

Abstract
Infantile spasms syndrome (ISS) is a catastrophic pediatric epilepsy with motor spasms, persistent seizures, mental retardation, and in some cases, autism. One of its monogenic causes is an insertion mutation [c.304ins (GCG)7] on the X chromosome, expanding the first polyalanine tract of the interneuron-specific transcription factor Aristaless-related homeobox (ARX) from 16 to 23 alanine codons. Null mutation of the Arx gene impairs GABA and cholinergic interneuronal migration but results in a neonatal lethal phenotype. We developed the first viable genetic mouse model of ISS that spontaneously recapitulates salient phenotypic features of the human triplet repeat expansion mutation. Arx(GCG)10+7 (“Arx plus 7”) pups display abnormal spasm-like myoclonus and other key EEG features, including multifocal spikes, electrodecremental episodes, and spontaneous seizures persisting into maturity. The neurobehavioral profile of Arx mutants was remarkable for lowered anxiety, impaired associative learning, and abnormal social interaction. Laminar decreases of Arx+ cortical interneurons and a selective reduction of calbindin-, but not parvalbumin- or calretinin-expressing interneurons in neocortical layers and hippocampus indicate that specific classes of synaptic inhibition are missing from the adult forebrain, providing a basis for the seizures and cognitive disorder. A significant reduction of calbindin-, NPY (neuropeptide Y)-expressing, and cholinergic interneurons in the mutant striatum suggest that dysinhibition within this network may contribute to the dyskinetic motor spasms. This mouse model narrows the range of critical pathogenic elements within brain inhibitory networks essential to recreate this complex neurodevelopmental syndrome.
Introduction
Infantile spasms syndrome (ISS) (or West syndrome) is a rare, complex seizure disorder appearing within the first year of life. The clinical features include brief motor spasms of the extremities and trunk, chaotic high-amplitude interictal EEG waves and multifocal spikes (“hypsarrhythmia”), and severe developmental delay. The motor spasms and hypsarrhythmia cease spontaneously in nearly 25% of cases per year and typically abate in most children by age 5; however, cortical seizures and mental retardation often persist into adulthood (Frost and Hrachovy, 2003). The disorder is poorly responsive to all medical therapy, and there is no effective treatment. Along with acquired etiologies of ISS and congenital brain malformations (Curatolo et al., 2002), inherited monogenic errors have been associated with ISS, including disruption of the CDKL5 gene for cyclin-dependent kinase-like 5/serine-threonine protein kinase 9 (Kalscheuer et al., 2003) and several mutations in the Aristaless-related homeobox (ARX) gene (for review, see Poirier et al., 2008).
ARX is one of several homeodomain-containing transcription factors controlling GABAergic interneuron migration and maturation in the brain (McManus and Golden, 2005; Friocourt et al., 2008). Human ARX mutations lead to a spectrum of severe neurobehavioral disorders, including ISS/epilepsy, dystonia, autism, and mental retardation (Bienvenu et al., 2002; Turner et al., 2002; Gécz et al., 2006; Guerrini et al., 2007). Mice with targeted deletions of Arx die at birth, and the neonatal brain shows an accumulation of newly born interneurons near their proliferative zones in the medial and caudal ganglionic eminences, resulting in major deficits of GABAergic interneuron number in neocortex, hippocampus, and striatum (Kitamura et al., 2002; Colombo et al., 2007). Cultured ARX-deficient interneuron precursors retain some degree of maturation potential, but show abnormal morphology and migration. Whether this defect is autonomous to ARX-expressing neurons or can be rescued in a permissive cellular environment is not yet clear; however, in vitro transplant evidence favors the conclusion that migration can be rescued in Arx−/− neurons (Colombo et al., 2007). The early death of Arx−/− mice prevents a determination of the full postnatal clinical and neuropathological pattern of ARX deficiency, as well as a viable model to gain insight into possible methods for rescuing impaired ARX function in the postnatal nervous system.
Here, we describe the generation and initial characterization of a nonlethal genetic mouse model of ISS engineered by targeted expansion of the first polyalanine tract in the X-linked Arx gene. This polyalanine tract expansion is the mutation most commonly associated with West syndrome and mental retardation in human ISS patients (Poirier et al., 2008) (see Online Mendelian Inheritance in Man 308350). Consistent with the predominant expression of Arx in GABAergic interneurons and neural precursor cell populations, the Arx(GCG)10+7 (“Arx plus 7”) mutant brain has reduced numbers of Arx-, calbindin-, and neuropeptide Y (NPY)-expressing interneurons, and striatal cholinergic cells, but spares other inhibitory subpopulations, including parvalbumin and calretinin. Metabolic abnormalities leading to early lethality in the Arx-null mutant are absent, and mice with this human pathogenic mutation survive to display infantile motor spasms, seizures, and distinctive EEG abnormalities, along with cognitive and behavioral abnormalities persisting into adulthood.
Materials and Methods
Development of Arx(GCG)10+7 polyalanine expansion knock-in mutant lines
The Arx(GCG)10+7 knock-in targeting construct was created in the pFloxFlpNeo vector (a gift from Dr. James Shayman, University of Michigan, Ann Arbor, MI) (Hiraoka et al., 2006). BAC clone RP23-53K18 (Invitrogen) (National Center for Biotechnology Information locus AL590876.20) was the PCR template for mouse Arx gene fragments. The 4.2 kb left homologous arm containing exon 1 was obtained using primers GCTCGAGTCGACTGTTGCTAAGTGTAGAGAAAATTAACTCAG and GCTCGAGCCTCTTTCTTTCTTGTAGTACTCCTTTCG to introduce 5′ tandem XhoI and SalI sites and a 3′ XhoI site. The 2.2 kb right homologous arm commencing 71 bases into intron 2 was made using GATTTAAATCAAGTATACTGGGGCTTTAAGTTTCTGTTG and GCATATGTAAGACTGATCTTTGCCTCTAGAATGCCTAC. Exon 2 includes the coding sequence for three of the four polyalanine tracts (see Fig. 1A). Wild-type (WT) exon 2 flanked by 205 and 71 bp of introns was produced as a BamHI fragment with GG-GATCCAGAAATGAAGGGACGAAGGTAAAAG and GGGATC-CGAGAGGTTCCTGGACTCTGTAGACC. It was cloned into the pGEM7 vector (Promega), which lacks NotI sites. By the method Nasrallah et al. (2004) used to expand the mouse ARX polyalanine tract 1, an oligo duplex with NotI overhangs created with GGCCGCTGCTGCTGCTGCTGCCGC and GGCCGCGGCAGCAGCAGCAGCAGC was inserted into the codon for amino acid 109. The resulting Arx(GCG)10+7 gene fragment was ligated into the loxP-flanked BamHI site of the vector. The herpes simplex thymidine kinase gene was obtained using the pPNT vector [a gift from Dr. Richard Mulligan, Children's Hospital, Boston, MA (Tybulewicz et al., 1991)] and primers GGTCGACTGCCAAGTTCTAATTCCATCAG and GGTCGACGGCCTTCACCCGAACTTG. It was ligated into the SalI site 5′ of the left homologous arm to complete the targeting vector. PCR products and the completed targeting vector were verified by full sequencing before proceeding (Lone Star Labs).
Figure 1.

Structure of the ARX protein and gene, and generation of Arx(GCG)10+7 mice. A, Schematic model of the 564 residue ARX protein showing the exon structure, position, and relative size of known protein domains and the four polyalanine tracts [tract 1 is indicated (arrow)]. B, Schematics of exon 1- and 2-containing (1, 2) region of the Arx gene at Xp21.3 relevant to the (GCG)10+7 knock-in into polyalanine tract 1. The long arrows show the AseI-based Southern blot strategy using a 3′-probe distinguishing wild-type embryonic stem cells from those with the entire knock-in construct recombined (Knock-in + Neo) (C). A second strategy with the 5′-probe used EcoRI fragments (E to E). Probe bars represent 1 kb. The small arrow pairs indicate where diagnostic PCR primers anneal. The knock-in plus Neo schematic demonstrates the left and right homologous arms in the targeting construct, loxP sites flanking the (GCG)10+7 knock-in into polyalanine tract 1, and the frt-flanked neomycin-resistance cassette. The knock-in schematic illustrates the remaining sequence in targeted embryonic stem cells after flp recombinase removed the neomycin-resistance cassette. C, Southern blot of AseI-digested genomic DNA from embryonic stem cells that are WT or contain the knock-in plus neomycin-resistance cassette (KI+Neo), labeled with the 3′-probe. D, Gel of the 2 kb versus 0.335 kb PCR products (small hooked arrow pairs in schematics) from targeted stem cells before (+Neo) and after (Flp-Neo) Flp-recombinase removed the neomycin-resistance cassette. The latter cells were used to produce the chimeras founding the Arx(GCG)10+7 lines. Molecular weight markers are indicated. E, Gel with the three possible genotyping PCR products. Female heterozygotes show both WT 0.277 kb and 0.335 kb (GCG)10+7 bands. Male hemizygous or female homozygous (GCG)10+7 show a 0.335 kb band (small hooked arrow pairs in B). Molecular weight markers are in 0.2 kb increments.
