconds and 72.0°C for 45 seconds, and a final extension at 72.0°C for 7 minutes. Products were resolved on a 1.25% agarose gel stained with ethidium bromide and imaged on a Typhoon Variable Mode Imager.
Immunohistochemistry
Human donor tissue was obtained from Tissue Transplant Services, UT Southwestern Medical Center, Dallas, TX. All tissue was obtained fresh and processed within 6 hours of death. To establish the localization of IGFBP3 in the human corneal epithelium, non-fixed tissue was embedded in tissue embedding medium (Electron Microscopy Services, Hatfield, PA) and snap frozen in liquid nitrogen. 10 μm cryostat-sectioned tissue was permeabilized in cold acetone (-20°C), washed in phosphate buffered saline (PBS), and blocked in 10% donkey serum at 37°C for 30 minutes. Sections were incubated with a goat polyclonal antibody directed against the C-terminus of human IGFBP3 (10 μg/ml, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) overnight at 4°C, followed by incubation in 30 μg/ml FITC-conjugated secondary donkey anti-goat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) at 37°C for 2 hours. Tissue sections were counter-stained with 10 μg/μl Propidium Iodide (PI, Sigma, St. Louis, MO) to label all epithelial nuclei and mounted To confirm antibody specificity, the antibody was incubated in five fold excess of an available blocking peptide in 500 μl of PBS (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 2 hours at 4°C with shaking. The primary antibody was omitted for a negative control. Sections were imaged on a Leica SP2 laser scanning confocal microscope (Leica Microsystems GmbH, Nussloch, Germany).
SDS PAGE and Western Blotting
Epithelial cell lysates were collected by lysing cells directly in plastic 6-well culture dishes using RIPA buffer containing a protease inhibitor cocktail tablet (Complete-Mini, Roche Diagnostics, Indianapolis, IN) on ice for 10 minutes. For collection of epithelial cells from tissue, an 8 mm circular trephine was used to separate cornea from limbus. Epithelial cells were removed by scraping using a sterile number 3 scalpel blade (Rotax International LTD, Sheffield, UK) and placed into RIPA/protease inhibitor cocktail. All lysates were snap frozen in liquid nitrogen and vortexed. For protein analysis from conditioned media, media was collected from T75 flasks after 48 hours and concentrated using iCON Protein Concentrators (Pierce, Rockford, IL). For analysis of resting, basal tears, tear samples were collected from the inferior tear meniscus at the lateral canthus of three non-dry eye patients using micro-pipettes (Drummond Scientific Co., Broomall, PA). Tear samples were added directly to 4× sample buffer. Cells transfected with pcDNA-IGFBP3, a plasmid encoding the cDNA for IGFBP3 (a gift of Dr. Sue Firth, the Kolling Institue, Sydney, Australia), and 0.5 μg rhIGFBP3 were used as a positive control. Transfections were performed using FuGene6 transfection reagent (Roche, Indianapolis, IN). All lysates and tear samples were boiled for 5 minutes in 4× sample buffer pH 6.8 containing 0.25 M tris, 8% lauryl sulfate, 40% glycerol, 20% mercaptoethanol and 0.04% bromophenol blue, resolved on a 12% SDS polyacrylamide gel and subsequently transferred to a nitrocellulose membrane. Membranes were blocked in 5% non-fat milk and blotted using a 1:5000 dilution of a rabbit antiserum raised against rhIGFBP3 (Diagnostic Systems Laboratories, Webster, TX) overnight at 4°C. Following incubation in a 1:5000 dilution of an HRP-conjugated anti-rabbit IgG secondary antibody (Amersham Biosciences, Piscataway, NJ), protein was visualized using ECL Plus Detection Reagents (Amersham Biosciences, Piscataway, NJ) and imaged on a Typhoon Variable Mode Imager. Quantitative protein analysis was performed using Image Quant Software, Amersham Biosciences, Piscataway, NJ).
Cell Surface Association Assay
To determine if recombinant human IGFBP3 associated with the cell membrane and/or internalized, 2.2 × 105 cells/well were seeded onto plastic 6-well tissue culture dishes and treated with 500 ng/ml rhIGFBP3 for up to 48 hours. Media was removed and cells were washed thoroughly with PBS. Cells were then lysed using RIPA buffer as described above and analyzed by SDS PAGE and western blotting.
Real Time PCR
Real Time RT-PCR was used to assess total message levels for IGFBP3. hTCEpi cells were seeded onto plastic 6-well tissue culture dishes at a density of 2.2 × 105 cells/well and treated with 3.31 μM TSA for up to 30 hours to assess the ability of TSA to upregulate IGFBP3 or were allowed to grow over 7 days to assess IGFBP3 mRNA levels as a function of confluence. mRNA was extracted from hTCEpi cells by homogenization using the standard RNA STAT 60 protocol (TEL-TEST, Inc., Friendswood, TX). Following homogenization, RNA was extracted using chloroform, precipitated with isopropanol, washed in 75% ethanol and air-dried. RT PCR with First-Strand Synthesis using Random Primers (Invitrogen, Carlsbad, CA) was used to generate cDNA. Real time PCR was performed with a 20 μl reaction using 10 μl of 2× Taqman Universal Master Mix (Applied Biosystems, Foster City, CA), 900 nM forward/reverse primer, 200 nM probe, and 4 μl cDNA. Gene specific primers were used to amplify a 131 bp fragment for IGFBP3 (forward primer 5′ GGT GTC TGA TCC CAA GTT CC 3′ and reverse primer 5′ CGG AGG AGA AGT TCT GGG TA 3′). The hybridization probe was designed using Primer Express software (MGB-CGCTACAAAGTTGACTAC, Applied Biosystems, Foster City, CA). For quantitation of mRNA, a standard curve was prepared using serial dilutions of a known concentration of hTCEpi RNA. RNA concentration was determined by measuring the optical density at 260 nm on a Beckman DU 530 spectrophotometer and converted to μg/μl. A pre-designed assay reagent kit for β-actin was used as an endogenous control (Applied Biosystems, Foster City, CA), along with a no template control. All reactions were performed in duplicate in a 384 well plate using an ABI Prism 7900-HTA Sequence Detection System (Applied Biosystems). All assays were performed three independent times to allow for statistical analysis.
