

Abstract
Polyphenolic natural products from green tea and red wine have been identified as metalloproteinase inhibitors. Members from the flavonoid and stilbene families found to possess metalloproteinase inhibitory activities include (−)-epigallocatechin gallate (EGCG), (−)-epicatechin gallate (ECG), and piceatannol, but their minimally active pharmacophores have not been evaluated. The present study has examined compounds that are structural components of or structurally related to EGCG, ECG, and piceatannol for inhibition of aggrecanases and four representative matrix metalloproteinases (MMPs). Piceatannol and pyrogallol were found to inhibit all aggrecanases and MMPs studied, indicating a crucial reliance on multiple hydroxyl groups for EGCG, ECG, and piceatannol activity. Differences in Ki values for pyrogallol as determined with two structurally distinct substrates indicated the likelihood that this compound binds in a noncompetitive modality. Further analysis showed that pyrogallol acts as an exosite inhibitor, consistent with the action of EGCG. In contrast, piceatannol was shown to be a competitive binding inhibitor and showed no differences in apparent Ki values as determined by distinct substrates, illustrating the benefits of using two structurally distinct substrates to assist the analysis of protease inhibitors. The compounds identified here could be utilized to develop novel metalloproteinase probes or as fragment components of more active inhibitors.
Keywords: Metalloproteinase, ADAMTS, matrix metalloproteinase, EGCG, inhibitor, flavonoid, FRET substrate
Introduction
Significant efforts have been devoted to the development of inhibitors for metalloproteinases implicated in the progression of arthritis, tumor metastasis, and vascular diseases. Within the last decade several members from the flavonoid family have been found to possess metalloproteinase inhibitory activities. For example, amongst a series of green tea catechin gallate esters tested against the aggrecanase activity of a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS) family members ADAMTS-1, ADAMTS-4, and ADAMTS-5, (−)-epigallocatechin gallate (EGCG) (Figure 1, compound 1) and (−)-epicatechin gallate (ECG) (Figure 1, compound 2) had IC50 values of 100–150 nM against ADAMTS-4 and ADAMTS-5 and 200–250 nM against ADAMTS-1 (1). EGCG was also found to be effective against several members of the matrix metalloproteinase (MMP) family. EGCG inhibited MMP-2, MMP-7, MMP-9, and MT1-MMP with IC50 values of 6–8, 1.6, 0.8–13, and 0.019 μM, respectively (2–6). EGCG and ECG were considerably less effective inhibitors of MMP-1, MMP-13, and ADAM-10, with only 14–30% inhibition occurring at inhibitor concentrations of 50 μM (1). On a cellular level, EGCG exhibited inhibitory activities against human umbilical vein endothelial cell (HUVEC) invasion, tube formation, and angiogenesis, as well as tumor cell invasion (2, 6).
Figure 1.

Structure of EGCG (1), ECG (2), and piceatannol (3).
Amongst 8 members of the flavonoid family tested, luteolin-7-O-glucoside was the most effective against MMP-9 (EC50 = 4.6 μM), while apigenin was the most effective against MMP-2 (EC50 = 7.5 μM) (7). Interestingly, apigenin was a very weak inhibitor of MMP-1 (IC50 > 100μM), whereas the flavonoids quercetin and kaempferol inhibited MMP-1 with IC50 values of 39.6 and 43.7 μM, respectively (8). Primary screening of ADAMTS-4 against the LOPAC™ inhibitor group, followed by RP-HPLC secondary screening, identified a member of the stilbene family, piceatannol (Figure 1, compound 3), as the best inhibitor of the protease (9). Piceatannol has been shown to inhibit proMMP-9 expression and tumor-induced angiogenesis (10). Piceatannol is structurally related to green tea polyphenols and flavonoids (Figure 1, compare 1 and 2 to 3). However, no studies have been performed to evaluate the minimally active pharmacophores that are commonly shared by green tea polyphenols, flavonoids, and stilbenes.
Circumstantial evidence suggests that flavonoid inhibition of metallproteinases occurs via interaction with regions distant from the active site (secondary binding sites; exosites). Inhibition of aggrecanases and MMP-2 by EGCG was not due to active site Zn2+ chelation (1, 4). EGCG inhibited MMP-2 noncompetitively (4). In similar fashion, luteolin inhibited MMP-9 by a noncompetitive mechanism with Ki = 5.4 μM (7). The action of EGCG on metalloproteinases is distinctly different from its inhibition of serine proteases such as human neutrophil elastase, whereby the later interactions are within the enzyme active site and are competitive with substrate (11).
Our laboratory recently observed that, by using fluorescence resonance energy transfer (FRET) substrates of different sizes and secondary structures, different MMP inhibitory values may be obtained with the same compound (12–14). In some cases, these different values are due to the inhibitor interactions with exosites and greater sensitivity of one substrate to this binding event (13). Exosites have been exploited to obtain inhibitors for caspases (15) and coagulation Factors VIIa, IXa, and Xa (16–18) and an activator for the serine protease HtrA (19). In addition, the collagenolytic activity of cathepsin K can be specifically inhibited by the addition of anionic polymers that displace binding of chondroitin sulfate to a highly cationic domain of the enzyme (20).
The present study has examined inhibition of members of the ADAMTS and MMP families by potential pharamacophores. The commercially available compounds are structural components of or structurally related to EGCG, ECG, and piceatannol. In addition, different FRET substrates have been utilized within each metalloproteinase family to determine if inhibitors exhibit different behaviors based on substrate size and secondary structure.
Materials and Methods
Reagents
All standard chemicals were purchased from VWR (West Chester, PA). 2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate, 1-hydroxybenzotriazole, and 9-fluorenylmethoxycarbonyl (Fmoc)-amino acid derivatives [including Fmoc-Lys(Mca) and Fmoc-Lys(Dnp)] were obtained from Novabiochem (San Diego, CA). Fluorescein-5-isothiocyanate and 5/6-carboxytetramethylrhodamine succinimidyl ester were purchased from AnaSpec (San Jose, CA). Piceatannol, [(E)-4-[2-(3,5-dihydroxyphenyl)ethenyl]1,2-benzediol; pyrocatechol, 1,2-dihydroxybenzene; pyrogallol, 1,2,3-trihydroxybenzene; trans-stilbene, trans-1,2-diphenylethylene; cis-stilbene, cis-1,2-diphenylethylene; deoxyrhapontin, 2-[3-hydroxy-5-[(E)-2-(4-methoxyphenyl)ethenyl]phenoxy]-6-(hydroxymethyl)oxane-3,4,5-triol; rhapontin, 2-[3-hydroxy-5-[2-(3-hydroxy-4-methoxyphenyl)ethenyl]phenoxy]-6-(hydroxymethyl)oxane-3,4,5-triol; and resveratrol, 5-[(1E)-2-(4-hydroxyphenyl)ethenyl]-1,3-benzenediol were obtained from Sigma (St. Louis, MO).