The NdeI-linearized targeting construct was electroporated into male129S5/SvEvBrd strain AB2.2 mouse embryonic stem cells. Cells were subjected to simultaneous positive and negative selection using G418 (180 μg/ml) and gangcyclovir (200 μm). Seven targeted clones were identified by duplicate Southern blots of EcoRI or AseI restriction fragments labeled with probes directed against 5′ or 3′ 900 bp sites (5′-probe obtained with GTTTTGAAGAGGTTGAGCAT and GTCGAAATTCAAAACCCAAC; 3′-probe with GGGGTATGGACTGGAAGAAGC and GTTGCTGTCACATCTCTGTTTGA). 129S5/SvEvBrd and C57BL/6J wild-type genomic DNA gave the same size of labeled restriction fragments. The frt-flanked neomycin resistance cassette was excised from targeted stem cells by Flp-recombinase expressed from plasmid pOG-Flpe6 (a gift from Dr. A. Francis Stewart, University of Technology, Dresden, Germany) (Buchholz et al., 1998). PCR with CGTCTTCACCAGGTATGGG and GTTTTAGAGCCAAACCGCCTC yielded a 2 kb neomycin-plus or a 335 bp neomycin-minus mutant band. This primer set was used for genotyping, yielding a wild-type band of 277 bp.
The Arx(GCG)10+7 knock-in was established in mice using standard procedures. C57BL/6J albino mice were purchased from The Jackson Laboratory, and 129S5/SvEvBrd mice were obtained from the Darwin Transgenic Core at Baylor. Experiments described here used mixed strain N2 mice (75% C57BL/6/25% 129S5/SvEvBrd). Mice were housed under constant temperature and humidity, with a light/dark cycle of 12 h (light, 7:00 A.M. to 7:00 P.M.). Animal care and use conformed to National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the Baylor College of Medicine Institutional Animal Care and Use Committee.
Gross anatomy and histology of major organs
Necropsy was performed on two wild-type and two Arx(GCG)10+7 6-week-old male littermates. Their major organs were prepared as hematoxylin- and eosin-stained sections and analyzed in Baylor's Comparative Pathology Laboratory.
Immunostaining brain sections and cell counts, Fluoro-Jade staining
Six to nine pairs of mutant and wild-type siblings were used for each immunostaining procedure. Cryosections (30 μm thick) were used for free-floating immunohistochemistry. The primary antibodies were rabbit anti-ARX (ARP36824_T200; Aviva Systems Biology) at 1:500, goat anti-doublecortin (Dcx) (C-18; Santa Cruz Biotechnology) at 1:200, goat anti-choline acetyltransferase (ChAT) (Millipore AB 144P at 1:200), rabbit polyclonal anti-neuropeptide Y (N 9528; Sigma-Aldrich) at 1:8000, mouse anti-parvalbumin and mouse anti-calbindin-D-28K (clones PARV-19 and CB-955, respectively; Sigma-Aldrich) at 1:1000, and mouse anti-calretinin (MAB 1568; Millipore) at 1:200. Avidin–biotin–peroxidase complex immunostaining was done with Elite ABC kits (Vector Laboratories). Substrate reactions were stopped simultaneously in control and mutant sections. Light microscopy was performed with an Olympus model IX71 inverted microscope. Photographs were imported into the Photoshop CS3 extended application (version 10.0; Adobe) for measurement of areas and thicknesses and counting immunostained cells. The boundaries of cortical layers I–IV were determined from sections immunostained for calbindin and applied to sections stained for other interneuron markers. All immunostained cells contained within the defined cortical areas were counted throughout equal numbers of sections at bregma −2.5 (Paxinos and Franklin, 2001) and from bregma 1.0 and 0.5 (counts from the latter two sections were pooled). Immunostained cells in the hippocampus were counted in sections at bregma −2.5. ChAT+, NPY+, and CR+ cells were counted in the entire caudate–putamen in sections at 0.5 and 1.0 bregma levels, and cell counts were pooled. The more abundant ARX+ and CB28K+ cells were counted in three 1 mm2 areas. All counts were converted to cells per square millimeter, and then expressed relative to wild-type values set at 100%. Significant differences were assessed by the unpaired two-tailed t test. Fluoro-Jade staining for dying cells was done on bregma −2.5 brain sections from pairs of 1 month olds of each genotype, using standard published methods (Schmued and Hopkins, 2000).
Blood glucose measurements for pancreatic function
Glucose levels were measured in blood from the tails of four wild-type and four mutant males aged 3 months, using an Accu-Chek Advantage monitor (Roche Diagnostics). For 4-h- and 24-h-fasting levels, food was removed but water was left.
Assessment of spontaneous movements of infant mice
The Arx(GCG)10+7 pups from four litters included mutant males and mutant homozygous females (n = 13), as well as nonmutant males and females (n = 24). Pups were marked on different toes and placed in numbered compartments of a plastic box, with a timer in view. Two sequential 30 min DVD recordings were made at approximately the same time during the lights-on period, in a humid, 35°C temperature-controlled environment. Quantitative analysis focused on pups 7–11 d postnatal. Mothers accepted their pups and pups achieved developmental markers at normal times. A 30 min recording in which the least voluntary movement occurred was reviewed pup by pup to score spontaneous movements during sleep and at the moment of awakening. Movements in three categories were counted: (1) high-amplitude movements included rapid, abrupt displacements of the entire body across the floor of the compartment, as well as (2) startles, defined as “sudden, spontaneous and simultaneous contractions of skeletal muscles throughout the body” (Karlsson et al., 2006), occurring as abdominal contractions bowing or twisting the body or as simultaneous strong movements of three or more appendages, and (3) low-amplitude movements included myoclonic twitches and kicks. Voluntary movements during obvious wakefulness were not scored.
Simultaneous video and electroencephalographic recording
Two- to 5-week-old Arx(GCG)10+7 mutant and wild-type littermate males were implanted for chronic EEG recordings following established methods. Mice were anesthetized by intraperitoneal injection of 0.02 ml/g Avertin. In mice older than 3 weeks, eight Teflon-coated 0.005-inch-diameter silver wire electrodes soldered to a microminiature connector were implanted into the subdural space over the temporal, parietal, and occipital cortices. A bipolar montage linking parietal to temporal and parietal to occipital electrodes on either hemisphere was used to localize EEG activity to specific brain regions. Only two bilateral electrodes (left temporal 3 and right temporal 4) were implanted in younger [postnatal day 11 (P11)] mice. Beginning 1 d after surgery, EEG activity was monitored with the mouse moving freely in a cage during random 3–4 h periods or overnight for 7–12 h. Behavior recorded with a digital video camera was correlated with EEG activity (Stellate Systems; Harmonie, version 5.0b). Recordings were made for up to 6 weeks after implantation. The EEG and behavioral data were evaluated by three expert observers.
Behavioral assessment
Groups of experimentally naive, 2-month-old mutants with their wild-type littermates were used for all behavioral tests, which were performed in an identical sequence over a period of 7 d for both genotypes.
Startle to an acoustic stimulus and prepulse inhibition of startle.
Prepulse inhibition is the reduction in a startle response caused by a low-intensity nonstartling sound, the prepulse, given before a 40-ms-long startling sound of 120 dB (Marubio and Paylor, 2004). Each mouse was placed in a Plexiglas cylinder on an accelerometer in a lighted, sound-attenuated box (SR-Lab System; San Diego Instruments) and left for 5 min with constant 70 dB white noise. Test sessions included 48 randomly generated trial types delivered over 10 min, with prepulse sounds of 74, 78, or 82 dB given 100 ms before the startle stimulus, or with the sounds given separately.
Nociception.
Mice were placed inside a Plexiglas chamber with the floor heated to 55°C. The latency to a hindlimb response, lifting or twitching or licking a foot, was recorded.
Motor function test.
Motor skills were evaluated with a rotating rod (Rota-Rod; model 7650; Ugo Basile) accelerating from 4 to 40 rpm in 5 min (McIlwain et al., 2001). Four trials, 30–60 min apart, were done on 2 d. Maximum trial time was 6 min, except for the last one, which was extended until the mice dropped off the rod or rode around twice.
Light–dark exploration test.
Mice were placed near the outer wall of the light end of a 44 × 21 × 21 cm Plexiglas box unequally divided into light and dark chambers. The number of entries during the 10 min period when a mouse had all feet in either chamber was recorded (McIlwain et al., 2001).
Open-field test.
Mice were placed in the center of an evenly lighted open-field space (40 × 40 × 30 cm) (McIlwain et al., 2001). Activities in the x-, y-, and z-axes were quantitated over 30 min by a computer-operated photo beam-based Versamax system (AccuScan Instruments). Data were collected over 2 min intervals.
Learning behavior: pavlovian conditioned fear.
Performance in a conditioned fear task was analyzed using the video-based motion monitor system (San Diego Instruments) (McIlwain et al., 2001). Training involved two events of a conditioned stimulus (CS) of 30 s of 80 dB white noise followed by a 0.4 mA mild 2 s footshock. A day later, mice were given 5 min in the context of the training environment. Sixty minutes later, the auditory CS test was done with altered contextual cues: the grid floor covered with Plexiglas, the square space changed with a 45° angled Plexiglas wall, and the smell changed by cleaning with ethanol rather than isopropyl alcohol and adding a cap of vanilla extract. Freezing behavior was noted during 3 min without CS, and then during 3 min with continuous CS. The CS-minus pre-CS values for 4 of the 21 subjects of each genotype were 2 SDs outside the mean, so were excluded.
Social behavior: tube test of social dominance.
Each Arx(GCG)10+7 mutant mouse encountered three different non-cage mate wild-type mice at approximately equal time intervals and from alternating ends of a clear Plexiglas cube. Retreat of one mouse was marked as a “lose” for that mouse and a “win” for the other. Most matches were won in <120 s; one lasting >600 s was excluded (Shahbazian et al., 2002).