Annexin V Assay
To measure early apoptotic changes, 2.2 × 105 cells/dish were plated onto delta T4 tissue culture dishes and allowed to adhere for 24 hours. Cells were treated with either 500 ng/ml non-glycosylated rhIGFBP3 or 3.31 μM Trichostatin-A (TSA, Sigma, St. Louis, MO) for 0, 2, 4, 6, 8, 18, and 24 hours. Cells were washed once in PBS and followed by the addition of 1× binding buffer containing 10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2. Cells were triple-labeled using an Annexin V Conjugate for Apoptosis Detection Alexa Fluor 647 (Molecular Probes, Invitrogen, Carlsbad, CA), 1 μg/ml PI, and 50 μM Calcein-AM (Molecular Probes) for 20 minutes at room temperature. Five regions of each culture dish were imaged on a Leica SP2 laser scanning confocal microscope. Cell counts were obtained using MetaMorph Software (Molecular Devices Corp., Downington, PA). The number of viable cells was determined by Calcein-AM staining. The percentage of Annexin V positive and PI positive cells was determined by dividing the number of positive cells by the total number of cells present in each field. All experiments were performed in duplicate and repeated twice.
TUNEL Labeling
To measure end stage apoptosis, TUNEL labeling was performed using the Apoptag Fluorescein In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA) according to manufacturer instructions. 5 × 104 hTCEpi cells per well were plated on collagen-coated coverslips and allowed to adhere for 24 hours. Cells were treated with either 3.3 μM TSA or 500 ng/ml rhIGFBP3 and collected at 0, 24, and 48 hours. Cells were fixed in 1% paraformaldehyde, washed with PBS, and post-fixed in 2:1 ethanol:acetic acid for 5 minutes at -20°C. Cells were washed in PBS and incubated in Equilibration buffer for 10 minutes at room temperature. Equilibration buffer was removed and cells were incubated in WS TdT Enzyme at 37°C for 1 hour, followed by washing with WS Stop/Wash buffer for 10 minutes at room temperature. Cells were washed again using PBS and then incubated in WS Anti-Digoxigenin Conjugate for 30 minutes at room temperature. Following a final wash in PBS, coverslips were mounted in a 50% (v/v) solution of PBS:glycerol containing 10 μg/μl PI, for visualization of epithelial cell nuclei. For a positive control, cells were incubated in DNase I (Invitrogen, Carlsbad, CA) for 20 minutes at room temperature prior to adding equilibration buffer. WS TdT enzyme was omitted in the negative control. Five regions of each culture dish were imaged on a Leica SP2 laser scanning confocal microscope. Cell counts were obtained using MetaMorph Software (Molecular Devices Corp., Downington, PA). The percentage of TUNEL positive cells was determined by dividing the number of positive cells by the total number of cells, assessed by PI staining, present in each field. All experiments were performed in duplicate and repeated twice.
Statistics
Statistical analysis was performed using SigmaStat 3.1 (Systat Software, Inc., San Jose, CA). All data are expressed in mean ± standard deviation. A One-way Anaylsis of Variance was used to determine if a significant difference existed. The multiple comparison Student-Newman-Keuls Test was performed to assess a significant difference between groups. Statistical significance was set at P<0.05.
Results
Intra- and Extracellular IGFBP3
To determine whether IGFPB3 was present in human corneal epithelial cells, RT PCR was performed on cDNA obtained from hTCEpi cells. RT PCR confirmed the presence of both transcripts, alpha and beta, for IGFBP3, which differ only within an intronic splice site (Figure 1). To establish the presence and localization of IGFBP3 in the human corneal epithelium, non-fixed human corneal tissue was snap frozen in liquid nitrogen within 6 hours of death. 10 μm cryosections were double-labeled with a goat polyclonal antibody recognizing the C terminus of IGFBP3. IGFBP3 localization varied from central cornea to limbus (Figure 2). In the central corneal epithelium, IGFBP3 expression was membrane bound in basal and suprabasal cells with expression most pronounced among the surface epithelial cells (2A). Moving peripherally, IGFBP3 again appeared membrane bound, with the strongest expression seen in the basal epithelial cells (2B). In the limbal epithelium, IGFBP3 appeared to localize to the surface of the plasma membrane in basal, wing, and superficial epithelial cells (2D). Interestingly, small clusters of cells in the basal and wing cell layers also demonstrated a cytoplasmic localization. This cytoplasmic pattern was only seen in limbal epithelia. No staining was seen after neutralization of the antibody by a blocking peptide (2E) or in the negative control (2F).
Figure 1.
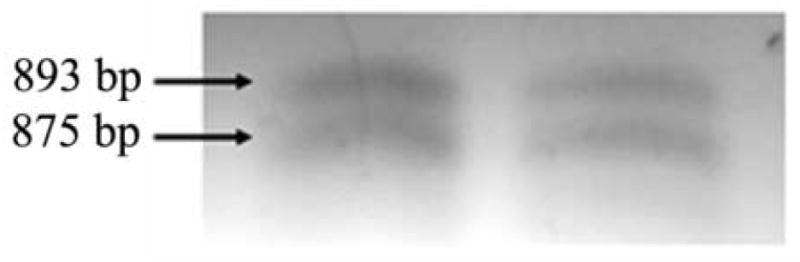
RT PCR demonstrating the presence of IGFPB3 transcriptional splice variants alpha and beta, 893 and 875 base pairs, respectively, in two independent samples of hTCEpi cells. A no RT template was used as a control (not shown).
Figure 2.
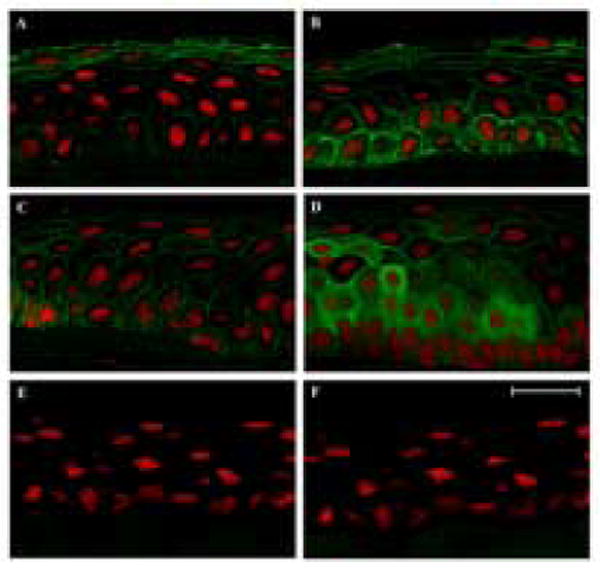
Cryostat-sectioned human corneal epithelium double-labeled with IGFBP3 (green) and PI (red). A: central; B: peripheral; C: transitional; D: limbal epithelium; E: Primary antibody incubated in 5 fold excess blocking peptide; F: negative control, primary antibody omitted. 63× magnification, scale: 24 μm.