Peptide Synthesis and Purification
The aggrecanase peptide substrates used were fTSSlong, fTSSshort, and fTSS1 (Table 1). The synthesis, purification, and characterization of fTSSlong, which is also referred to as fSSPa, have been described (21). Syntheses of the other two substrates were performed by Fmoc solid-phase methodology on a Protein Technology PS3 Peptide Synthesizer. For fTSS1 (Table 1), Fmoc-Lys(Dde) and Boc-Lys(Fmoc) were utilized during chain assembly in the positions where Lys(Tamra) and Lys(Fam) were desired. Following removal of the ε-amino Fmoc group by standard deprotection conditions, Fam was added using fluorescein-5-isothiocyanate. The ε-amino Dde group was then removed as described previously (22), and Tamra was added by acylation with 5/6-carboxytetramethylrhodamine succinimidyl ester. Peptides were cleaved from the resin using thioanisole–water–trifluoroacetic acid (TFA) (5:5:90) for 2 h. Cleavage solutions were extracted with methyl tert-butyl ether prior to purification. RP-HPLC purification was performed on a Rainin AutoPrep System with a Vydac 218TP152022 C18 column (15–20 μm particle size, 300 Å pore size, 250 × 22 mm) at a flow rate of 10.0 mL/min. Eluants were 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B). The elution gradient of was 0% B for the first 5 min, 0–25% B for additional 5 min followed by 25–70% B in 50 min. Detection was at λ = 220 nm. Fractions were analyzed by MALDI-TOF MS and the ones with the correct mass were pooled together and lyophilized. The purity of peptide was confirmed by analytical RP-HPLC and MALDI-TOF MS. Analytical RP-HPLC was performed on a Hewlett-Packard 1100 liquid chromatograph equipped with a Vydac 218TP5415 C18 RP column (5 μm particle size, 300 Å pore size, 150 × 4.6 mm). Eluants were 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B). The elution gradient was 0–100% B in 20 min with a flow rate of 1.0 mL/min. Detection was at λ = 220, 324, and 363 nm. MALDI-TOF MS was performed on an ABD DE-STR Voyager mass spectrometer using α-cyano-4-hydroxycinnamic acid matrix. Determined peptide mass values were [M + H]+ 1631.5 Da (theoretical, 1631.0 Da) for fTSSshort (Table 1), and [M + H]+ 2862.1 Da (theoretical, 2863.3 Da) for fTSS1. The KM value for ADAMTS-4 hydrolysis of fTSSlong is 16.1 ± 2.6 μM (Table 4), while the KM value for ADAMTS-4 hydrolysis of fTSS1 is ~35 μM (23).
Table 1.
Substrates described in the present study.
| Enzymes | Substrate Name | Sequence |
|---|---|---|
| Aggrecanases | fTSSlong | C6-(Gly-Pro-Hyp-Pro-Hyp-Gly)2-Gly-Pro-Hyp-Gly-Thr-Lys(Mca)-Gly-Glu~Leu~Glu~Gly-Arg~Gly-Thr-Lys(Dnp)-Gly-Ile-Ser-(Gly-Pro-Hyp-Pro-Hyp-Gly)2-Gly-Pro-Hyp-NH2 |
| fTSSshort | Lys(Mca)-Lys-Gln-Glu-Phe-Arg-Gly-Gln-Thr-Lys(Dnp)-NH2 | |
| fTSS1 | Lys(Fam)-Asp-Val-Gln-Glu-Phe-Arg-Gly-Val-Thr-Ala-Val-Ile-Arg-Lys(Tamra)-Lys-Gly-Lys-NH2 | |
| fTSS2 | Abz-Thr-Glu-Gly-Glu~Ala-Arg-Gly-Ser-Val-Ile-Dap(Dnp)-Lys-Lys-NH2 | |
| fTSS3 | Fam-Ala-Glu~Leu-Gln-Gly-Arg-Pro-Ile-Ser-Ile-Ala-Lys(Tamra) | |
| MMPs | fMMPSlong | (Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Gln-Gly~Leu-Arg-Gly-Gln-Lys(Dnp)-Gly-Val-Arg-(Gly-Pro-Hyp)5-NH2 |
| fMMPSshort | Mca-Lys-Pro-Leu-Gly-Leu-Lys(Dnp)-Ala-Arg-NH2 |
Table 4.
Results of non-linear regression analysis of ADAMTS-4 inhibition of fTSSlong hydrolysis by compounds 3 and 10.
| Inhibitor | Concentration (μM) | Apparent Vmax (RFU/h) | Apparent KM (μM) |
|---|---|---|---|
| 3 | 0 | 177.2 ± 10.7 | 16.1 ± 2.6 |
| 2 | 196.9 ± 16.9 | 39.8 ± 6.6 | |
| 3.5 | 177.9 ± 34.7 | 39.5 ± 14.9 | |
| 5 | 184.3 ± 27.3 | 48.9 ± 13.1 | |
| 10 | 0 | 177.2 ± 10.7 | 16.1 ± 2.6 |
| 2 | 128.4 ± 5.6 | 13.3 ± 1.8 | |
| 3.5 | 87.1 ± 7.5 | 13.0 ± 3.2 | |
| 5 | 56.3 ± 1.6 | 11.7 ± 1.0 |
The MMP substrates were fMMPSshort and fMMPSlong (Table 1). Both were synthesized and characterized by methods described previously, where fMMPSshort is often referred to as Knight SSP and fMMPSlong is referred to as fTHP-4/fTHP-15 (14, 24–26). The KM values for MMP hydrolysis of fMMPSlong range from 7.6 μM (MMP-8) to 17.2 μM (MMP-2) (12, 26, 27), while the KM values for MMP hydrolysis of fMMPSshort ranges from 27.7 μM (MMP-9) to 130 μM (MMP-12) (12, 28).
Enzymes
ADAMTS-4 (aggrecanase-1) was expressed as previously described (29). ADAMTS-4–2 was used in the present study. This form represents the full length ADAMTS-4 minus the C-terminal spacer domain, and it retains the maximum peptidase activity of the enzyme (21). ADAMTS-5 was purchased from Chemicon International, and was expressed without the C-terminal spacer and second thrombospondin domains, making it structurally comparable to ADAMTS-4–2 (30). The amount of active ADAMTS-4 and ADAMTS-5 was determined by titration with recombinant N-TIMP-3 (31). MMPs were obtained as previously described (14).