Statistical analysis
Data are expressed as mean ± SEM. Statistical analyses were performed using one-way ANOVA (genotype by trial) and were followed by the unpaired two-tailed Student t test if variances were unequal. The χ2 test was used on the tube test results.
Results
Generation of a mouse genetic model for infantile spasms syndrome
The human ARX (GCG)10+7 mutation most closely associated with West syndrome/X-linked infantile spasms syndrome (ISSX) expands the first polyalanine tract of amino acids 100–115 from 16 to 23 residues through the insertion of seven GCG alanine codons within 10 consecutive GCG alanine codons (Fig. 1A) (Strømme et al., 2002a) (for review, see Guerrini et al., 2007; Poirier et al., 2008). Alanine is encoded by GCX, where X is any nucleotide. The mouse Arx gene (locus NP_031518; UniGene Mm.275547) has 15 alanine codons in the first polyalanine tract, but the longest GCG repeat is 4, not 10. In keeping with the mixed alanine codon usage, the mouse Arx expansion knock-in mutation was generated with GCT repeats, resulting in a total of 23 alanine codons in tract 1. This mouse Arx knock-in reproduces the 23 alanine codons in human ISSX-ARX (GCG)10+7, so we have designated the resultant mouse line Arx(GCG)10+7.
The original construct has homologous arms of 4.2 and 2.2 kb, a TK cassette, and an frt-flanked neomycin resistance cassette (Fig. 1B, Knock-in + Neo). Exon 2 with the knock-in is flanked by loxP sites. Recombination into embryonic stem cells was confirmed by Southern blotting of Ase-digested DNA and screening with a 3′-probe (Fig. 1C). Eight of 210 clones had a 3.5 kb Ase fragment resulting from recombination. Confirmatory Southern blot screening used a 5′-probe for an EcoR1 fragment increased by 2 kb because of the neomycin-resistance cassette. To ensure the mutant transcript would properly splice, the frt-flanked neomycin-resistance cassette was recombined out of intron 2 by Flp recombinase. PCR for a product that was 2 kb smaller confirmed this event (Fig. 1D). PCR with the same primers distinguished cells with wild-type genomic DNA or knock-in DNA and was also used for genotyping (Fig. 1E). The larger knock-in PCR product is attributable to the presence of the loxP and frt sites 3′ of exon 2. Their presence in the knock-in stem cell clones was confirmed by cloning and sequencing the PCR product. Mating heterozygous Arx(GCG)10+7 females to wild-type males yielded Arx(GCG)10+7 males at the expected 25% mendelian frequency. The presence of the alanine expansion in three mutant males used for experimental analysis was verified by sequencing the PCR product of exon 2.
Normal development of non-neuronal organ systems, pancreatic metabolism, and fertility of the Arx(GCG)10+7 mutant
Arx is expressed in the developing nervous system, pancreas, and testes, as well as in adult brain, skeletal muscle, heart, and liver (Kitamura et al., 2002; Ohira et al., 2002; Colombo et al., 2004; Poirier et al., 2004). Arx knock-out mice developed by two strategies have severe defects in organs in which the gene is expressed during development and die within days of birth (Kitamura et al., 2002; Collombat et al., 2003). In contrast, Arx(GCG)10+7 males and heterozygous as well as homozygous Arx(GCG)10+7 females are viable, attain normal adult body weight, and are fertile. In adult male and female Arx(GCG)10+7 mutants, muscle and bone and all organs are normal in gross appearance. The heart, lungs, kidney, liver, stomach, small and large intestine, pancreas, spleen, thymus, and lymph nodes have normal histological appearance (n = 2 each genotype). Arx(GCG)10+7 testicles, epididymis, ductus deferens, prostate, coagulating gland, and seminal vesicle are also structurally normal. The adrenal glands appear normal in both cortical and medullary layers (data not shown). Since Arx-null mutants die perinatally of apparent dehydration and hypoglycemia because of skewed differentiation of pancreatic exocrine cells (Kitamura et al., 2002; Collombat et al., 2005, 2007), feeding and fasting glucose levels of Arx(GCG)10+7 mutants and wild-type littermates were measured (n = 4 each). Consistent with their viability and the normal histological appearance of pancreatic islets, blood glucose levels were within the normal range in fed, 4-h-fasted, and 24-h-fasted mutant mice (fed: WT, 176 ± 4, and mutant, 162 ± 9; 4 h fasted: 166 ± 14 and 146 ± 5; 24 h fasted: 126 ± 2 and 140 ± 9; p = 0.16–0.23). The finding that the mutant mice show normal development, fertility, and pancreatic function suggests that the mutant ARX protein retains at least partial function in some non-CNS pathways.
Infant Arx(GCG)10+7 mutants show spontaneous spasm-like myoclonic events
Rodent pups display spontaneous limb and tail movements during normal sleep or on awakening, including brief focal myoclonic twitches, kicks, and generalized startles (Blumberg et al., 2007). Startles are sudden, spontaneous, and simultaneous contractions of skeletal muscles throughout the body and have been defined behaviorally as “abrupt, high-amplitude, synchronous movements of at least three limbs” (Karlsson et al., 2006). We designed a “littermate array” for our behavioral videomonitoring studies to reproducibly sample and analyze aberrant motor movements; pups were placed unrestrained in separate wells of a transparent plate that allowed full range of motion and simultaneous observation of all littermates under the same temperature-controlled conditions (Fig. 2A–C). In addition to motor startles as defined above, we observed another distinct category of spontaneous high-amplitude movements: sustained spasm-like movements so strong as to cause the pup to flip or fall over, axial contractions that bowed or twisted the body, and abrupt lateral displacements of the pup across the floor of the compartment.
Figure 2.

Infant Arx(GCG)10+7 pups display twice as many spontaneous, severe spasm-like movements as do wild-type littermates. A, Still image from a videorecording of a wild-type (no. 1) and three mutant 9-d-old male mouse littermates. Mutant pup no. 9 in lower right quadrant is in the midst of major spasm-like movements. B, Six seconds later, pup no. 9 tonically extends all limbs. C, Eleven seconds later, spasm-like movement in pup no. 9 is complete. D, Spontaneous high-amplitude movements, including startles and displacement of the entire body, occur twice as often in Arx(GCG)10+7 pups as in nonmutant littermates. Arx(GCG)10+7 pups (n = 13) are mutant males and females. Nonmutant pups (n = 24) are wild-type males and heterozygous females. ***p < 0.001 by one-way ANOVA. E, Mutants displayed spontaneous low-amplitude movements, including myoclonic twitches and short-distance kicks, at approximately the same rate as did nonmutant littermates (p = 0.24–0.36). Group data shown are average ± SEM.
The most severe spasm-like movements, those causing a pup to flip or fall over (supplemental Video 1, available at www.jneurosci.org as supplemental material), were increased nearly fourfold in mutants at all ages compared with wild-type siblings. Mutant pups at 7 d had between 0 and 11 such episodes in a 30 min period, whereas nonmutant pups had a maximum of 2 (means of 2.8 ± 0.8 vs 0.6 ± 0.02; p < 0.002). At 9 d of age, 31% of mutant pups had one to two severe spasms, whereas only one nonmutant pup had a single episode in a 30 min period. At 11 d of age, 38% of mutant pups displayed one to three severe spasms per 30 min, whereas none were observed in their nonmutant counterparts. Overall, when compared with their nonmutant littermates, the Arx(GCG)10+7 mutant pups showed twice as many total spontaneous, high-amplitude movements (including the severe spasms described above as well as startles, body displacement, and rapid trunk flexion) at all three of the ages monitored (Fig. 2D). The frequency of these monitored events declined between days 9 and 11, to approximately one-half. Hemizygous males and homozygous females showed similar rates at the three ages (p = 0.49, 0.29, 0.67). Similarly, and consistent with the normal development of females heterozygous for the Arx-null mutation, the spectrum and frequency of spontaneous myoclonic movements in wild-type males and female heterozygotes did not significantly differ (p = 0.42, 0.12, 0.52 for the three ages).
The final category of movements scored was low-amplitude, phasic movements, including myoclonic twitches and short-distance kicks that did not disturb body posture. These are exhibited by every pup at all ages. There was no significant difference between the rate of low-amplitude movements in mutant and nonmutant pups at the three tested ages (Fig. 2E) (p = 0.29–0.36). The incidence of movements in this category declined steadily and at a similar rate from postnatal day 7 to day 11 in both mutant and nonmutant pups. Unlike the high-amplitude spasm movements, the Arx mutation had no significant effect on the frequency of low-amplitude motor activities at infantile stages.
Spontaneous epileptic EEG activity in Arx(GCG)10+7 mutants
To determine whether the Arx(GCG)10+7 mutation alters cortical excitability, we performed prolonged video-electroencephalographic recordings from chronically implanted mutant pups 11–21 d of age (n = 5), mutant juveniles and adults between 3.5 and 10 weeks of age (n = 12), and age-matched wild-type littermates (n = 7). Because of their small size, it was not possible to reliably record from mice as young as those monitored for spasm-like movements (7–11 d of age). Recordings for periods exceeding 4 h on pups 16–20 d old revealed multiple 4–5 s episodes that began with a high-voltage (up to 3.9 mV) slow wave transient followed by attenuation of the background EEG amplitude and a transient increase of higher frequency activity (Fig. 3A). The pups exhibited a very brief myoclonic jerk involving the head and body at the onset of the attenuation event (Fig. 3A, arrow). These stereotyped “electrodecremental” episodes associated with myoclonic jerks are similar to those ictal events seen in infantile spasm patients (Hrachovy and Frost, 2003). High-amplitude cortical spikes and sharp waves also occurred frequently, up to 20 times per hour. These discharges were multifocal in origin (arising independently in both hemispheres) and were more frequent during non-rapid eye movement sleep. In addition, high-voltage slow waves associated with a spike or sharp component occurred independently over both hemispheres, usually infrequently but sometimes 10–20 times per hour. These high-voltage patterns resemble those in Figure 3A except that the activity before and after the high-voltage event are similar; there is no attenuation or increase in frequency and no obvious motor behavior linked to the discharge.