IGFBP3 expression was further assessed by western blotting from epithelial scrapes of human corneal and limbal epithelium using a rabbit polyclonal antibody. In human corneal epithelium, multiple isoforms were detected ranging from 32 to 64 kDa (Figure 3A). In central cornea, the majority of IGFBP3 was present at 47 kDa, with a doublet noted at 40 kDa. Interestingly, in both limbal and corneal epithelium, we noted the presence of a 64 kDa dimer.
Figure 3.
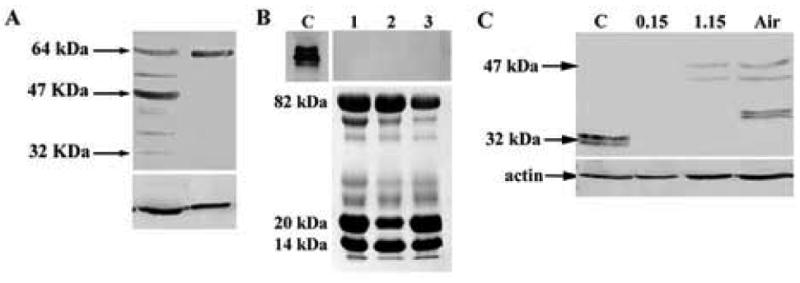
(A) Western blotting of cell lysates obtained by epithelial scrape from human corneal epithelium and limbus for IGFBP3 using a goat polyclonal antibody. Blot representative of repeated experiments (n=2). (B) Western blot for IGFBP3 from basal, resting tear samples. IGFBP3 was not detectable in human tear samples (collected from 3 independent patients, represented 1, 2, and 3, top). rhIGFPB3 served as a positive control, C. Sypro orange staining confirmed presence of protein in tear samples (bottom). (C) Western blotting for IGFBP3 in hTCEpi cells grown in submersed/airlifted culture. IGFBP3 protein was undetectable in hTCEpi cells under 0.15 mM calcium culture conditions. Appearance of IGFBP3 isoforms correlated with an increase in calcium (1.15 mM) levels and subsequent air-lifted (Air) differentiation. B-actin was used as a loading control (n=2).
Due to the secretory nature of IGFBP3, IGFBP3 protein levels were also probed in human tears samples. Western blotting of normal, unstimulated human tears failed to demonstrate detectable levels of IGFBP3 (Figure 3B). Conversely, sypro orange stained SDS PAGE gels confirmed the presence of proteins migrating at 82, 20, and 14 kDa, corresponding to lactoferrin, lipocalin-1, and lysozyme, major protein components in the normal, resting pre-ocular tear film.
IGFBP3 expression was further assessed as a function of differentiation. hTCEpi cells were grown in submersed/air-lifted culture and protein lysates collected. By western blotting, IGFBP3 was not detectable under low calcium culture conditions (Figure 3C). After 7 days of culture in submersed, high calcium media, two isoforms were present at 47 and 44 kDa. Following continued air-lifted culture, isoforms were detected at 47, 44 and 40 kDa.
To evaluate the effects of confluence on IGFBP3, hTCEpi cells were plated on collagen-coated glass coverslips and grown for up to 7 days. Phase contrast microscopy was used to demonstrate relative confluence of epithelial cells (Figure 4A-D). Real time PCR demonstrated relatively low levels of IGFBP3 throughout cell growth and became upregulated following confluence (P<0.001, One-way ANOVA, SNK multiple comparison test) (Figure 4E). The upregulation was transient in nature as mRNA levels were not maintained following continued culture.
Figure 4.
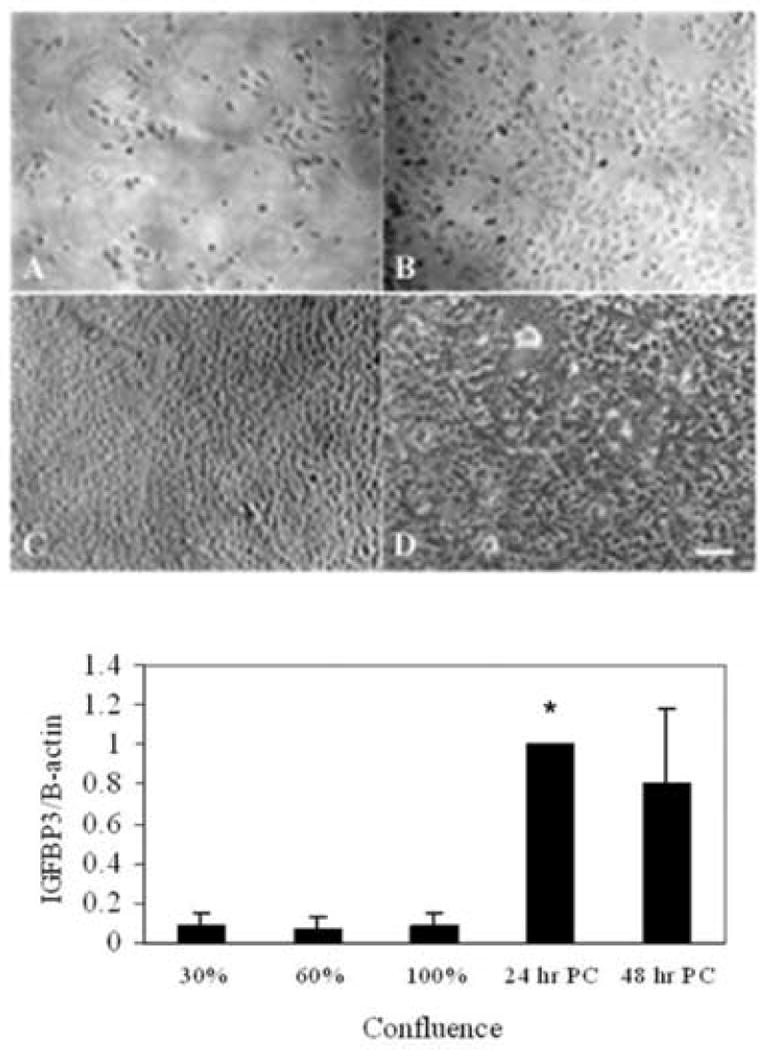
(A-D) Phase contrast microscopy showing representative areas of confluence of hTCEpi cells. A: 20% confluence; B: 60% confluence; C: 100% confluence; D: 24 hour post-confluence. 10× magnification, scale 95 μm. E: Real time PCR demonstrated low levels of IGFBP3 in proliferating hTCEpi cells grown in serum free culture. Following confluence, IGFBP3 mRNA levels demonstrated robust transcriptional upregulation (n=3, mean ± standard deviation of 3 repeated experiments, One-way ANOVA, P<0.001). 30, 60, and 100% represent percentage confluence; 24 hr PC: 24 hour post confluence; 48 hr PC: 48 hour post confluence.