Inhibition Assay
Stock solutions of peptides were prepared at 100 μM concentrations in TSB buffer (50 mM Tris HCl, pH = 7.5, 100 mM NaCl, 10 mM CaCl2, 0.05% Brij-35, 0.02% NaN3). Inhibitors were prepared as 2 mM solutions in DMSO and than further diluted with TSB. Enzyme assays were conducted in TSB by incubating 10 μM substrate (ADAMTS-4) or 5 μM substrate (ADAMTS-5, MMPs) with 10 nM enzyme for 24 h in the presence or absence of inhibitors. Final concentrations of inhibitors were 0.1, 1, 10, and 100 μM. For ADAMTSs, fluorescence readings (λexcitation = 324 nm and λemission = 393 nm for Mca) were obtained at 0, 1, 2, 3, 4, 5, and 19 h. For MMPs, fluorescence readings were taken at minimal intervals for 10–30 min, assuring all readings were taken within the linear portion of the hydrolysis curve. The enzyme and inhibitor were pre-incubated for 1 h at 37 °C before the addition of a substrate. The change in relative fluorescence units (ΔRFU) was calculated by ΔRFU = RFU(S+E+I)24 h − RFU(S+E+I)0 h. To determine Ki values, inhibition assays were repeated using a narrower concentration range of inhibitors producing approximately 40–80% inhibition with 10 μM substrate. Ki values were determined by SigmaPlot non-linear regression analysis, using the equation for reversible inhibitors, f = 1/[1+(I/Ki)], fitting f to y where y = Vi/Vo, with a constraint of Ki > 0. GraphPad Prism software was used to determine inhibitor binding modality. Data were analyzed using non-linear regression with fitting to the Michaelis-Menton equation to determine apparent Vmax and KM in the presence or absence of inhibitor. The data were further analyzed by non-linear regression analyses with curve fitting to each modality of inhibitor binding (competitive, noncompetitive, mixed noncompetitive, or uncompetitive). In each case, unmodified software equations were utilized.
Analysis of Cleavage Products
After the final reading at 24 h, the enzyme reaction was stopped by addition of 15 μL of 50 mM EDTA. The reaction solution was analyzed by RP-HPLC. Analytical RP-HPLC was performed with a Vydac 208TP5415 protein and peptide C8 column (15–10 μm particle size, 300 Å pore size, 150 × 4.1 mm). Eluants were 0.1 % TFA in water (A) and 0.1 % TFA in acetonitrile (B). The elution gradient was 0–50 % B in 20 min with a flow of 1 mL/min. Detection was at λ = 220, 324, and 363 nm. Reaction yields in the presence of inhibitors were evaluated by the integration of the RP-HPLC peaks formed compared to the enzyme reaction without inhibitor present. Integrations were averaged from two injections. Product identification was achieved by MALDI-TOF MS.
Results
A prior high throughput screening effort identified piceatannol ([(E)-4-[2-(3,5-dihydroxyphenyl)ethenyl]1,2-benzediol; Figure 1, compound 3) as an inhibitor of ADAMTS-4, with an IC50 value of 1 μM (9). Similarly, EGCG and ECG (Figure 1, compound 1) inhibit ADAMTS-4 and ADAMTS-5 with IC50 values of approximately 100–150 nM (1). Thus, the compounds examined herein represented common or analogous components of piceatannol, EGCG, and/or ECG. Piceatannol itself captures many of the catechin features of EGCG and ECG. Resveratrol (5-[(1E)-2-(4-hydroxyphenyl)ethenyl]-1,3-benzenediol) (4) (Table 2) possesses one fewer hydroxyl group than piceatannol. Trans-stilbene (trans-1,2-diphenylethylene) (5) (Table 2) represents the non-hydroxylated backbone of piceatannol and resveratrol, while cis-stilbene (cis-1,2-diphenylethylene) (6) (Table 2) is its isomer. Two glycosylated stilbenes related to resveratrol were also included, deoxyrhapontin (2-[3-hydroxy-5-[(E)-2-(4-methoxyphenyl)ethenyl]phenoxy]-6-(hydroxymethyl)oxane-3,4,5-triol) (7) and rhapontin (2-[3-hydroxy-5-[2-(3-hydroxy-4-methoxyphenyl)ethenyl]phenoxy]-6-(hydroxymethyl)oxane-3,4,5-triol) (8) (Table 2). Modulation of MMP activity and selectivity had been observed previously for methylated hydroxyl and glycosylated flavanoids (7, 32) and methylated hydroxyl green tea polyphenols (5). Finally, pyrocatechol (1,2-dihydroxybenzene) (9) (Table 2), which represents a potentially minimally active component of picetannol and ECG, and pyrogallol (1,2,3-trihydroxybenzene) (10) (Table 2), which represents a potentially minimally active component of EGCG, were evaluated.
Table 2.
Initial screening of ADAMTS-4 inhibitors using the substrate fTSSlong.
| Inhibitor Structure | Inhibitor Name | Compound Number | FRET Assay IC50 (μM) | RP-HPLC Assay IC50(μM) |
|---|---|---|---|---|
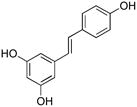 |
resveratrol | 4 | 50 | >100 |
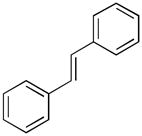 |
trans-stillbene | 5 | >100 | >100 |
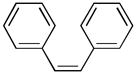 |
cis-stillbene | 6 | ~100 | >100 |
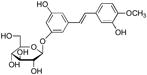 |
deoxyrhapontin | 7 | 20 | >100 |
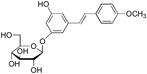 |
rhapontin | 8 | 50 | >100 |
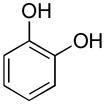 |
pyrocatechol | 9 | 20 | >100 |
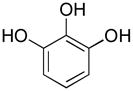 |
pyrogallol | 10 | 5 | 5 |
Assays were performed in duplicate.
Initially, assays were performed by monitoring ADAMTS-4 hydrolysis of a collagen-model FRET substrate for aggrecanase members of the ADAMTS family, fTSSlong (Table 1) (9, 21). Compounds were initially screened using concentrations of 0.1, 1.0, 10, and 100 μM. Hydrolysis was monitored using both fluorescence and RP-HPLC analyses to avoid “false positive” results (9). Only compound 10 showed consistent inhibition in both assays (Table 2). The importance of utilizing a secondary screen is illustrated by the differences in IC50 values between the FRET assay and RP-HPLC assay for compounds 4, 7, 8, and 9 (Table 2). For each of these compounds there was a difference in inhibitory capacity as measured by the assays. Substrate hydrolysis as measured by RP-HPLC analysis of cleavage products is entirely unambiguous, whereas fluorescence measurements are susceptible to interference by alterations in temperature, buffer composition, or the presence of the inhibitor itself. For example, the inhibitor could interact with the product in such a way that it quenches fluorescence. This masking of fluorescence would give a lower than expected IC50 value in the FRET assay, but not in the RP-HPLC assay.