Figure 3.

EEG abnormalities in Arx(GCG)10+7 mice. A, Mutants under 21 d of age display sharp spike-slow wave transients followed by attenuation of background activity and an increase in high-frequency background rhythmic activity. The arrow indicates the beginning of a myoclonic twitch associated behaviorally with sudden head drop. This pattern is not seen in older mutants. The record is from temporal electrodes. B, EEG recording of a representative 29 s spontaneous generalized seizure seen in 19-d-old (and older) adult mutants. Slow versive movements of the head accompany these EEG seizures. C, The 6/s spike wave bursts accompanied by behavioral arrest are seen in mutants starting at 14 d through late adulthood. Recording electrode montage: L, left; R, right; F, frontal; T, temporal; P, parietal; O, occipital.
Arx(GCG)10+7 mutants between the ages of 3.5 and 10 weeks show spontaneous electrographic seizures characterized by generalized attenuation of the background activity and the appearance of low-voltage fast activity, followed by generalized high-frequency and high-amplitude spike and polyspike activity dissipating at different times in different brain regions (Fig. 3B). The episodes of high-frequency spiking lasted 18–29 s, during which the mice generally made 4–10 slow versive movements of the head and or trunk, and clonic movements followed by vigorous grooming suggestive of limbic seizures with hippocampal involvement. After generalized seizure discharges, there was prolonged attenuation of the EEG voltage. One juvenile mutant male was observed to have spontaneous tonic/clonic seizures (two in a 7 h period). A second type of seizure, also seen in pups, was distinguished by 6 Hz spike wave bursts with amplitudes of 150–400 μV (Fig. 3C), and accompanied by behavioral arrest. These episodes last up to 4 s, with an average of 1.2 per second, and occur between 3 and 28 times per hour (average rate of 10.6 per hour). These spike wave bursts occurred in one-half (2 of 4) of the mice that had other seizure patterns during the monitoring period, and in 75% (8 of 12) of the mutant adults monitored. The third type of abnormality observed in the majority of the mutant mice, whether awake or asleep, were frequent (up to 118 per hour) multifocal single spike and sharp wave patterns of complex morphology with amplitudes ranging between 600 and 1200 μV. Rare (one to three per hour) low-voltage spike or sharp waveforms were seen in wild-type mice and were typically <600 μV in amplitude.
Cognitive impairment and abnormal behavior in Arx(GCG)10+7 mutant mice
Large groups of mutant and wild-type control 2-month-old male littermates were screened with a battery of standardized behavioral tests (n = 21 of each genotype). In a prepulse inhibition test to assess sensorimotor gating, Arx(GCG)10+7 and wild-type mice responded to the loudest sound, 120 dB, with a startle movement of similar maximum amplitude (669 ± 150 vs 703 ± 94). The acoustic startle response in both genotypes was inhibited by ∼10, 25, or 44% when warning prepulse sounds of 74, 78, or 82 dB, respectively, preceded the 120 dB startling sound (Fig. 4A). These results demonstrate intact hearing and unaltered brainstem reflexes to a startle stimulus, distinguishing the spontaneous startle events from the hypekplexia caused by dysinhibition with the brainstem and spinal cord as seen in mice with glycine receptor α-subunit mutations (Ryan et al., 1994). Mutant mice were significantly more sensitive than wild-type mice to heat-induced pain, with a 30% shorter latency to exhibiting a hindlimb response (7.6 vs 9.95 s; p = 0.003). Coordination and the ability to learn motor skills were assayed on the accelerating rotarod, in four trials on 2 successive days. Mutant mice performed significantly better than wild-type mice in most trials (p = 0.006–0.036), and slightly better but insignificantly in the other trials (Fig. 4B). Mutant mice seemed less fearful, often turning around on the rod while it rotated slowly.
Figure 4.

Arx(GCG)10+7 mutants behave with abnormally low anxiety, are cognitively impaired, and display an autism-like characteristic. A, The startle response of mice of both genotypes habituates similarly as louder prepulse sounds repeatedly precede the 120 dB startling sound. B, Rotarod tests show that mutant mice perform significantly better than wild-type mice in most trials, and both sets of mice improve their motor skills at a similar rate. C, Mutant mice behave abnormally in the light/dark exploration test, spending twofold longer in the lighted area and making 67% more transitions from dark to light. D, The conditioned fear test reveals impaired associative learning from contextual cues (environment and conditioning sound stimulus) with an aversive stimulus (footshock) in Arx(GCG)10+7 mice, as assessed by freezing behavior. Data are average ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus wild type by one-way ANOVA.
In a more direct measure of anxiety, Arx(GCG)10+7 mice behaved abnormally in the light/dark exploration test, spending more than twice as long in the light and 20% less in the dark as did wild-type mice (light time, 38 vs 17%; dark time, 62 vs 83%; p < 0.0001) (Fig. 4C) with an average of 35% longer in the light at each interval (p = 0.007). The mutants made 67% more light/dark transitions (46.2 vs 27.6; p < 0.0001). The latency until the first entry into the darkened chamber was similar in both genotypes. These results indicate that mutant mice initially behave like wild-type mice, but then explore with less anxiety. In the open-field test, Arx mutant mice showed similar speeds and amounts of locomotor activity during a 30 min period of exploration of a novel arena. There was no difference in horizontal, vertical, or circling activity, and a slight but insignificant increase in the total number of movements by the mutants. The striking difference was that mutant mice spent 40% more time in the center than did wild-type littermates (0.316 vs 0.226 center/total distance ratio; p = 0.004). The abnormal behavioral profiles in the light/dark and open-field exploration tests reveal subnormal anxiety levels in the Arx mutants.
Reduced anxiety may also contribute to cognitive impairments in the Arx(GCG)10+7 mutant mice, as demonstrated by pavlovian conditioned fear training involving a brief footshock applied after a conditioning sound (CS) cue (Fig. 4D). Trained mutant mice placed in the training context/environment the next day adopted a fear-induced “frozen” posture 48% less often than did wild-type mice (context, 21 vs 44%; p < 0.0001). After many variables of the training context were changed, including the chamber floor, shape, and scent, the freezing responses by mutant mice were only 27% as frequent as those of wild-type mice (PreC-cue, 4.6 vs 16.9%; p = 0.018). When the conditioning sound cue was delivered in this altered environment, mutants froze only 47% as frequently as control littermates (CS, 32.9 vs 69.7%; p < 0.00001). The relative lack of learned fear in mutants was also revealed by a major reduction in freezing responses in mutants post-sound cue versus pre-sound cue (CS–pre-CS) compared with control mice (28.4 vs 52.8%; p < 0.0003). These results indicate an impairment of associative learning and memory in the mutants, with deficits in both context- and sound cue-dependent aspects, despite their normal acoustic response and hypersensitivity to painful stimuli. Finally, we performed the tube test for social dominance, often used as an indicator of autism-like characteristics (Shahbazian et al., 2002; Spencer et al., 2005). In three separate trials, each mouse was confronted with a different non-cage mate mouse of the opposite genotype. In 79% of trials, Arx(GCG)10+7 mice demonstrated the autistic-like behavior of retreating, whereas wild-type mice retreated in only 21% of the trials (49 of 60 matches; p < 0.00001).
Reduction of mature Arx-positive interneuron populations in the Arx(GCG)10+7 mutant
ARX+ GABAergic interneurons are expressed in the mature mouse brain in multiple forebrain regions, with the highest populations present within the neocortex, hippocampus, and adult neural progenitor zones. Using a commercial antibody specific for ARX, in wild-type mice ARX+ interneurons were labeled throughout the neocortex, especially in the deeper layers (Fig. 5A,C). Compared with adult male wild-type siblings, only ∼68% of ARX+ cells remained in layers I–IV of somatosensory and motor cortex in Arx(GCG)10+7 mutants, and only 53% as many ARX+ cells were present in the deeper layers V–VI (Fig. 5B,D, Table 1). The total reduction throughout the cortex was to ∼58% of wild-type values. At the cellular level, ARX protein was localized in the nuclei of 72 ± 5% of the wild-type and 45 ± 11% of the mutant interneurons (n = 5 each genotype; p < 0.05), therefore remaining in the cytoplasm of a larger percentage of mutant interneurons (Fig. 5D, large arrows). There was no difference in ARX+ cell distribution in the parietal cortex (n = 3 each genotype) (data not shown). In the hippocampal formation, approximately one-half as many ARX+ cells were present in the hilar region of the mutant dentate gyrus (Fig. 5E,F). Likewise, ARX+ cell populations in the mutant striatum were reduced to approximately one-half of the wild-type level (Fig. 5G,H). Nuclear ARX localization in hippocampal and striatal interneurons appeared similar in both genotypes.
Figure 5.