Trichostatin A (TSA), an inhibitor of histone deacetylase, has been shown to upregulate IGFBP3 in other epithelial systems, presumably by facilitating recruitment of RNA polymerase and the requisite transcriptional machinery. To establish whether TSA upregulates IGFBP3 in hTCEpi cells, cells were seeded onto plastic 6-well tissue culture plates and treated with TSA for up to 30 hours. Real time PCR demonstrated an increase in IGFPB3 mRNA at 24 and 30 hours (P<0.001, One-way ANOVA, SNK multiple comparison test) (Figure 5A).
Figure 5.
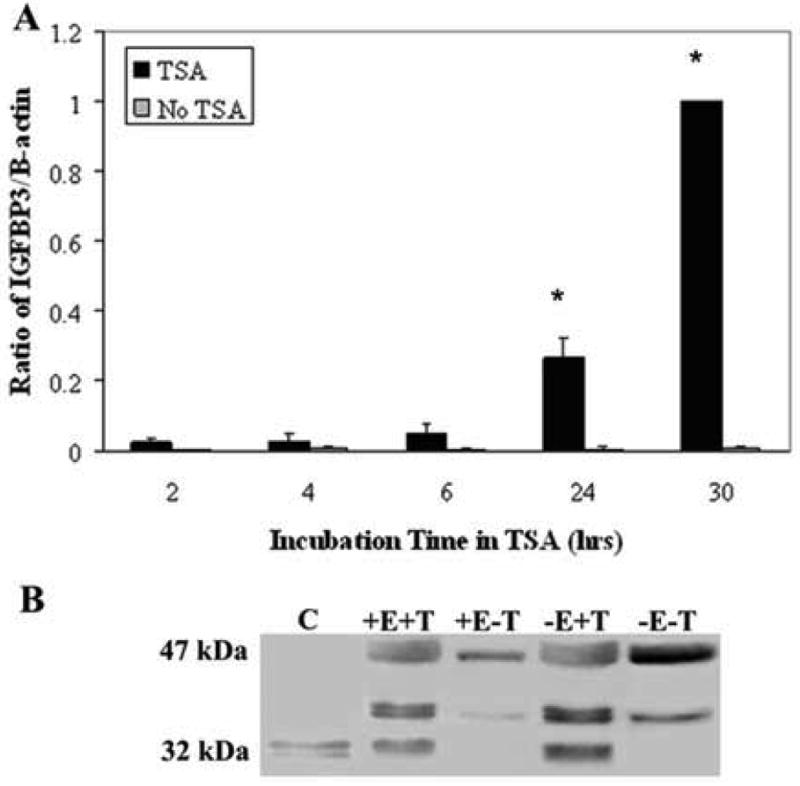
(A) Real time PCR for IGFBP3 in hTCEpi cells following incubation in 3.31 μM TSA for up to 30 hours and a non-treated control. There was a significant increase in IGFBP3 mRNA at 24 and 30 hours (n=3, mean ± standard deviation of 3 repeated experiments, One-way ANOVA, P<0.001). B: Conditioned media from hTCEpi cells treated with/without rhEGF (+/- E) and TSA (+/- T) for 48 hours. Treatment with TSA increased IGFBP3 secretion in conditioned media.
IGFBP3 protein levels were also assessed in vitro in conditioned media of hTCEpi cells. Conditioned media was collected from hTCEpi cells in T75 flasks 48 hours after plating. Conditioned media was concentrated and equal volumes loaded for SDS PAGE. In addition, conditioned media was also collected from cells treated with TSA and in the presence and absence of hEGF, a component of the supplemental bullet kit which has been reported to dampen IGFBP3 levels (Edmondson et al., 1999). Membranes were blotted with an IGFBP3 rabbit anti-serum. hTCEpi cells transfected with a plasmid encoding the cDNA for IGFBP3 was used as a control (Figure 5B). Treatment with TSA resulted in an increase in IGFBP3 in both the rhEGF and non-rhEGF groups. The increase in protein was mostly noted in the non-glycosylated IGFBP3 (32 kDa). The removal of rhEGF from the KGM2 media did not produce a detectable change in either group, suggesting that the concentration of rhEGF used in the serum-free cell culture media is not high enough to effect IGFBP3 signaling.
Apoptosis
To evaluate the effects of IGFBP3 on apoptosis, hTCEpi cells plated in 6-well plastic tissue culture dished were treated with 500 ng/ml rhIGFBP3 and incubated for up to 48 hours. Western blotting for IGFBP3 demonstrated an increase in rhIGFBP3 protein as a function of time up to 6 hours, decreasing following longer incubation periods (P<0.001, One-way ANOVA, SNK multiple comparison test) (Figure 6). We further compared the ability of exogenous rhIGFBP3 to induce apoptosis in hTCEpi cells by triple-labeling with Calcein-AM, Annexin V and PI. Despite patchy areas of membrane blebbing, rhIGFBP3 did not appear to induce positive Annexin V staining in the time points evaluated (Figure 7A). Occasional PI positive cells were noted. We have previously shown that treatment of hTCEpi cells with Trichostatin A results in an upregulation of endogenous IGFBP3 in conditioned media (Figure 5). To determine the effect of Trichostatin A on apoptosis, hTCEpi cells were seeded onto delta T4 tissue culture dishes and allowed to adhere for 24 hours. Cells were treated with TSA, incubated for up to 24 hours, and triple-labeled with Calcein-AM, Annexin V and PI (Figure 7B). By 18 hours, cells demonstrated the formation of blebs and there was a significant increase in the number of apoptotic cells measured by Annexin V and PI (P<0.001, One-way ANOVA, SNK multiple comparison test) (Figure 8). The number of dead cells were further increased at 24 hours (P<0.001).