The inhibitory activities of compound 3, the compound identified from the previous high-throughput screening effort, and 10, the novel compound identified herein, were tested against ADAMTS-5 another aggrecanase family member. Both compounds inhibited substrate hydrolysis by both fluorescence- and RP-HPLC analyses (data not shown).
Historically, small molecule and peptidomimetic MMP inhibitors have often utilized Zn-binding components to generate active-site binding compounds. This pathway has created numerous inhibitors effective in vitro, but have yet to yield a clinically useful compound. Potential problems have been cited as; binding structurally related family members, as well as dramatically off-target effects related to binding structurally unrelated Zn2+-metalloenzymes (33). Recently, the process of metzincin protease inhibitor screening, design, and optimization has delved into targeting exosites. An exosite is a region important for regulating enzyme behavior, but found outside of the active site. Binding an exosite offers a compound a few advantages, such as being able to distinguish amongst family members with greater selectivity because regions outside of the active site differ more widely amongst metzincin clan members. Additionally, it has also been shown that an exosite binding inhibitor had the capability of modulating hydrolysis of a subset of potential substrates. For example, a triple-helical model of an MMP-2 and MMP-9 fibronectin-like domain targeted (exosite binding) inhibitor was shown to inhibit the hydrolysis of gelatin and a moderately large triple-helical substrate, but did not inhibit the hydrolysis of a short random coil substrate (12). Interestingly, this inhibitor was unable to modulate the hydrolysis of a second triple-helcial substrate of similar size but different primary structure. This highlights the importance of targeting exosites for the development of selective inhibitors because one can not only distinguish amongst family members more selectively, but also one could potentially modulate the activity towards different substrates allowing a fine-tuned protease regulation not currently produced by exogenous metzincin-binding inhibitors. Behaviors such as these should be very helpful in producing a clinically effective inhibitor. Towards this end, two structurally different substrates were used for the analyses of inhibitors 3 and 10 with ADAMTS-4 and ADAMTS-5.
Apparent Ki values were determined for inhibition of ADAMTS-4 and ADAMTS-5 activity towards the polyPro II-like helical FRET substrate (fTSSlong) as well as a “short” substrate (Table 3). Three short ADAMTS-4 FRET substrates have been described previously, fTSS1, fTSS2, and fTSS3 (Table 1) (23, 34, 35). Although the fTSS1 sequence was initially reported to be selective for ADAMTS-4 over ADAMTS-5 (23), we found that the substrate fTSS1 (Table 1), in which the original Cys(Qsy-9) was replaced with Lys(Tamra), was hydrolyzed well by both ADAMTS-4 and ADAMTS-5 (data not shown). It was subsequently reported that the fTSS1 sequence was not ADAMTS-4/ADAMTS-5 selective (36). Due to its poor performance as a FRET substrate (23), fTSS1 was not further pursued. However, the core sequence of this substrate (Gln-Glu~Phe-Arg-Gly-Xxx-Thr) was retained in our design of the “short” aggrecanase substrate fTSSshort (Table 1), which was shorter than any of the previously described ADAMTS- 4/ADAMTS-5 substrates. The P3 subsite does not appear to be critical for aggrecanase activity towards synthetic substrates (35), and thus Lys was added there to improve peptide solubility, while inclusion of Gln the P4′ subsite was based on phage display motifs for ADAMTS-4 epitope sequences (23). MALDI-TOF MS analysis indicated that fTSSshort was hydrolyzed by both ADAMTS-4 and ADAMTS-5 primarily at the Glu~Phe bond.
Table 3.
Inhibition of ADAMTS-4 and ADAMTS-5 by compounds 3 and 10.
| Enzyme | Substrate | Inhibitor Structure | Inhibitor | Ki (μM) |
|---|---|---|---|---|
| ADAMTS-4 | fTSSshort | 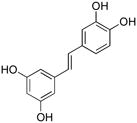 |
3 | 5.06 ± 0.54 |
| fTSSlong | 2.68 ± 0.28 | |||
| ADAMTS-5 | fTSSshort | 11.08 ± 3.95 | ||
| fTSSlong | 1.18 ± 0.17 | |||
| ADAMTS-4 | fTSSshort | 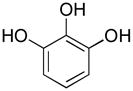 |
10 | 41.12 ± 9.77 |
| fTSSlong | 6.34 ± 0.53 | |||
| ADAMTS-5 | fTSSshort | 36.90 ± 8.82 | ||
| fTSSlong | 3.63 ± 0.61 |
Compound 10 was a weaker inhibitor of both ADAMTS-4 and ADAMTS-5 activity towards the short aggrecanase (fTSSshort) substrate compared with the longer (fTSSlong) substrate (Table 3). The different inhibitory activities suggest that compound 10 may act as an exosite inhibitor. To investigate this possibility, the mode of inhibition was compared for compounds 3 and 10. Inhibition of ADAMTS-4 and ADAMTS-5 was repeated for these compounds over a narrower concentration range, optimizing the number of concentrations producing 40–80% inhibition. To identify the modality of inhibitor binding, the analyses were repeated using varying fTSSlong substrate and inhibitor concentrations with ADAMTS-4. Non-linear regression analysis was used to determine apparent Vmax and apparent KM for inhibitors 3 and 10 with ADAMTS-4 (Table 4). Interaction with inhibitor 3 caused the apparent KM to increase, whereas the apparent Vmax was unchanged, suggesting a competitive interaction (Table 4). In contrast, inhibitor 10 caused no change in the apparent KM, but a decrease in apparent Vmax, suggesting noncompetitive inhibition of ADAMTS-4 (Table 4). The data was also analyzed using global nonlinear regression analyses to determine which model of inhibitor modality is best described by the data. Inhibitor 3 produced a slightly better fit as a competitive inhibitor, with R2 = 0.986, sum of squares = 573.7, and sy.x = 5.23. Inhibitor 10 produced equivalent fit with both noncompetitive and mixed noncompetitive modalities with R2 = 0.972, sum of squares = 845.1, and sy.x = 6.34; and R2 = 0.973, sum of squares = 824.2, and sy.x = 6.42, respectively. The α value for inhibitor 10 is predicted to be 1.419 ± 0.711, which is not surprising considering equivalent fit between the two noncompetitive modalities of inhibition. It should be noted that Prism utilizes a strict definition of noncompetitive in which α = 1 and mixed noncompetitive applies for finite α values not equal to 1. The authors prefer a looser definition for the noncompetitive modality of binding in which the sole limitation on α is that it is a finite number. The IC50 values for both compounds were determined over a range of fTSSlong substrate concentrations. The resulting IC50 versus substrate concentration curves produced linear curve fitting with equations of y = 0.126x + 3.049 for compound 3 and y = 0.0039x + 3.324 for compound 10. This suggests a competitive mode of interaction for inhibitor 3 and a noncompetitive interaction for inhibitor 10, with an α value equal to 1. Clearly, the mode of interaction with ADAMTS-4 is different for inhibitors 3 and 10, with inhibitor 3 most likely binding the enzyme competitively and inhibitor 10 binding noncompetitively.