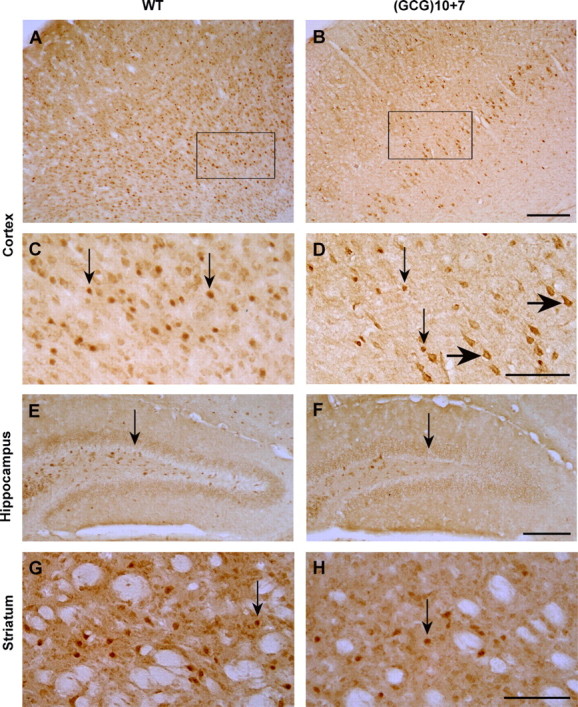
ARX-positive interneurons are significantly reduced from normal in the neocortex, hippocampus, and striatum of the Arx(GCG)10+7 mutant. A, B, Low-magnification views of sections of cortex spanning from the pia to the white matter, immunostained with anti-ARX antibody, show a reduction of ARX+ cells throughout the Arx(GCG)10+7 mutant motor/somatosensory cortex compared with that of Arx+/+ WT sibling, but more so in the deeper layers. C, D, Higher magnification views (A, B, rectangles) demonstrate that, although ARX is localized in the nuclei of wild-type neurons and some mutant neurons, it is present in the cytoplasm of many mutant cortical neurons (D, large arrows). Note that the selected areas have the highest concentration of ARX+ interneurons for each genotype. E, F, A decrease in ARX+ interneurons in the Arx mutant is also evident in the hilus of the hippocampal dentate gyrus (arrows) compared with that of wild type. G, H, ARX+ interneurons in the mutant caudate–putamen are reduced to approximately one-half the wild-type level; ARX is localized in nuclei (arrows) in both genotypes. Scale bars: A, B, E, F, 200 μm; C, D, G, H, 100 μm.
Table 1.
Relative presence of neuron types that are affected in Arx(GCG)10+7 mutants (WT, 100%)
| Cortical layers I–IV | Cortical layers V–VI | All cortical layers | Hippocampus | Striatum | |
|---|---|---|---|---|---|
| ARX+ | 68 ± 5* | 53 ± 13* | 58 ± 9* | 69 ± 15* | 49 ± 7* |
| CB28K+ | 64 ± 9** | 77 ± 14 | 67 ± 9** | 56 ± 16 | 47 ± 6*** |
| NPY+ | 100 ± 5 | 100 ± 11 | 100 ± 7 | 100 ± 19 | 69 ± 8** |
| ChAT+ | — | — | — | — | 39 ± 16*** |
The unit of measure is percentage of immunostained cells present in defined areas in brain sections of bregma 1.0 and 0.5, relative to WT values that were set at 100%. CB28K, Calbindin 28K; —, absence of ChAT + cells. Values are average ± SEM.
*p < 0.05, **p < 0.01, ***p < 0.001 versus wild type.
Selective reductions of interneuron subtypes
We used antibodies specific for the interneuron markers parvalbumin, calretinin, calbindin, and NPY to examine specific subsets of interneurons in the adult mutant, and found that not all were equally affected by the Arx(GCG)10+7 mutation. The density and location of parvalbumin- and calretinin-expressing GABAergic interneurons in mutant brains was similar to that in wild-type littermates in all regions. In contrast, other subtype populations were reduced in specific regions.
Reduction of calbindin-expressing interneurons in Arx(GCG)10+7 mutant forebrain
The most striking loss was in calbindin+ interneurons in multiple regions, including layers I–IV of the neocortex with a lesser reduction in the deeper layers (Fig. 6A–D), accompanied by an approximately equal reduction in these cells in the granule cell layer of the dentate gyrus (Fig. 6E,F), and striatum (Fig. 6G,H, Table 1). The Arx(GCG)10+7 mutation had little effect on calbindin expression itself, as evidenced by strong immunopositivity of other calbindin-expressing structures, such as the mossy fiber axons of granule cells in the dentate hilar region (Fig. 6F) (n = 4 each genotype).
Figure 6.
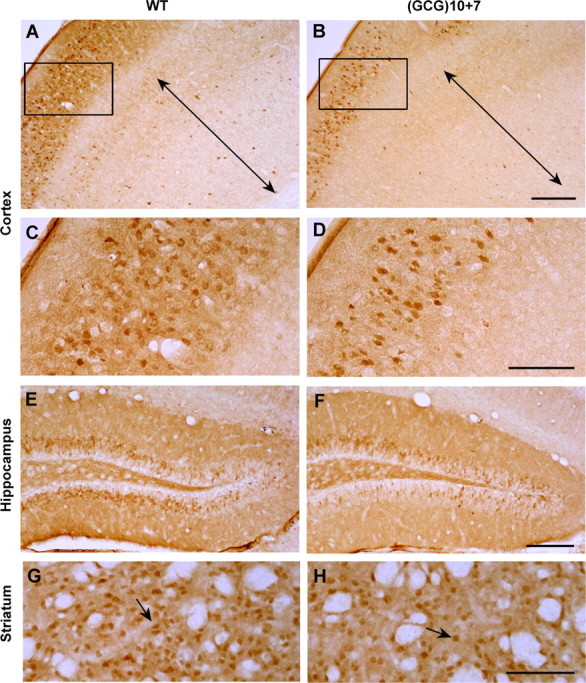
Calbindin D28K-positive interneurons are less abundant in the neocortex, hippocampus, and striatum of the Arx(GCG)10+7 mutant. A, B, Neocortex of WT and mutant motor/somatosensory cortex immunostained with anti-calbindin D28K antibody, showing loss of calbindin+ cells throughout the Arx(GCG)10+7 cortex compared with a WT sibling. Layers V–VI are indicated by the two-headed arrows. C, D, Higher magnification images (A, B, rectangles) show calbindin+ interneurons in layers I–IV. E, F, Calbindin+ interneurons are also decreased in the granule cell layer of the mutant dentate gyrus. G, H, Like the ARX+ interneurons, calbindin+ projection neurons (arrows) in the mutant striatum are reduced to approximately one-half the WT level. Scale bars: A, B, E, F, 200 μm; C, D, G, H, 100 μm.
Reduction of NPY-expressing interneurons in the Arx(GCG)10+7 mutant striatum
In contrast, NPY+ interneuron loss was region specific. A subset of GABAergic interneurons express NPY, and likewise, a subset of NPY+ cells express ARX. NPY+ cells were absent from the Arx-null mouse (Kitamura et al., 2002), but in the Arx(GCG)10+7 there was no difference in the number of NPY+ cells in layers I–V or layers V–VI of the motor/somatosensory cortex or the parietal cortex (Fig. 7A,B), and no discernible difference in NPY+ cell numbers in hippocampal sections (Table 1). However, the mutant striatum showed a 31% loss of NPY+ cells compared with wild-type siblings, although the cell morphology appeared normal (Fig. 7C,D, Table 1) (n = 3 each genotype).
Figure 7.
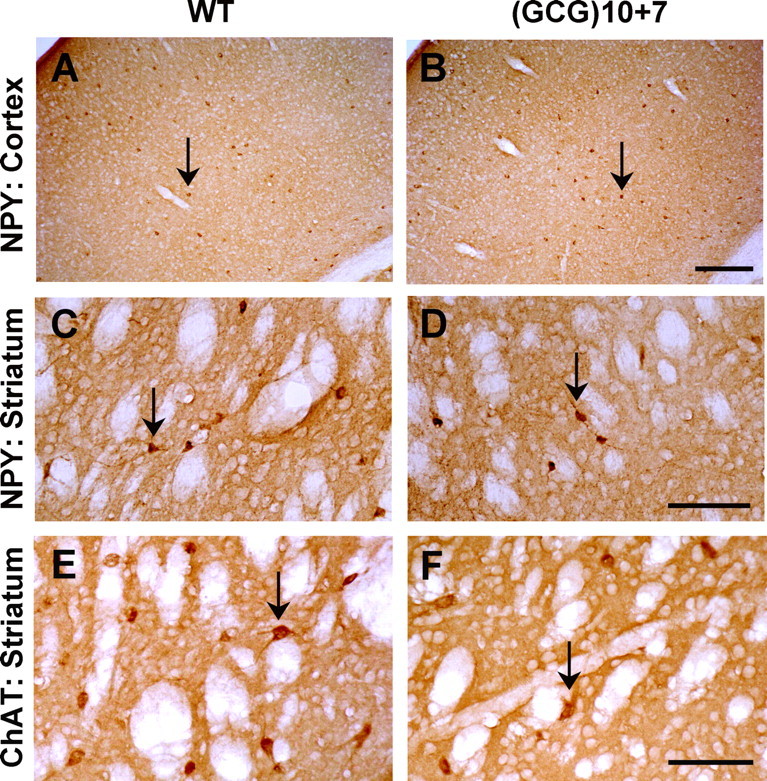
Severe reduction of striatal interneurons. The NPY+ interneuron population is reduced in the striatum of the Arx(GCG)10+7 mutant, although present at WT levels in the neocortex. A, B, Full-thickness views of somatosensory cortex immunostained with anti-NPY antibody show that NPY+ cells (arrows) are not altered in number or location in the Arx(GCG)10+7 mutant. C, D, NPY+ interneurons (arrows) are decreased by 31% in the mutant caudate–putamen compared with that of a WT sibling. Scale bars: A, B, 200 μm; C, D, 100 μm. The population of cholinergic interneurons in the striatum is reduced in mutant mice to 39% of wild-type values. E, Section of WT striatum showing numerous interneurons labeled with antibodies to ChAT. F, Markedly fewer cholinergic cells are detected in the same region of the Arx(GCG)10+7 striatum. The arrows indicate immunostained cells. Scale bar, 100 μm.