Figure 6.
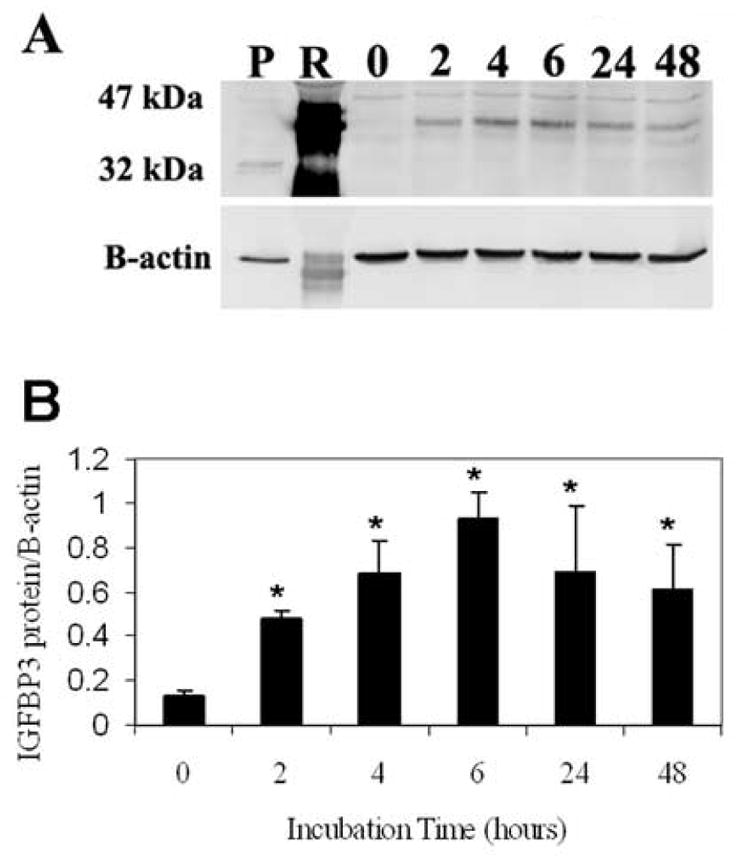
(A) hTCEpi cells grown on plastic and incubated with 20 μg of rhIGFBP3 demonstrated a corresponding increase in IGFBP3 protein levels. B-actin was used as a loading control. P: IGFBP3 plasmid expressed in hTCEpi cells as a positive control; R: 20 μg of recombinant human protein loaded into lane 2 as a positive control; 0 – 48 represent incubation times in hours. (B) Quantitative analysis of IGFBP3 as a function of incubation time. The addition of rhIGFBP3 resulted in an increase in cellular associated IGFBP3 compared to the control (n=3, mean ± standard deviation of 3 repeated experiments, One-way ANOVA, P<0.001).
Figure 7.
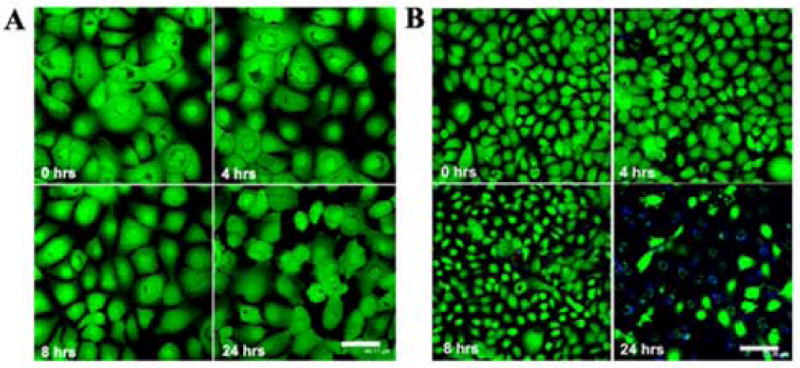
(A) hTCEpi cells grown on glass-bottom tissue culture dishes triple-labeled with Calcein-AM (live cells, green), Annexin V (early apoptotic cells, blue), and PI (dead cells, red) following incubation in rhIGFBP3. Membrane blebbing was seen in occasional cells at 24 hours. 20× magnification, scale: 44 μm. (B) hTCEpi cells grown on glass-bottom tissue culture dishes triple-labeled with Calcein-AM (live cells, green), Annexin V (early apoptotic cells, blue), and PI (dead cells, red) following incubation in TSA. Significant apoptosis was visible at 24 hours. 20× magnification, scale: 75 μm.
Figure 8.
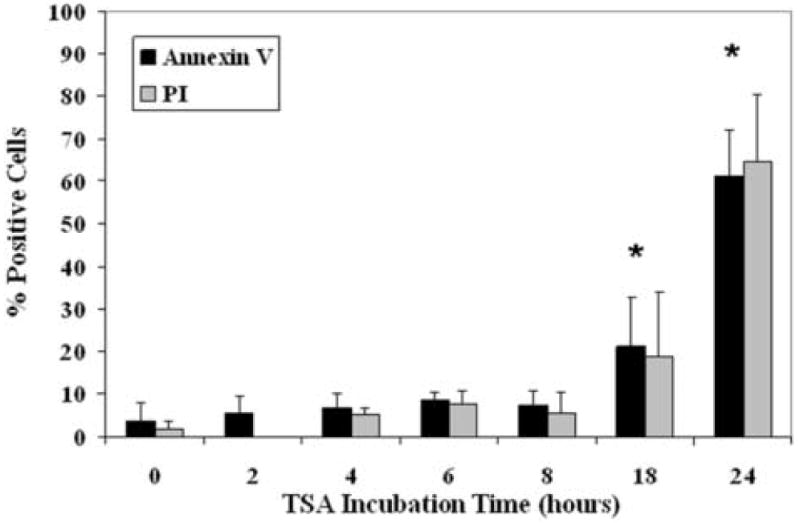
hTCEpi cells grown on glass-bottom tissue culture dishes triple-labeled with Calcein-AM, Annexin V, and PI following incubation in TSA. A significant number of dead cells was seen at 18 and 24 hours (n=2, graph representative of repeated experiments, One-way ANOVA, P<0.001).