To determine relative selectivity of these inhibitors amongst metzincin clan members, the activity of compounds 3 and 10 was then examined using representative members of the MMP family. Two different substrates, one short peptide (fMMPSshort) and a longer one possessing the triple-helical super-secondary structure of collagen (fMMPSlong) (Table 1), were used for MMP analysis. Compounds 3 and 10 inhibited MMP-2, MMP-8, MMP-9, and MMP-12 activity towards both substrates (Table 5). Thus, neither compound 3 nor compound 10 was selective for the aggrecanases. However, there were different effects on the activities depending on the substrate and the enzyme. Inhibition of triple-helical peptidase (fMMPSlong) activity by compound 10 was better than inhibition of hydrolysis of the short substrate (fMMPSshort) for MMP-2, MMP-8, and MMP-12, while no difference in inhibition was observed with MMP-9 (Table 5). This suggests that inhibitor 10 might bind noncompetitively with MMP-2, MMP-8, and MMP-12. Conversely, compound 3 showed little difference in inhibitory potency based on substrate for MMP-2 and MMP-8, while the triple-helical peptidase activity towards fMMPSlong by MMP-9 or MMP-12 was inhibited more effectively by compound 3 than activity towards fMMPSshort (Table 5). These results differ than those for the aggrecanases, but overall suggest that compound 10 is an exosite binding MMP inhibitor, with respect to MMP-2, MMP-8, and MMP-12.
Table 5.
Inhibition of MMPs by compounds 3 and 10.
| Enzyme | Substrate | Inhibitor Structure | Inhibitor | Ki (μM) |
|---|---|---|---|---|
| MMP-2 | fMMPSshort | 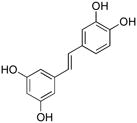 |
3 | 12.18 ± 1.82 |
| fMMPSlong | 10.04 ± 0.89 | |||
| MMP-8 | fMMPSshort | 11.44 ± 2.11 | ||
| fMMPSlong | 8.71 ± 2.07 | |||
| MMP-9 | fMMPSshort | 8.69 ± 1.09 | ||
| fMMPSlong | 4.77 ± 0.56 | |||
| MMP-12 | fMMPSshort | 34.09 ± 2.47 | ||
| fMMPSlong | 5.57 ± 0.95 | |||
| MMP-2 | fMMPSshort | 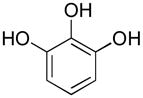 |
10 | 11.31 ± 0.52 |
| fMMPSlong | 3.89 ± 0.40 | |||
| MMP-8 | fMMPSshort | 10.34 ± 0.94 | ||
| fMMPSlong | 4.43 ± 0.71 | |||
| MMP-9 | fMMPSshort | 7.01 ± 0.58 | ||
| fMMPSlong | 8.29 ± 0.63 | |||
| MMP-12 | fMMPSshort | 34.37 ± 3.99 | ||
| fMMPSlong | 2.03 ± 0.19 |
Discussion
The pursuit of novel inhibitors for metalloproteinases had lead to the identification of active natural products found in a variety of plant species (1–3, 5, 32, 37, 38). However, prior studies have not examined the minimally active pharmacophores in these compounds, nor determined if common pharmacophores are found across natural product classes. Additionally, the mode of action of natural product metalloproteinase inhibition suggests some unique binding behaviors, but the mechanistic details have not been extensively examined. The present work utilizes distinct substrates with differing three-dimensional structures as tools to identify potential exosite binding molecules. Piceatannol and its structural analogs offered the opportunity to determine: (a) which structural features within the compound frameworks were important for aggrecanase or MMP inhibition; and (b) if these substrates can be utilized to differentiate active site binding molecules from exosite binding molecules.
The present and a prior study (9) demonstrated piceatannol inhibition of aggrecanases and MMPs. The inhibition of these proteases by piceatannol requires all four hydroxyl groups to be present and unmodified. This result is consistent with a prior study demonstrating that resveratrol, which possesses one fewer hydroxyl group than piceatannol, is not an inhibitor of MMP-2 or MMP-9 (3). In addition, piceatannol appears to inhibit metalloproteinases by a different mode of action than EGCG and ECG. Piceatannol is a competitive inhibitor, while EGCG is a noncompetitive inhibitor (1, 4). Thus, piceatannol, although similar in structure to the EGCG/ECG catechin unit, does not represent a minimally active EGCG/ECG.
In contrast to piceatannol, pyrogallol (1,2,3-trihydroxybenzene) captured some of the inhibitory features of EGCG. Pyrogallol inhibited aggrecanases in a noncompetitive fashion, as does EGCG (1). Ki values for ADAMTS-4 and ADAMTS-5 inhibition by pyrogallol were in the low μM range, while IC50 values for inhibition of the same enzymes by EGCG was in the 0.10–0.15 μM range (1). Thus, pyrogallol may represent a minimally active component of EGCG.
The compounds tested here were not selective for aggrecanases compared to MMPs. This result is not surprising, as many members of the flavonoid family inhibit both aggrecanases and MMPs, although EGCG may be slightly more potent against aggrecanases (1, 3, 4). The catalytic domains of aggrecanases and MMPs show a similar α/β structure (central five-stranded β-sheet flanked by α-helices) (36, 39). Inhibition of these metalloproteinase families by piceatannol and pyrogallol suggests that the inhibitors recognize structural elements common to aggrecanases and MMPs within their catalytic domains, as opposed to inhibitor binding to the structurally divergent ancillary domains found in each protease (i.e., disintegrin-like domain in aggrecanases and hemopexin-like domain in MMPs) (39).
MMP sensitivity to the number of hydroxyl groups present in an inhibitor was different based on the enzyme studied. Compound 3 was a moderately better inhibitor of MMP-9 then MMP-2 regardless of substrate, perhaps due to the presence of four hydroxyl groups. Inhibition of MMP-2, MMP-9, and MMP-13 by anthraquinones revealed a reasonable sensitivity to the 6-hydroxy group for MMP-9, less so for MMP-2, and not at all for MMP-13 (32).