Reduction of striatal cholinergic interneurons in the Arx(GCG)10+7 mutant striatum
Basal ganglia pathology is found in children with ARX mutations who display marked and prolonged dystonia (Juhász et al., 2001; Guerrini et al., 2007), and Arx-null mice show a severe lack of cholinergic interneurons in the basal ganglia (Colombo et el., 2007). The latter defect may be attributable to limited migration of cholinergic precursor cells from the ventral subpallium during early development, and a strong reduction in expression of transcription factors such as Lhx7 that are linked to the expression of ARX. The effect of the Arx(GCG)10+7 mutation on striatal cholinergic interneurons was assessed by quantification of ChAT-expressing cells in the caudate–putamen. Mutants contained only 39% as many striatal ChAT+ cells compared with their wild-type littermates (Table 1) (n = 3 each genotype) (Fig. 7E,F).
Presence of Dcx-positive neural progenitors
Finally, we examined cerebral progenitor zones in adult mutants and +/+ mice (n = 4 each genotype) for other Arx-related defects, since ARX is found in adult neural stem cells (Colombo et al., 2004). Dcx is a microtubule-associated protein marker for neural stem cells in germinal zones, expressed shortly after mitosis during the commitment to a neuronal fate and in the early stage of migration (Overstreet-Wadiche and Westbrook, 2006; von Bohlen und Halbach, 2007). Prolonged hippocampal seizure activity induces neurogenesis and the expression of Dcx+ cells within these zones (Parent et al., 2006). The density and location of Dcx+ cells appeared unchanged in the subgranular layer of the dentate gyrus, the tertiary germinal matrix in which adult stem cell proliferative activity coexists with GABAergic interneurons. In addition, there was no obvious decrease of Dcx+ cells in the cortical subventricular germinal zone and rostral migratory stream (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The lack of a clear expansion in the size of this population is consistent with the phenotype of brief seizures observed in these mutants. Consistent with this interpretation, we found no evidence of cell death using the Fluoro-Jade technique (data not shown) in either the mutants or wild-type siblings.
Discussion
We have generated a viable mouse knock-in developmental model of a human ARX mutation, a (GCG)10+7 expansion of polyalanine tract 1 encoded by exon 2, associated with ISSX/West syndrome. This is a catastrophic form of childhood epilepsy, with mental retardation, and in ∼70% of cases (Kato, 2006; Shinozaki et al., 2009) infantile spasms, progressing to other types of seizures by 3–4 years of age. The ISSX syndrome is highly resistant to medical treatment. The Arx(GCG)10+7 mouse model is the first genetic mouse model of Arx deficiency that survives into adulthood and recapitulates major features of the human disease, in particular, abnormal infantile motor spasms, seizures, and neurobehavioral deficits. The major cellular features include a selective loss of cortical, hippocampal, and striatal Arx+ and calbindin+ interneurons, and a striking reduction of NPY+ and cholinergic interneurons in the striatum. Additional molecular analysis of the Arx(GCG)10+7 mouse model may be helpful in determining the downstream changes in the cellular basis of infantile spasms syndrome, and in identifying novel therapeutic strategies for patients with this disorder.
Mode of action of ARX mutant protein in cells
The molecular details of how the multifunctional ARX transcription factor regulates maturation and migration of interneurons are still unclear, and additional work will be required to understand the precise effects of the expanded repeat mutation. The highly conserved octapeptide domain and the fourth polyalanine tract have transcriptional repressor activity, whereas the aristaless-related domain has transcriptional activator activity (McKenzie et al., 2007). The paired-class (bicoid subfamily) homeodomain may also regulate transcription. The abnormal function of ARX with the (GCG)10+7 mutation that expands the first polyalanine tract from 16 to 23 residues can have several molecular origins. In a transcription assay, ARX protein fragments with the (GCG)10+7 mutation have 10-fold greater repressor activity than wild type. At the cellular level, in vitro studies show that the (GCG)10+7 expansion causes a partial loss of function through protein aggregation, as evidenced by ARX accumulation in nuclear aggregates in a subpopulation of transfected COS or 293T cells or brain cells (Nasrallah et al., 2004; Friocourt et al., 2006), and aggregates in the cytoplasm of transfected SH-SY5Y and PC12 cells (Shoubridge et al., 2007). However, even normal ARX protein forms nuclear and cytoplasmic aggregates in a fraction of these cells. The ARX IVS4-816_EX5701del mutation, which truncates ARX before the aristaless domain, has been detected in two males with infantile spasms and mental retardation (Strømme et al., 2002b). This truncated protein did not aggregate when expressed in COS cells (Nasrallah et al., 2004), suggesting that one form of ISSX/mental retardation occurs in the absence of aggregation of mutant ARX protein. In our mouse model, ARX(GCG)10+7 protein was detected in the cytoplasm of a greater percentage of neurons than was the wild-type protein (Fig. 7D). Limited genotype–phenotype correlations suggest that the overall severity of the phenotype correlates with the size of the first polyalanine repeat tract expansion; 1–3 extra alanines produce mental retardation (Bienvenu et al., 2002), 7 additional alanines are associated with infantile spasms/West syndrome (Guerrini et al., 2007), and 11 extra alanines causes a severe developmental disorder (Ohtahara syndrome) with an EEG suppression burst pattern (Kato et al., 2007).
Role of Arx(GCG)10+7 mutant cells in brain development
Analysis of neuroanatomical defects in males from several lines of mice with Arx gene disruption demonstrated that Arx contributes to all fundamental processes of brain development: neuronal proliferation, patterning, cell migration, and axonal outgrowth (Kitamura et al., 2002; Colombo et al., 2007; Colasante et al., 2008; Friocourt et al., 2008). Arx is expressed at levels detectable by in situ hybridization at the 3-somite stage and by the 10-somite stage is expressed in the dorsal telencephalon in regions that coexpress Nkx2.1 and Dlx-1 and -2 (Miura et al., 1997; Colombo et al., 2004). Arx expression is regulated by several members of the Dlx family of homeobox proteins, as shown by ectopic induction after electroporation of plasmids expressing Dlx-1, -2, or -5, even in Dlx1/2 knock-out brains lacking most interneurons or interneuron precursors (Cobos et al., 2005a; Colasante et al., 2008).
Wild-type Arx alleles contribute to migration of GABAergic interneurons from the lateral ganglionic eminence (LGE) and the medial ganglionic eminence (MGE) and to proliferation and migration of cortical pyramidal progenitor cells in the subventricular zone (SVZ) of the developing forebrain (Kitamura et al., 2002; Colombo et al., 2007; Friocourt et al., 2008). In Arx-null embryos, migration from the LGE to the striatum and other structures and from the MGE to the cortical intermediate zone and the marginal zone is nearly absent, and migration through the SVZ is partially impaired. As a consequence, in the Arx-null mutant, glutamate-expressing pyramidal cells are mislocalized, calbindin+ and calretinin+ cells are severely reduced, and NPY+ [NPY/nNOS+ (neuronal nitric oxide synthase-positive)] interneurons are nearly absent throughout the brain. Arx also plays a role in regionalization of the brain by its effect on expression of other transcription factors, so that in its absence, cholinergic striatal interneurons are completely lacking and thalamic development is aberrant. In our Arx(GCG)10+7 model, we found a striking and selective interneuronopathy with a major loss of ARX+ interneurons in the neocortex, hippocampus, and striatum, reflected mostly in a reduction of calbindin+ cells, although NPY+ interneurons are also less abundant in the striatum. Parvalbumin- and calretinin-expressing cells appeared unaffected. The mutant striatum also shows less than one-half the normal content of cholinergic interneurons. These decreases are less severe than those described in the null mutant. Since subtypes of cholinergic and GABAergic inhibitory defects in this network play a pivotal role in the expression of both fleeting and sustained dyskinetic movements (for review, see Wilson, 2007; Breakefield et al., 2008), additional exploration of the role of striatal function in this model may lead to a better understanding of the motor spasms in this syndrome.
Comparison of Arx(GCG)10+7 mutants with human ISS and other models of West syndrome/infantile spasms syndrome
Even single human ARX mutations lead to a spectrum of severe neurobehavioral disorders, including epilepsy, infantile spasms, dystonia, autism, and mental retardation (Turner et al., 2002). The Arx(GCG)10+7 mouse mutant shares critical phenotypic features with the most common presentation of human ISS, including infantile spasm-like movements, electrodecremental discharges, and multifocal EEG spikes, and displays seizures in juvenile and older mice. Persistent seizures occur in children with the Arx(GCG)10+7 mutation that have outgrown the infantile spasm stage (Guerrini et al., 2007). One EEG feature not detected in the mice we examined is hypsarrhythmia. Hypsarrhythmia is defined as high-amplitude, irregular slow waves with superimposed discharges of multifocal spikes. Since this abnormality is present early in the syndrome and diminishes with maturity, it remains possible that it was missed because of the technical difficulty of recording EEG in unanesthetized mice younger than P12. Like their human counterparts, Arx(GCG)10+7 mutant animals also have a significantly abnormal neurobehavioral profile consistent with mental retardation and autistic-like features.