As a final measure of apoptotic cell death, we assessed hTCEpi cells grown on collagen-coated glass coverslips by double-labeling with TUNEL and PI (Figure 9A). hTCEpi cells were treated with either TSA or rhIGFBP3 and evaluated at 0, 24, and 48 hours. At 24 hours, 60% of the cells treated with TSA labeled positive for TUNEL (P<0.001, One-way ANOVA, SNK multiple comparison test), decreasing to 50% at 48 hours (Figure 9B). Likewise, there was a significant reduction in the number of cells present after 48 hours in TSA. No TUNEL-positive cells were seen following treatment with exogenous rhIGFBP3 at the time points examined. Cells pre-treated with DNase I for use as a positive control demonstrated greater than 98% positive TUNEL labeling. No positive TUNEL labeling was seen in the negative control.
Figure 9.
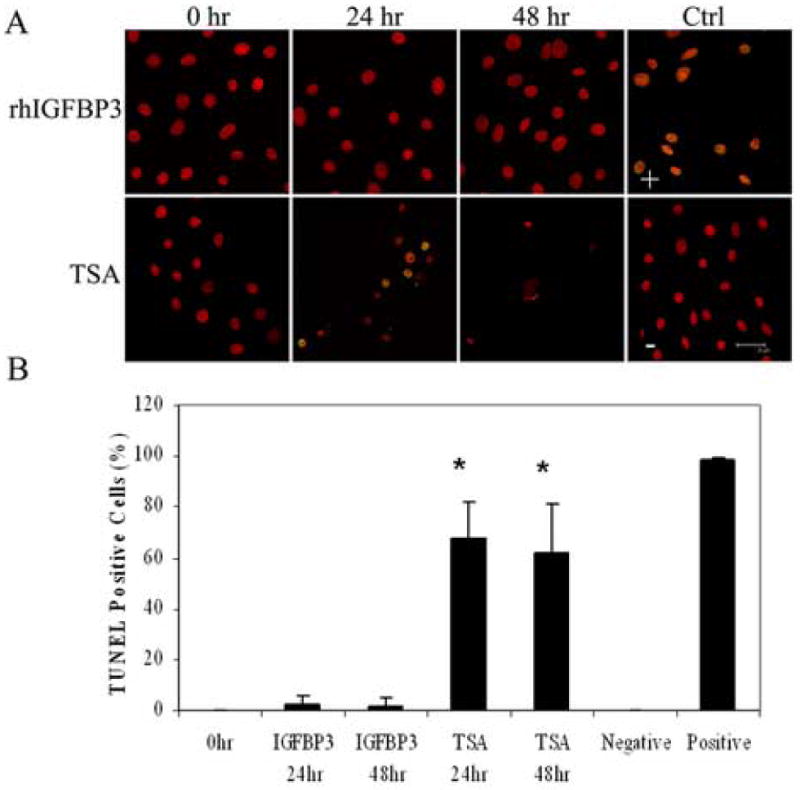
(A) Double-labeling of hTCEpi cells with TUNEL (green) and PI (red) on collagen-coated glass coverslips and treated with 500 ng/ml rhIGFBP3 or 3.31 μM TSA for 0, 24, and 48 hours. Positive control (+): cells treated with DNase; negative control (-): TdT Enzyme omitted. Scale: 28 μm. (B) Percentage of TUNEL positive cells following treatment with TSA and rhIGFBP3 protein. TSA induced significant cell death (n=2, graph representative of repeated experiments, One-way ANOVA, P<0.001). At 48 hours, robust cell loss was seen due to apoptosis, making quantification difficult. 63× magnification, scale: 28 μm.
Discussion
Extracellular IGFBP3
IGFBP3 is a secretory, high-affinity binding protein with known growth inhibitory and apoptotic functions. The mechanism by which IGFBP3 exerts these effects, autocrine, intracrine, or paracrine, is presently unknown. Using indirect immunofluorescence, we report the presence of IGFBP3 in normal human cornea, localizing IGFBP3 to the plasma membrane of corneal epithelial cells. This extracellular localization pattern of IGFBP3 is consistent with a secretory role for this protein, which was confirmed by our studies using conditioned media. Since it has been previously established that apoptosis occurs in surface cells of the central cornea (Ren and Wilson, 1996), the pronounced staining of IGFBP3 in the superficial cells of the central corneal epithelium is suggestive of a regulatory role in corneal surface cell shedding (Li et al., 2002; Yamamoto et al., 2002). Surprisingly, the non-detectable level of IGFBP3 in basal, resting human tears demonstrates that IGFBP3 is not derived from the lacrimal system and is not present in the native tear film, suggesting that it functions in an auto- or intracrine-mediated feedback loop within the corneal epithelium.
In contrast, basal cells in the peripheral cornea appeared to demonstrate an increase in plasma membrane staining compared to central corneal epithelium. This increase in peripheral basal cell staining may stem from differential regulation during early epithelial differentiation and migration. Other IGF-binding proteins, IGFBP1 and 2, have been shown to bind directly to integrins through an RGD binding motif, effecting roles in adhesion and cell motility (Firth and Baxter, 2002). In addition, IGFBP3 and IGFBP5 share a conserved 18 amino acid sequence at the C-terminus, which has been reported to have a stimulatory effect on cell migration (Firth and Baxter, 2002). Therefore, the strong staining for IGFBP3 in the basal layer of the peripheral epithelium may indicate a direct role in the centripetal migration of epithelial cells.
Glycosylation and proteolysis likely plays a key role in the tissue-specific activity of IGFBP3. Previous reports have established that IGFBP3 undergoes N-linked glycosylation at residues Asn89, Asn109 and Asn172, allowing for single and multi-site glycosylation (Firth and Baxter, 1999). Moreover, the authors of that study demonstrated that each carbohydrate moiety adds 4 – 5 kDa to the molecular weight. IGFBP3 is normally reported to be present as at 40-42 kDa and 45 – 47 kDa when glycosylated at two or three residues, respectively. Additional de-glycosylation experiments by the same group using sequential non-reducing and reducing conditions further demonstrated the presence of a 37 kDa form of IGFBP3 under non-reducing conditions and a 30 kDa non-glycosylated protein under reducing conditions. Thus, the multi-isoform expression seen in our experiments using human corneal tissue at 32 kDa, 37 kDa, 42 kDa and 47 kDa are suggestive of various stages of glycosylated protein. The significance of variable levels of glycosylation is unknown; however, the absence of these carbohydrate chains does not appear to prevent IGFBP3 secretion, as evident by the presence of a 32 kDa isoform in our conditioned media studies and those reported elsewhere.