In addition to their roles in metalloproteinase inhibition, flavonoids possess anti-angiogenic and anti-cell invasion activities. One wonders if these activities are interrelated. EGCG inhibits HUVEC invasion and tube formation, as well as tumor cell invasion (2, 6), while both piceatannol and resveratrol inhibit HUVEC motility, migration, proliferation, and formation of capillary-like tubes (angiogenesis) (10). The lack of MMP inhibition by resveratrol observed here suggests that the HUVEC behaviors described above are not MMP dependent. This illustrates a limitation to the usage of these compounds as metalloproteinase inhibitors in their present form, as their binding has been linked to a variety of cellular events. It does not, however, detract from the further development of piceatannol or pyrogallol derivatives as metzincin inhibitors. Alternatively, obtaining a small molecule metalloproteinase inhibitor such as pyrogallol presents several future options. Pyrogallol represents a unique probe that, through traditional medicinal chemistry efforts, could be modified to create an inhibitor of higher affinity. Alternatively, pyrogallol could be used in a fragment-based approach for assembly of novel inhibitory compounds, as fragment-based approaches have had substantial prior and recent successes in metalloproteinase inhibitor development (40–42).
Interestingly, piceatannol (compound 3) is a reasonably effective aggrecanase and MMP inhibitor, while resveratrol (compound 4), which differs by one hydroxyl group, was not. If one assumes that the entire molecule is involved in the binding interaction, this would be surprising. Since the only conclusion we can make based on the data herein is that compound 3 binds ADAMTS-4 in a competitive manner, one plausible answer is that both phenolic rings are not critical for binding. Rather, one may be important for binding (region not conserved between piceatannol and resveratrol) and the region conserved between the molecules might not be involved in the ADAMTS-4 interaction. Alternatively, one could propose that two vicinal hydroxyls present on piceatannol are utilized as a Zn2+-binding group and the single hydroxyl present on resveratrol produces inadequate Zn2+ binding. However, a review of known Zn2+-binding groups suggests that efficient Zn2+-binding is mediated by a thio-alcohol, but not hydroxyls (43). The binding mode of a thiol is known to be monodentate, but the thio-alcohol binding mode is unknown (43). Thus, the hydroxyl may not participate appreciably in Zn2+-binding.
Another interesting observation is that pyrogallol (compound 10) was determined to be an effective inhibitor of the aggrecanases, whereas pyrocatechol (compound 9), differing by only one hydroxyl, was not. Since our data support pyrogallol as an exosite binding inhibitor, but no data exists to determine where it binds the enzyme, it would be difficult to speculate as to why this difference exists. It is possible that the 2-hydroxyl modified pyrocatechol does bind ADAMTS-4, albeit weaker than the 3-hydroxyl modified pyrogallol. It should be noted that a limiting factor for aggrecanase analysis is their relatively slow rate of hydrolysis (21). The reaction rate must be measured over a time frame of hours rather than minutes necessary for MMP analysis. Thus, for the analysis of pyrocatechol the binding affinity might be altered such that it is difficult to measure rather than being entirely abolished.
In the present study, distinctly different Ki values were observed for pyrogallol (compound 10) inhibition of both ADAMTS-4 and ADAMTS-5 depending on substrate, with better inhibition observed with the longer substrate. Pyrogallol may well act as an exosite binding inhibitor of MMP-2, MMP-8, and MMP-12, as differences in Ki values between the fMMPSlong and fMMPSshort substrates were observed for these enzymes. We have previously documented different Ki values for inhibition of MMP-13 by an exosite inhibitor based on the use of triple- helical and single-stranded substrates (13). Alternatively, the apparent differences in binding affinity could be an artifact due to the inflation of IC50 or Ki through the use of substrate concentrations nearing or surpassing KM. While substrate concentration is an important consideration when optimizing experimental design, the differences herein are not likely artifactual because enzyme and substrate concentrations were fixed for Ki determination. Thus any overestimation of the Ki value for compound 10 would be mirrored for compound 3. Since the Ki value as determined by different substrates for ADAMTS-4 with compound 3 are not appreciably different, but the Ki values for compound 10 are 4–6 fold higher with the shorter substrate, the differences are unlikely to be linked to experimental parameters (Table 3). For MMPs, KM values with shorter substrates are generally higher than longer substrates, and thus it is possible that Ki values determined here with the longer substrate (fMMPSlong), could be overestimated. In the case of aggrecanases, the KM values for both short and long substrates are approximately equal, and therefore any overestimation of Ki values is consistent for all cases.
The search for unique, exosite binding inhibitors could proceed in a rapid fashion by screening substrate pairs, where one substrate is larger, retaining native protein three-dimensional structure and the other is not (9, 14). Alternatively, screening could be done in the presence of two different yet similarly sized, topologically constrained peptide substrates modeling different proteins to identify exosite-binding inhibitors modulating hydrolysis of one protein while sparing another. Previous analysis of a fibronectin-like domain binding exosite inhibitor of MMP-2 and MMP-9 illustrated this point (12). This inhibitor modulated activity towards gelatin and a triple-helical model of the type V collagen, but did not modulate hydrolysis of a short unstructured peptide or a triple-helical model of types I, II, and III collagen. Thus, topologically constrained peptides could be used to identify novel protease modulators, which bind more selectively amongst family members as well as alter hydrolysis of a subset of potential protein substrates. The identification of exosite binding regulators, both inhibitors and activators, is difficult using the currently available technology, yet the potential benefit of these compounds remains (44). This multifaceted enzyme regulation will undoubtedly advance the field of inhibitor design, and can potentially identify and develop clinically useful metalloproteinase inhibitors.
Supplementary Material
Acknowledgments
We gratefully acknowledge support by the National Institutes of Health (CA98799), the Robert A. Welch Foundation, the Texas Higher Education STAR Program (all to GBF), and the National Institutes of Health NIDCR (DE14318) for the COSTAR Program (to JLF).
Abbreviations
- Abz
2-aminobenzoyl
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motif
- Fam
carboxyfluorescein
- Dap
2,3-diaminopropionic acid
- Dde
1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl
- Dnp
2,4-dinitrophenyl
- ECG
(−)-epicatechin gallate
- EGCG
(−)-epigallocatechin gallate
- Fmoc
9-fluorenylmethoxycarbonyl
- FRET
fluorescence resonance energy transfer
- fTSSlong
fluorescent “long” a disintegrin and metalloproteainase with thromboxpondin motif substrate
- fTSSshort
fluorescent “short” a disintegrin and metalloproteainase with thromboxpondin motif substrate
- fMMPSlong
fluorescent “long” matrix metalloproteinase substrate
- fMMPSshort
fluorescent “short” matrix metalloproteinase substrate
- HUVEC
human umbilical vein endothelial cell
- MALDI-TOF MS
matrix-assisted laser desorption/ionization mass spectrometry
- MMP
matrix metalloproteinase
- SSP
single-stranded peptide
- Tamra
tetramethylrhodamine
- TFA
trifluoroacetic acid
Footnotes
A sampling of SigmaPlot graphs derived from ADAMTS-4 and ADAMTS-5 inhibition by compounds 3 and 10 is presented.