Several pharmacological models of epileptic spasms that express varying degrees of the full phenotypic spectrum have been developed in rats; one induced by intracortical tetrodotoxin injection (Lee et al., 2008) that displays hypsarrhythmia and spasms in the adolescent animal, another by prenatal treatment with betamethasone followed by postnatal administration of NMDA (Velísek et al., 2007). Clusters of acute extensor flexions accompanied by EEG decremental responses can also be induced by acute administration of the GABAB receptor agonist prodrug GBL (γ-butyrolactone) into transgenic Ts65Dn mice (Cortez et al., 2009). These postnatal treatments do not impair interneuronal migration, but disturb patterns of excitability that are regulated by synaptic inhibition. A report of a conditional deletion of Arx in Dlx5/6-expressing neurons in the mouse describing a loss of calbindin+ neurons and multiple seizure types appeared as this paper went to press (Marsh et al., 2009).
A growing list of mouse genetic models of epilepsy have been identified with selective patterns of dysmigration of interneuron subtypes, including Cdk5 (Chae et al., 1997), Upar (Powell et al., 2003), Lis1 (McManus et al., 2004), Dlx proteins (Cobos et al., 2005b), semaphorins, and neuropilin (Sahay et al., 2005; Gant et al., 2009). Our finding of reductions in specific forebrain interneuron subtypes, with presumed secondary rearrangement of somatic and dendritic inhibitory inputs, favors the view that impaired local circuit control of both network synchrony and synaptic plasticity (Magloczky and Freund, 2005) mediate the primary elements of the Arx syndrome. Since at least 84 transcriptional targets of Arx have been identified in the developing brain (Fulp et al., 2008), the downstream patterns of Arx regulated genes also contribute extensive molecular diversity to the mechanisms underlying the complex infantile spasms syndrome phenotype. Continuing analysis of the Arx(GCG)10+7 mouse mutant at specific developmental stages and into adulthood with a focus on brain regions in which specific interneuronal subtypes have failed to migrate, coupled with comparisons of excitability in those regions among other phenocopies of the disorder, will help highlight necessary and sufficient lesions underlying this catastrophic form of epilepsy.
Footnotes
Funds were provided by the Baylor Department of Neurology (Peter Kellaway Research Fund), Blue Bird Circle Foundation, National Institute of Neurological Disorders and Stroke Grant NS29709 (J.L.N.), National Institute of Child Health and Human Development Eunice Shriver Intellectual and Developmental Disabilities Research Center Grant HD024064, and People Against Childhood Epilepsy (M.G.P.). We are grateful for the advice and assistance of Drs. Kristen Senechal, Corinne Spenser, Sara McCann Ernst, and Meenaski B. Bhattacharjee, and that of Isabel Lorenzo, Kaiping Xu, Allison Patrick, and Kevin Kline. The pFloxFlpNeo, pPNT, and pOG-Flpe6 vectors were gifts from Drs. James Shayman, Richard Mulligan, and A. Francis Stewart, respectively.
References
- Bienvenu et al., 2002.Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, Ben Jeema L, Zemni R, Vinet MC, Francis F, Couvert P, Gomot M, Moraine C, van Bokhoven H, Kalscheuer V, Frints S, Gécz J, Ohzaki K, Chaabouni H, Fryns JP, et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet. 2002;11:981–991. doi: 10.1093/hmg/11.8.981. [DOI] [PubMed] [Google Scholar]
- Blumberg et al., 2007.Blumberg MS, Coleman CM, Johnson ED, Shaw C. Developmental divergence of sleep-wake patterns in orexin knockout and wild-type mice. Eur J Neurosci. 2007;25:512–518. doi: 10.1111/j.1460-9568.2006.05292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breakefield et al., 2008.Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- Buchholz et al., 1998.Buchholz F, Angrand PO, Stewart AF. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat Biotechnol. 1998;16:657–662. doi: 10.1038/nbt0798-657. [DOI] [PubMed] [Google Scholar]
- Chae et al., 1997.Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- Cobos et al., 2005a.Cobos I, Broccoli V, Rubenstein JL. The vertebrate ortholog of Aristaless is regulated by Dlx genes in the developing forebrain. J Comp Neurol. 2005a;483:292–303. doi: 10.1002/cne.20405. [DOI] [PubMed] [Google Scholar]
- Cobos et al., 2005b.Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci. 2005b;8:1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- Colasante et al., 2008.Colasante G, Collombat P, Raimondi V, Bonanomi D, Ferrai C, Maira M, Yoshikawa K, Mansouri A, Valtorta F, Rubenstein JL, Broccoli V. Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J Neurosci. 2008;28:10674–10686. doi: 10.1523/JNEUROSCI.1283-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat et al., 2003.Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17:2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat et al., 2005.Collombat P, Hecksher-Sorensen J, Broccoli V, Krull J, Ponte I, Mundiger T, Smith J, Gruss P, Serup P, Mansouri A. The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the alpha- and beta-cell lineages in the mouse endocrine pancreas. Development. 2005;13:2969–2980. doi: 10.1242/dev.01870. [DOI] [PubMed] [Google Scholar]
- Collombat et al., 2007.Collombat P, Hecksher-Sørensen J, Krull J, Berger J, Riedel D, Herrera PL, Serup P, Mansouri A. Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest. 2007;117:961–970. doi: 10.1172/JCI29115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo et al., 2004.Colombo E, Galli R, Cossu G, Gécz J, Broccoli V. Mouse orthologue of ARX, a gene mutated in several X-linked forms of mental retardation and epilepsy, is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev Dyn. 2004;231:631–639. doi: 10.1002/dvdy.20164. [DOI] [PubMed] [Google Scholar]
- Colombo et al., 2007.Colombo E, Collombat P, Colasante G, Bianchi M, Long J, Mansouri A, Rubenstein JL, Broccoli V. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J Neurosci. 2007;27:4786–4798. doi: 10.1523/JNEUROSCI.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez et al., 2009.Cortez MA, Shen L, Wu Y, Aleem IS, Trepanier CH, Sadeghnia HR, Ashraf A, Kanawaty A, Liu CC, Stewart L, Snead OC., 3rd Infantile spasms and Down syndrome: a new animal model. Pediatr Res. 2009 doi: 10.1203/PDR.0b013e31819d9076. [DOI] [PubMed] [Google Scholar]
- Curatolo et al., 2002.Curatolo P, Verdecchia M, Bombardieri R. Tuberous sclerosis complex: a review of neurological aspects. Eur J Pediatr Neurol. 2002;6:15–23. doi: 10.1053/ejpn.2001.0538. [DOI] [PubMed] [Google Scholar]
- Friocourt et al., 2006.Friocourt G, Poirier K, Rakić S, Parnavelas JG, Chelly J. The role of ARX in cortical development. Eur J Neurosci. 2006;23:869–876. doi: 10.1111/j.1460-9568.2006.04629.x. [DOI] [PubMed] [Google Scholar]
- Friocourt et al., 2008.Friocourt G, Kanatani S, Tabata H, Yozu M, Takahashi T, Antypa M, Raguénès O, Chelly J, Férec C, Nakajima K, Parnavelas JG. Cell-autonomous roles of ARX in cell proliferation and neuronal migration during corticogenesis. J Neurosci. 2008;28:5794–5805. doi: 10.1523/JNEUROSCI.1067-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost and Hrachovy, 2003.Frost JD, Jr, Hrachovy RA. New York: Kluwer; 2003. Infantile spasms: diagnosis, management and prognosis. [Google Scholar]
- Fulp et al., 2008.Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA. Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet. 2008;17:3740–3760. doi: 10.1093/hmg/ddn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant et al., 2009.Gant JC, Thibault O, Blalock EM, Yang J, Bachstetter A, Kotick J, Schauwecker PE, Hauser KF, Smith GM, Mervis R, Li Y, Barnes GN. Decreased number of interneurons and increased seizures in neuropilin 2 deficient mice: implications for autism and epilepsy. Epilepsia. 2009;50:629–645. doi: 10.1111/j.1528-1167.2008.01725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gécz et al., 2006.Gécz J, Cloosterman D, Partington M. ARX: a gene for all seasons. Curr Opin Genet Dev. 2006;16:308–316. doi: 10.1016/j.gde.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Guerrini et al., 2007.Guerrini R, Moro F, Kato M, Barkovich AJ, Shiihara T, McShane MA, Hurst J, Loi M, Tohyama J, Norci V, Hayasaka K, Kang UJ, Das S, Dobyns WB. Expansion of the first polyA tract of ARX causes infantile spasms and status dystonicus. Neurology. 2007;69:427–433. doi: 10.1212/01.wnl.0000266594.16202.c1. [DOI] [PubMed] [Google Scholar]
- Hiraoka et al., 2006.Hiraoka M, Abe A, Lu Y, Yang K, Han X, Gross RW, Shayman JA. Lysosomal phospholipase A2 and phospholipidosis. Mol Cell Biol. 2006;26:6139–6148. doi: 10.1128/MCB.00627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrachovy and Frost, 2003.Hrachovy RA, Frost JD., Jr Infantile epileptic encephalopathy with hypsarrhythmia (infantile spasms/West syndrome) J Clin Neurophysiol. 2003;20:408–425. doi: 10.1097/00004691-200311000-00004. [DOI] [PubMed] [Google Scholar]