Significantly, we have not yet been able to localize IGFBP3 to the nuclei of human corneal epithelium in situ or in vitro; however, previous studies in human breast cancer cells demonstrate that nuclear localization of IGFBP3 is not required for growth inhibition and apoptosis and suggests that cytoplasmic IGFBP3 may regulate gene activities indirectly by binding to transcription factors and modulating their nuclear translocation (Butt et al., 2002). Additional studies supporting the nuclear independent effects of IGFBP3 have reported that IGFBP3 can induce signaling events in an intracrine signaling pathway, adding another potential level of signaling complexity to this protein (Bhattacharyya et al., 2006).
Intracellular IGFBP3
A surprising finding is the localization of IGFBP3 throughout the limbal epithelium in vivo. In the limbus IGFBP3 localized to the cytoplasm in clusters of basal and suprabasal limbal epithelial cells. This subcellular localization and distribution of IGFBP3 is suggestive of individual tissue-compartment specific effects and support a role for IGFBP3 in mediating early onset differentiation of epithelial cells as they migrate out of the limbus and onward into the central cornea. While the recruitment of daughter cells from the “niche” may drive an upregulation of locally produced IGFBP3 needed to balance the effects of IGF-I in maintaining growth down-regulation, the distribution of IGFBP3 in the vascularized limbus, as seen in Figure 2, may reflect a potential role for serum proteins and their affect on cellular growth and tissue maintenance. IGFBP3 is the most predominant insulin-like binding protein in serum and the high levels of IGFBP3 may be a direct result of extravasation from the limbal arcades (Baxter and Martin, 1989). The potential for systemic effects of IGFPB3 in the limbus in regulation of proliferation and differentiation raises interesting questions regarding the differences in binding partners and post-translational modifications between systemic and locally produced IGFBP3 and their effects on tissue homeostasis. Not surprisingly, our western blotting data demonstrated the presence of different IGFBP3 isoforms between the limbus and central cornea. Significantly, we identified a 64 kDa isoform of IGFBP3 in human tissue, which was not observed in conditioned media or lysates from in vitro cell culture experiments. The presence of a 64 kDa dimer of IGFBP3 has been previously reported and was shown to be associated with the cell surface (Leal et al., 1999). Thus, the presence of a cell surface associated dimer is consistent with our immunofluorescent findings and suggests that dimerization of IGFBP3 is necessary for cell surface binding and may be a prerequisite for internalization of this protein.
Given the critical role of IGFBP3 in mediating IGF-I induced proliferation; we evaluated IGFBP3 in cells in their log growth phase and following confluence. By western blotting, intracellular IGFBP3 expression was not detectable in proliferating hTCEpi cells and mRNA levels remained relatively low. However, both mRNA and protein levels were elevated following confluence and calcium induced differentiation. The upregulation of IGFBP3 following confluence and differentiation is suggestive of a growth inhibitory role for IGFBP3 in the corneal epithelium. While not yet investigated in the corneal epithelium, matrix metalloproteinases (MMPs) have been shown to cleave IGFBP3 in epidermal keratinocytes, freeing bound IGF-1 to induce proliferation, representing a potential mechanism that may account for the low levels of IGFBP3 seen in growing cells (Manes et al., 1999; Sadowski et al., 2003).
IGFBP3 Binding, Internalization, and Apoptosis
In our study, exogenously applied rhIGFBP3 appeared to associate with cells in vitro, suggestive of membrane binding and/or internalization; however, the addition of rhIGFBP3 to cell culture media did not appear to have the same direct affect on apoptosis as TSA. This difference could be attributed to cell surface receptor specificity or failure of the purified, recombinant protein to internalize and initiate the apoptotic cascade due to differences in glycosylation. The role of glycosylation and various glycoforms in the ability of proteins to bind to and activate receptors has been demonstrated previously in studies evaluating gonadotrophin hormone signaling. Specifically, deglycosylated human chorionic gonadotrophin retains the ability to bind to the appropriate receptor but fails to activate and initiate signal transduction (Thotakura and Blithe, 1995). Thus various glycoforms of IGFBP3 may mediate intracellular signaling in a compartment-specific manner. Further studies are underway to evaluate the significance of these glycosylation variants of IGFBP3 in the human corneal epithelium and the signaling specificity of TSA to mediate apoptosis.
Conclusions
These findings demonstrate for the first time the presence of IGFPB3 in the human corneal epithelium. The differential localization of IGFBP3 in human tissue suggests that the effects of IGFBP3 may be tissue and cell type specific. Further, the ability of IGFBP3 to associate with the cell surface and initiate a signaling cascade, either through a cell surface receptor or through internalization, may be tightly regulated by glycosylation and dimerization. Significantly, IGFBP3 in superficial cells of the central cornea may mediate apoptosis directly through an intrinsic apoptotic mechanisms or may function to sensitize the apical surface to extrinsic apoptotic signaling events induced by various cytokines and chemokines present in the ocular surface milieu. The susceptibility to apoptosis of the surface epithelia from pro-inflammatory cytokines may account for the increased rates in desquamation associated with inflammatory conditions such as dry eye. While additional study is necessary to elucidate the role of IGFBP3 in the limbal and corneal epithelium, the results from these studies support a potential role for IGFBP3 in growth down regulation, differentiation, and apoptosis in the corneal epithelium.
Acknowledgments
Supported in Part by NIH Grant K08 EY15713 (DMR), EY10738 (HDC), Infrastructure Grant EY016664, W.C. Ezell Fellowship (DMR), The Pearle Vision Foundation, Dallas, Texas, and a Lew R. Wasserman Merit Award (WMP) and an unrestricted grant from Research to Prevent Blindness, Inc., New York, New York.