References
- 1.Vankemmelbeke MN, Jones GC, Fowles C, Ilic MZ, Handley CJ, Day AJ, et al. Selective inhibition of ADAMTS-1, -4 and -5 by catechin gallate esters. Eur J Biochem; 2003;270:2394–403. doi: 10.1046/j.1432-1033.2003.03607.x. [DOI] [PubMed] [Google Scholar]
- 2.Garbisa S, Biggin S, Cavallarin N, Sartor L, Benelli R, Albini A. Tumor invasion: molecular shears blunted by green tea. Nat Med; 1999;5:1216. doi: 10.1038/15145. [DOI] [PubMed] [Google Scholar]
- 3.Demeule M, Brossard M, Page M, Gingras D, Béliveau R. Matrix metalloproteinase inhibition by green tea catchins. Biochim Biophys Acta; 2000;1478:51–60. doi: 10.1016/s0167-4838(00)00009-1. [DOI] [PubMed] [Google Scholar]
- 4.Garbisa S, Sartor L, Biggin S, Salvato B, Benelli R, Albini A. Tumor gelatinases and invasion inhibited by the green tea flavanol epigallocatechin-3-gallate. Cancer; 2001;91:822–32. doi: 10.1002/1097-0142(20010215)91:4<822::aid-cncr1070>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 5.Oku N, Matsukawa M, Yamakawa S, Asai T, Yahara S, Hashimoto F, et al. Inhibitory effect of green tea polyphenols on membrane-type 1 matrix metalloproteinase, MT1-MMP. Biol Pharm Bull; 2003;26:1235–8. doi: 10.1248/bpb.26.1235. [DOI] [PubMed] [Google Scholar]
- 6.Yamakawa S, Asai T, Uchida T, Matsukawa M, Akizawa T, Oku N. (−)-Epigallocatechin gallate inhibits membrane-type 1 matrix metalloproteinase, MT1-MMP, and tumor angiogenesis. Cancer Lett; 2004;210:47–55. doi: 10.1016/j.canlet.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Ende C, Gebhardt R. Inhibition of matrix metalloproteinase-2 and -9 activities by selected flavonoids. Planta Med; 2004;70:1006–8. doi: 10.1055/s-2004-832630. [DOI] [PubMed] [Google Scholar]
- 8.Lim H, Kim HP. Inhibition of mammalian collagenase, matrix metalloproteinase-1, by naturally-occurring flavonoids. Planta Med. 2007:1267–74. doi: 10.1055/s-2007-990220. [DOI] [PubMed] [Google Scholar]
- 9.Lauer-Fields JL, Spicer TP, Chase PS, Cudic M, Burstein GD, Nagase H, et al. Screening of potential ADAMTS-4 inhibitors utilizing a collagen-model FRET substrate. Anal Biochem. 2008;373:43–51. doi: 10.1016/j.ab.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura Y, Sumiyoshi M, Baba K. Antitumor activities of synthetic and natural stilbenes through antiangiogenic action. Cancer Sci. 2008;99:2083–96. doi: 10.1111/j.1349-7006.2008.00948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuccioloni M, Mozzicafreddo M, Bonfili L, Cecarini V, Eleuteri AM, Angeletti M. Natural occuring polyphenols as template for drug design: Focus on serine proteases. Chem Biol Drug Des. 2009;74:1–15. doi: 10.1111/j.1747-0285.2009.00836.x. [DOI] [PubMed] [Google Scholar]
- 12.Lauer-Fields JL, Whitehead JK, Li S, Hammer RP, Brew K, Fields GB. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J Biol Chem. 2008;283:20087–95. doi: 10.1074/jbc.M801438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gooljarsingh LT, Lakdawala A, Coppo F, Luo L, Fields GB, Tummino PJ, et al. Characterization of an Exosite Binding Inhibitor of Matrix Metalloprotease 13. Protein Sci. 2008;17:66–71. doi: 10.1110/ps.073130208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauer-Fields JL, Minond D, Chase PS, Baillargeon PE, Saldanha SA, Stawikowska R, et al. High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triple-helical FRET substrate. Bioorg Med Chem. 2009;17:990–1005. doi: 10.1016/j.bmc.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheer JM, Romanowski MJ, Wells JA. A common allosteric site and mechanism in caspases. Proc Natl Acad Sci USA. 2006;103:7595–600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dennis MS, Eigenbrot C, Skelton NJ, Ultsch MH, Santell L, Dwyer MA, et al. Peptide exosite inhibitors of factor VIIa as anticoagulants. Nature. 2000;404:465–70. doi: 10.1038/35006574. [DOI] [PubMed] [Google Scholar]
- 17.Roberge M, Santell L, Dennis MS, Eigenbrot C, Dwyer MA, Lazarus RA. A novel exosite on coagulation factor VIIa and its molecular interactions with a new class of peptide inhibitors. Biochemistry. 2001;40:9522–31. doi: 10.1021/bi010592d. [DOI] [PubMed] [Google Scholar]
- 18.Izaguirre G, Rezale AR, Olson ST. Engineering functional antithrombin exosites in 〈1-proteinase inhibitor that specifically promote the inhibition of factor Xa and factor IXa. J Biol Chem. 2008;284:1550–8. doi: 10.1074/jbc.M807340200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meltzer M, Hasenbein S, Hauske P, Kucz N, Merdanovic M, Grau S, et al. Allosteric activation of HtrA protease DegP by stress signals during bacterial protein quality control. Angew Chem Int Ed Eng. 2008;47:1332–4. doi: 10.1002/anie.200703273. [DOI] [PubMed] [Google Scholar]
- 20.Selent J, Kaleta J, Li Z, Lalmanach G, Brömme D. Selective inhibition of the collagenase activity of cathepsin K. J Biol Chem. 2007;282:16492–501. doi: 10.1074/jbc.M700242200. [DOI] [PubMed] [Google Scholar]
- 21.Lauer-Fields JL, Sritharan T, Kashiwagi M, Nagase H, Fields GB. Substrate Conformation Modulates Aggrecanase (ADAMTS-4) Affinity and Sequence Specificity: Suggestion of a Common Topological Specificity of Functionally Diverse Proteases. J Biol Chem. 2007;282:142–50. doi: 10.1074/jbc.M605236200. [DOI] [PubMed] [Google Scholar]
- 22.Fields CG, Lovdahl CM, Miles AJ, Matthias-Hagen VL, Fields GB. Solid-phase synthesis and stability of triple-helical peptides incorporating native collagen sequences. Biopolymers. 1993;33:1695–707. doi: 10.1002/bip.360331107. [DOI] [PubMed] [Google Scholar]