- Juhász et al., 2001.Juhász C, Chugani HT, Muzik O, Chugani DC. Neuroradiological assessment of brain structure and function and its implication in the pathogenesis of West syndrome. Brain Dev. 2001;23:488–495. doi: 10.1016/s0387-7604(01)00295-9. [DOI] [PubMed] [Google Scholar]
- Kalscheuer et al., 2003.Kalscheuer VM, Tao J, Donnelly A, Hollway G, Schwinger E, Kübart S, Menzel C, Hoeltzenbein M, Tommerup N, Eyre H, Harbord M, Haan E, Sutherland GR, Ropers HH, Gécz J. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet. 2003;72:1401–1411. doi: 10.1086/375538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson et al., 2006.Karlsson KA, Mohns EJ, di Prisco GV, Blumberg MS. On the co-occurrence of startles and hippocampal sharp waves in newborn rats. Hippocampus. 2006;16:959–965. doi: 10.1002/hipo.20224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, 2006.Kato M. A new paradigm for West syndrome based on molecular and cell biology. Epilepsy Res. 2006;70(Suppl 1):S87–S95. doi: 10.1016/j.eplepsyres.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Kato et al., 2007.Kato M, Saitoh S, Kamei A, Shiraishi H, Ueda Y, Akasaka M, Tohyama J, Akasaka N, Hayasaka K. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome) Am J Hum Genet. 2007;81:361–366. doi: 10.1086/518903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura et al., 2002.Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo S, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- Lee et al., 2008.Lee CL, Frost JD, Jr, Swann JW, Hrachovy RA. A new animal model of infantile spasms with unprovoked persistent seizures. Epilepsia. 2008;49:298–307. doi: 10.1111/j.1528-1167.2007.01377.x. [DOI] [PubMed] [Google Scholar]
- Magloczky and Freund, 2005.Magloczky Z, Freund TF. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005;28:334–340. doi: 10.1016/j.tins.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Marsh et al., 2009.Marsh E, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, Christian SL, Mancini G, Labosky P, Dobyns W, Brooks-Kayal A, Golden JA. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain. 2009;132:1563–1576. doi: 10.1093/brain/awp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marubio and Paylor, 2004.Marubio LM, Paylor R. Impaired passive avoidance learning in mice lacking central neuronal nicotinic acetylcholine receptors. Neuroscience. 2004;129:575–582. doi: 10.1016/j.neuroscience.2004.09.003. [DOI] [PubMed] [Google Scholar]
- McIlwain et al., 2001.McIlwain KL, Merriweather MY, Yuva-Paylor LA, Paylor R. The use of behavioral test batteries: effects of training history. Physiol Behav. 2001;73:705–717. doi: 10.1016/s0031-9384(01)00528-5. [DOI] [PubMed] [Google Scholar]
- McKenzie et al., 2007.McKenzie O, Ponte I, Mangelsdorf M, Finnis M, Colasante G, Shoubridge C, Stifani S, Gécz J, Broccoli V. Aristaless-related homeobox gene, the gene responsible for West syndrome and related disorders, is a Groucho/transducin-like enhancer of split dependent transcriptional repressor. Neuroscience. 2007;146:236–247. doi: 10.1016/j.neuroscience.2007.01.038. [DOI] [PubMed] [Google Scholar]
- McManus and Golden, 2005.McManus MF, Golden JA. Neuronal migration in developmental disorders. J Child Neurol. 2005;20:280–286. doi: 10.1177/08830738050200040301. [DOI] [PubMed] [Google Scholar]
- McManus et al., 2004.McManus MF, Nasrallah IM, Pancoast MM, Wynshaw-Boris A, Golden JA. Lis1 is necessary for normal non-radial migration of inhibitory interneurons. Am J Pathol. 2004;165:775–784. doi: 10.1016/S0002-9440(10)63340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura et al., 1997.Miura H, Yanazawa M, Kato K, Kitamura K. Expression of a novel aristaless related homeobox gene ‘Arx’ in the vertebrate telencephalon, diencephalon and floor plate. Mech Dev. 1997;65:99–109. doi: 10.1016/s0925-4773(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Nasrallah et al., 2004.Nasrallah IM, Minarcik JC, Golden JA. A polyalanine tract expansion in Arx forms intranuclear inclusions and results in increased cell death. J Cell Biol. 2004;167:411–416. doi: 10.1083/jcb.200408091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira et al., 2002.Ohira R, Zhang YH, Guo W, Dipple K, Shih SL, Doerr J, Huang BL, Fu LJ, Abu-Khalil A, Geschwind D, McCabe ER. Human ARX gene: genomic characterization and expression. Mol Genet Metab. 2002;77:179–188. doi: 10.1016/s1096-7192(02)00126-9. [DOI] [PubMed] [Google Scholar]
- Overstreet-Wadiche and Westbrook, 2006.Overstreet-Wadiche LS, Westbrook GL. Functional maturation of adult-generated granule cells. Hippocampus. 2006;16:208–215. doi: 10.1002/hipo.20152. [DOI] [PubMed] [Google Scholar]
- Parent et al., 2006.Parent JM, von dem Bussche N, Lowenstein DH. Prolonged seizures recruit caudal subventricular zone glial progenitors into the injured hippocampus. Hippocampus. 2006;16:321–328. doi: 10.1002/hipo.20166. [DOI] [PubMed] [Google Scholar]
- Paxinos and Franklin, 2001.Paxinos GM, Franklin KBJ. Ed 2. San Diego: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Poirier et al., 2004.Poirier K, Van Esch H, Friocourt G, Saillour Y, Bahi N, Backer S, Souil E, Castelnau-Ptakhine L, Beldjord C, Francis F, Bienvenu T, Chelly J. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Brain Res Mol Brain Res. 2004;122:35–46. doi: 10.1016/j.molbrainres.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Poirier et al., 2008.Poirier K, Eisermann M, Caubel I, Kaminka A, Peudonnierg S, Boddaerth N, Sailloura Y, Dulac O, Souville I, Beldjord C, Lascelles K, Plouin P, Chelly J, Bahi-Buisson N. Combination of infantile spasms, non-epileptic seizures and complex movement disorder: a new case of ARX-related epilepsy. Epilepsy Res. 2008;2–3:224–228. doi: 10.1016/j.eplepsyres.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Powell et al., 2003.Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J Neurosci. 2003;23:622–631. doi: 10.1523/JNEUROSCI.23-02-00622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan et al., 1994.Ryan SG, Buckwalter MS, Lynch JW, Handford CA, Segura L, Shiang R, Wasmuth JJ, Camper SA, Schofield P, O'Connell P. A missense mutation in the gene encoding the alpha 1 subunit of the inhibitory glycine receptor in the spasmodic mouse. Nat Genet. 1994;7:131–135. doi: 10.1038/ng0694-131. [DOI] [PubMed] [Google Scholar]
- Sahay et al., 2005.Sahay A, Kim CH, Sepkuty JP, Cho E, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J Neurosci. 2005;25:3613–3620. doi: 10.1523/JNEUROSCI.5255-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued and Hopkins, 2000.Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Shahbazian et al., 2002.Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Shinozaki et al., 2009.Shinozaki Y, Osawa M, Sakuma H, Komaki H, Nakagawa E, Sugai K, Sasaki M, Goto Y. Expansion of the first polyalanine tract of the ARX gene in a boy presenting with generalized dystonia in the absence of infantile spasms. Brain Dev. 2009;31:469–472. doi: 10.1016/j.braindev.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Shoubridge et al., 2007.Shoubridge C, Cloosterman D, Parkinson-Lawerence E, Brooks D, Gécz J. Molecular pathology of expanded polyalanine tract mutations in the Aristaless-related homeobox gene. Genomics. 2007;90:59–71. doi: 10.1016/j.ygeno.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Spencer et al., 2005.Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R. Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav. 2005;4:420–430. doi: 10.1111/j.1601-183X.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Strømme et al., 2002a.Strømme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SM, Bruyere H, Lütcherath V, Gedeon AK, Wallace RH, Scheffer IE, Turner G, Partington M, Frints SG, Fryns JP, Sutherland GR, Mulley JC, Gécz J. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002a;30:441–445. doi: 10.1038/ng862. [DOI] [PubMed] [Google Scholar]
- Strømme et al., 2002b.Strømme P, Mangelsdorf ME, Scheffer IE, Gécz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev. 2002b;24:266–268. doi: 10.1016/s0387-7604(02)00079-7. [DOI] [PubMed] [Google Scholar]
- Turner et al., 2002.Turner G, Partington M, Kerr B, Mangelsdorf M, Gécz J. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am J Med Genet. 2002;112:405–411. doi: 10.1002/ajmg.10714. [DOI] [PubMed] [Google Scholar]
- Tybulewicz et al., 1991.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Velísek et al., 2007.Velísek L, Jehle K, Asche S, Velísková J. Model of infantile spasms induced by N-methyl-d-aspartic acid in prenatally impaired brain. Ann Neurol. 2007;61:109–119. doi: 10.1002/ana.21082. [DOI] [PubMed] [Google Scholar]
- von Bohlen und Halbach, 2007.von Bohlen , Halbach O. Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 2007;329:409–420. doi: 10.1007/s00441-007-0432-4. [DOI] [PubMed] [Google Scholar]
- Wilson, 2007.Wilson CJ. GABAergic inhibition in the neostriatum. Prog Brain Res. 2007;160:91–110. doi: 10.1016/S0079-6123(06)60006-X. [DOI] [PubMed] [Google Scholar]