Footnotes
CR: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baxter RC, Martin JL. Structure of the Mr 140,000 Growth Hormone-Dependent Insulin-Like Growth Factor Binding Protein Complex: Determination by Reconstitution and Affinity-Labeling. Proc Natl Acad Sci USA. 1989;86:6898–6902. doi: 10.1073/pnas.86.18.6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya N, Pechhold K, Shahjee H, Zappala G, Elbi C, Raaka B, Wiench M, Hong J, Rechler MM. Nonsecreted Insulin-like Growth Factor Binding Protein-3 (IGFBP-3) Can Induce Apoptosis in Human Prostate Cancer Cells by IGF-independent Mechanisms without Being Concentrated in the Nucleus. J Biol Chem. 2006;281:24588–24601. doi: 10.1074/jbc.M509463200. [DOI] [PubMed] [Google Scholar]
- Butt AJ, Fraley KA, Firth SM, Baxter RC. IGF-Binding Protein-3-Induced Growth Inhibition and Apoptosis Do Not Require Cell Surface Binding and Nuclear Translocation in Human Breast Cancer Cells. Endocrinology. 2002;143:2693–2699. doi: 10.1210/endo.143.7.8876. [DOI] [PubMed] [Google Scholar]
- Butt AJ, Williams AC. IGFBP-3 and apoptosis - a license to kill? Apoptosis. 2001;6:199–205. doi: 10.1023/a:1011388710719. [DOI] [PubMed] [Google Scholar]
- Edmondson SR, Murashita MM, Russo VC, Wraight CJ, Werther GA. Expression of Insulin-Like Growth Factor Binding Protein-3 (IGFBP-3) in Human Keratinocytes Is Regulated by EGF and TGFB-1. J Cell Physiol. 1999;179:201–207. doi: 10.1002/(SICI)1097-4652(199905)179:2<201::AID-JCP10>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Firth SM, Baxter RC. Characterisation of recombinant glycosylation variants of insulin-like growth factor binding protein-3. J of Endocrinology. 1999;160 doi: 10.1677/joe.0.1600379. [DOI] [PubMed] [Google Scholar]
- Firth SM, Baxter RC. Cellular Actions of the Insulin-Like Growth Factor Binding Proteins. Endocrine Reviews. 2002;23:824–854. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- Izumi K, Kurosaka D, Iwata T, Oguchi Y, Tanaka Y, Mashima Y, Tsubota K. Involvement of Insulin-like Growth Factor-I and Insulin-like Growth Factor Binding Protein-3 in Corneal Fibroblasts during Corneal Wound Healing. Invest Ophthalmol Vis Sci. 2006;47:591–598. doi: 10.1167/iovs.05-0097. [DOI] [PubMed] [Google Scholar]
- Leal SM, Huang SS, Huang JS. Interactions of High Affinity Insulin-like Growth Factor-binding Proteins with the Type V Transforming Growth Factor-Beta Receptor in Mink Lung Epithelial Cells. J Biol Chem. 1999;274:6711–6717. doi: 10.1074/jbc.274.10.6711. [DOI] [PubMed] [Google Scholar]
- Lee JH, Ryu IH, Kim EK, Lee JE, Hong SW, Lee HK. Induced Expression of Insulin-like Growth Factor-1 by Amniotic Membrane-Conditioned Medium in Cultured Human Corneal Epithelial Cells. Invest Ophthalmol Vis Sci. 2006;47:864–872. doi: 10.1167/iovs.05-0596. [DOI] [PubMed] [Google Scholar]
- Li L, Ren DH, Ladage PM, Yamamoto K, Petroll WM, Jester JV, Cavanagh HD. Annexin V Binding to Rabbit Corneal Epithelial Cells Following Overnight Contact Lens Wear or Eyelid Closure. CLAO J. 2002;28:48–54. [PubMed] [Google Scholar]
- Manes S, Llorente M, Lacalle RA, Gomez-Mouton C, Kremer L, Mira E, Martinez-A C. The Matrix Metalloproteinase-9 Regulates the Insulin-like Growth Factor-triggered Autocrine Response in DU-145 Carcinoma Cells. J Biol Chem. 1999;274:6935–6945. doi: 10.1074/jbc.274.11.6935. [DOI] [PubMed] [Google Scholar]
- Nagama T, Nakamura M, Nakata K, Yamaguchi T, Takase K, Okahara A, Ikuse T, Nishida T. Effects of Substance P and IGF-1 on Corneal Epithelial Barrier Function and Wound Healing in a Rat Model of Neurotrophic Keratopathy. Invest Ophthalmol Vis Sci. 2003;44:3810–3815. doi: 10.1167/iovs.03-0189. [DOI] [PubMed] [Google Scholar]
- Nishida T. Neurotrophic Mediators and Corneal Wound Healing. The Ocular Surface. 2005;3:194–202. doi: 10.1016/s1542-0124(12)70206-9. [DOI] [PubMed] [Google Scholar]
- Rajah R, Lee KW, Cohen P. Insulin-like Growth Factor Binding Protein-3 Mediates Tumor Necrosis Factor-alpha-induced Apoptosis: Role of Bcl-2 Phosphorylation. Cell Growth and Differentiation. 2002;13:163–171. [PubMed] [Google Scholar]
- Ren H, Wilson G. Apoptosis in the Corneal Epithelium. Invest Ophthalmol Vis Sci. 1996;37:1017–1025. [PubMed] [Google Scholar]
- Rocha EM, Cunha DA, Carneiro EM, Boschero AC, Saad MJA, Velloso LA. Identification of Insulin in the Tear Film and Insulin Receptor and IGF-I Receptor on the Human Ocular Surface. Invest Ophthalmol Vis Sci. 2001;43:963–967. [PubMed] [Google Scholar]
- Sadowski T, Dietrich S, Koschinsky F, Sedlacek R. Matrix Metalloproteinase 19 Regulates Insulin-like Growth Factor-mediated Proliferation, Migration, and Adhesion in Human Keratinocytes through Proteolysis of Insulin-like Growth Factor Binding Protein-3. Mol Cell Biol. 2003;14:4569–4580. doi: 10.1091/mbc.E03-01-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thotakura NR, Blithe DL. Glycoprotein hormones: glycobiology of gonadotrophins, thyrotrophin and free alpha subunit. Glycobiology. 1995;5:3–10. doi: 10.1093/glycob/5.1.3. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ladage PM, Ren DH, Li L, Jester JV, Cavanagh HD. Epitope Variability of Bcl-2 Immunolocalization in the Human Corneal Epithelium. CLAO J. 2001a;27:221–224. [PubMed] [Google Scholar]
- Yamamoto K, Ladage PM, Ren DH, Li L, Petroll WM, Jester JV, Cavanagh HD. Bcl-2 Expression in the human cornea. Exp Eye Res. 2001b;73:247–255. doi: 10.1006/exer.2001.1027. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ladage PM, Ren DH, Li L, Petroll WM, Jester JV, Cavanagh HD. Effect of Eyelid Closure and Overnight Contact Lens Wear on Viability of Surface Epithelial Cells in Rabbit Cornea. Cornea. 2002;21:85–90. doi: 10.1097/00003226-200201000-00018. [DOI] [PubMed] [Google Scholar]