- 23.Hills R, Mazzarella R, Fok K, Liu M, Nemirovskiy O, Leone J, et al. Identification of an ADAMTS-4 cleavage motif using phase display leads to the development of fluorogenic peptide substrates and reveals matrillin-3 as a novel substrate. J Biol Chem. 2007;282:11101–9. doi: 10.1074/jbc.M611588200. [DOI] [PubMed] [Google Scholar]
- 24.Nagase H, Fields CG, Fields GB. Design and characterization of a fluorogenic substrate selectively hydrolyzed by stromelysin 1 (matrix metalloproteinase-3) J Biol Chem. 1994;269:20952–7. [PubMed] [Google Scholar]
- 25.Neumann U, Kubota H, Frei K, Ganu V, Leppert D. Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and tumor necrosis factor converting enzyme. Anal Biochem. 2004;328:166–73. doi: 10.1016/j.ab.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 26.Minond D, Lauer-Fields JL, Cudic M, Overall CM, Pei D, Brew K, et al. The roles of substrate thermal stability and P2 and P1′ subsite identity on matrix metalloproteinase triple-helical peptidase activity and collagen specificity. J Biol Chem. 2006;281:38302–13. doi: 10.1074/jbc.M606004200. [DOI] [PubMed] [Google Scholar]
- 27.Lauer-Fields JL, Broder T, Sritharan T, Nagase H, Fields GB. Kinetic analysis of matrix metalloproteinase triple-helicase activity using fluorogenic substrates. Biochemistry. 2001;40:5795–803. doi: 10.1021/bi0101190. [DOI] [PubMed] [Google Scholar]
- 28.Bhaskaran R, Palmier MO, Lauer-Fields JL, Fields GB, Van Doren SR. MMP-12 catalytic domain recognizes triple-helical peptide models of collagen V with exosites and high activity. J Biol Chem. 2008;283:21779–88. doi: 10.1074/jbc.M709966200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kashiwagi M, Enghild JJ, Gendron C, Hughes C, Caterson B, Itoh Y, et al. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J Biol Chem. 2004;279:10109–19. doi: 10.1074/jbc.M312123200. [DOI] [PubMed] [Google Scholar]
- 30.Gendron C, Kashiwagi M, Lim NH, Enghild JJ, Thogersen IB, Hughes C, et al. Proteolytic activities of human ADAMTS-5: comparative studies with ADAMTS-4. J Biol Chem. 2007;282:18294–306. doi: 10.1074/jbc.M701523200. [DOI] [PubMed] [Google Scholar]
- 31.Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5) J Biol Chem. 2001;276:12501–4. doi: 10.1074/jbc.C000848200. [DOI] [PubMed] [Google Scholar]
- 32.Wierzchacz C, Su E, Kolander J, Gebhardt R. Differential inhibition of matrix metalloproteinases-2, -9, and -13 activities by selected anthraquinones. Planta Med. 2009;75:327–9. doi: 10.1055/s-0028-1112205. [DOI] [PubMed] [Google Scholar]
- 33.Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Activity-based probes for the proteomic profiling of metalloproteases. Proc Natl Acad Sci USA. 2004;101:10000–5. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Xu J, Levin J, Hegen M, Li G, Robertshaw H, et al. Identification and Characterization of 4-[[4-(2-Butynyloxy)phenyl]sulfonyl]-N-hydroxy-2,2-dimethyl-(3S)thiomorpholinecarboxamide (TMI-1), a Novel Dual Tumor Necrosis Factor--Converting Enzyme/Matrix Metalloprotease Inhibitor for the Treatment of Rheumatoid Arthritis. J Pharmacol Exp Ther. 2004;309:348–55. doi: 10.1124/jpet.103.059675. [DOI] [PubMed] [Google Scholar]
- 35.Wayne GJ, Deng S-J, Amour A, Borman S, Matico R, Carter HL, et al. TIMP-3 inhibition of ADAMTS-4 (aggrecanase-1) is modulated by interactions between aggrecan and the C-terminal domain of ADAMTS-4. J Biol Chem. 2007;282:20991–8. doi: 10.1074/jbc.M610721200. [DOI] [PubMed] [Google Scholar]
- 36.Shieh H-S, Mathis KJ, Williams JM, Hills RL, Wiese JF, Benson TE, et al. High resolution crystal structure of the catalytic domain of ADAMTS-5 (aggrecanase-2) J Biol Chem. 2008;283:1501–7. doi: 10.1074/jbc.M705879200. [DOI] [PubMed] [Google Scholar]
- 37.Yeh L-A, Chen JM, Baculi F, Gingrich DE, Shen TY. Inhibition of metalloproteinase by futoenone derivatives. Bioorg Med Chem Lett. 1995;5:1637–42. [Google Scholar]
- 38.Barrantes E, Guinea M. Inhibition of collagenase and metallproteinases by aloins and aloe gel. Life Sci. 2003;72:843–50. doi: 10.1016/s0024-3205(02)02308-1. [DOI] [PubMed] [Google Scholar]
- 39.Gerhardt S, Hassall G, Hawtin P, McCall E, Flavell L, Minshull C, et al. Crystal structures of human ADAMTS-1 reveal a conserved catalytic domain and a disintegrin-like domain with a fold homologous to cysteine-rich domains. J Mol Biol. 2007;373:891–902. doi: 10.1016/j.jmb.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 40.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discovery. 2007;6:211–9. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 41.Fattori D, Squarcia A, Bartoli S. Fragment-based approach to drug lead discovery: Overview and advances in various techniques. Drugs R D. 2008;9:217–27. doi: 10.2165/00126839-200809040-00002. [DOI] [PubMed] [Google Scholar]
- 42.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51:3661–80. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 43.Puerta DT, Cohen SM. A bioinorganic perspective on matrix metalloproteinase inhibition. Curr Topics Med Chem. 2004;4:1551–73. doi: 10.2174/1568026043387368. [DOI] [PubMed] [Google Scholar]
- 44.Hauske P, Ottmann C, Meltzer M, Ehrmann M, Kaiser M. Allosteric regulation of proteases. ChemBioChem. 2008;9:2920–8. doi: 10.1002/cbic.200800528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.