

Abstract
Cellular signaling pathways respond to external inputs to drive pivotal cellular decisions. Far from being mere data relay systems, signaling cascades form complex interacting networks with multiple layers of feedback and feed-forward control loops regulated in both space and time. While it may be intuitively obvious that this complexity allows cells to assess and respond appropriately to a myriad of external cues, untangling the wires to understand precisely how complex networks function as control and computational systems presents a daunting challenge to theoretical and experimental biologists alike. In this review we have focused on activation of the canonical MAP kinase cascade by receptor tyrosine kinases (RTKs) in order to examine some of the fundamental design principles used to build biological circuits and control systems. In particular, we explore how cells can reconfigure signaling cascades to generate distinct biological outputs by utilizing the unique spatial constraints available in biological membranes.
Keywords: Ras, Raf, MEK, ERK, MAP kinase, nanocluster, lipid raft, digital, analog, scaffold, robust, noise, stochastic
Introduction to MAPK Signaling
Receptor tyrosine kinases (RTKs) regulate a plethora of critical biological functions throughout embryogenesis, growth and development. RTKs are cell surface proteins that span the plasma membrane. The extracellular N-terminal domain binds to the ligand, the majority of which are soluble peptides, while the intracellular C-terminus contains a tyrosine kinase domain. In general RTKs exist as inactive monomers that form dimers following ligand binding.1-3 Receptor dimerization leads to autophosphorylation of tyrosine residues in the intracellular domain by the tyrosine kinase domain of the receptor. These phosphorylation sites serve as specific binding sites for proteins that contain either Src-homology 2 (SH2) or phosphotyrosine binding (PTB) domains. Ras proteins transmit signals from RTKs to intracellular networks. In mammals the three main Ras isoforms, H-Ras, N-Ras and K-Ras are tethered to the inner plasma membrane by carboxy-terminal lipid anchors.4,5 Ras activation is tightly regulated by guanine nucleotide exchange factors that catalyze GDP release and allow GTP binding and activation. The canonical pathway of Ras activation envisages recruitment of the exchange factor SOS-Grb2 complex from the cytosol to the activated RTKs at the membrane.6,7 Recruitment occurs through protein-protein interactions between the SH2 domain of Grb2 and the tyrosine phosphorylated receptor or with docking proteins recruited to the membrane by the RTK.3 Recent work has identified an alternative, more important mechanism of Sos recruitment that involves protein-lipid interactions between the PH domain of Sos and phosphatidic acid (PA).8-10 The PA is generated from phosphatidylcholine by phospholipase D2, a plasma membrane localized lipase that is activated by the RTK. Recruiting SOS to the plasma membrane generates Ras-GTP, which can then bind to the REM domain of Sos inducing higher GEF activity.11,12 This positive feedback loop modulates RTK signal amplitude and duration in response to ligand stimulation.13
One of the core signaling pathways activated by Ras is the mitogen activated protein kinase cascade (MAPK) comprised of Raf, MEK and ERK.14 All eukaryotic organisms possess MAPK pathways that are used multiple times during growth and development.15 MAPK pathways are three-tiered kinase cascades. At the top of the cascade is a MAP Kinase Kinase Kinase (MKKK) that phosphorylates and activates a MAP Kinase Kinase (MKK) that in turn phosphorylates and activates a MAP kinase (MAPK).15 Active Ras proteins interact directly with the Raf family of MKKKs (comprised of three isoforms in mammals, A-Raf, B-Raf and C-Raf). The Ras-Raf interaction is the crucial first step in Raf activation, however Raf activation is a complex process that involves Ras binding, interaction with lipids, de-phosphorylation of inhibitory residues and phosphorylation of activating residues and changes in protein-protein interactions.3,16,17 Inactive MEK is in a complex with ERK in the cytosol.18,19 Raf activates MEK by phosphorylating two serine residues within the MEK activation loop. Once activated MEK phosphorylates ERK at threonine and tyrosine residues in a ‘TEY’ motif in the ERK activation loop. ERK phosphorylation occurs by a distributive mechanism, where ERK dissociates from MEK between the first and second phosphorylation.20,21 Active ERK dissociates from MEK and phosphorylates >150 substrates throughout the cell.22
One Pathway, Multiple Outputs
A central questions concerning the MAP kinase pathway is how a single kinase cascade can simultaneously regulate multiple biological functions within the same cell. One solution to the ‘specificity problem’ may be found in how MAP kinase modules are wired to create different network configurations in vivo. In all cells MAP kinase modules are tightly controlled by positive and negative feedback loops, as well as a variety feed-forward loops (Fig. 1). Different feedback control systems can theoretically allow the same module to generate different signal outputs, including graded (analog), ultrasensitive (switch-like), all or nothing (digital) and oscillations23-29 (Fig. 2). Arguably the MAPK module is not a single pathway at all but rather a diverse set of different signaling circuits, with the basic three-tiered module operating as a ‘plug-and-play’ template used over and again to control different biological outcomes. For the remainder of this review, we will focus on digital and analog signaling and illustrate how cells use spatial regulation to create different network configurations.
Figure 1.
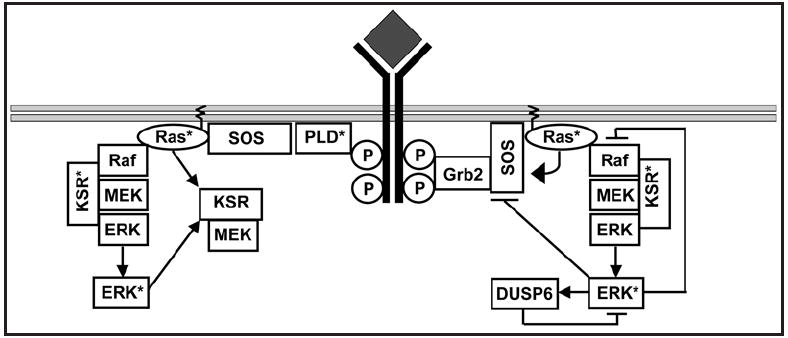
Multiple regulatory loops exist within the EGF signaling cascade. After the EGF receptor is activated by ligand two exchange factors generate active Ras, the classical Grb2-SOS complex and the recently discovered PLD mediated SOS recruitment. SOS activates Ras, which in turn recruits and activates the MAP kinase module. Multiple regulatory loops exist with the cascade, including a positive feedback loop from active Ras to SOS,13 a positive feed-forward loop from Ras to KSR,74 a positive feedback loop from ERK to KSR,59 a negative feedback loop from ERK to Raf,75 and a negative feedback loop from ERK back onto itself via the phosphatase DUSP6.76 The presence of so many regulatory loops provides the pathway with a variety of control systems, which may be used to create different types of output from the Ras pathway in vivo.
Figure 2.

One pathway, many outputs. The MAP kinase cascade can produce multiple outputs, including: (A) Graded. Here the system displays a hyperbolic curve, at low-level stimulus the output increases linearly with the input. (B) Switchlike or ultrasensitive. These systems exhibit sigmoidal input-output curves where the first increments produce little response, then when the system does respond it rapidly reaches maximum output. The activation of the MAP kinase module in vitro is switch-like due to the distributive phosphorylation of ERK.26 (C) Bistable or Digital. These systems represent the extreme conclusion of switch-like kinetics, in which the input-output curve is also sigmoidal and in ideal systems the transition from the off to on state is instantaneous (i.e. the slope of the curve between the off state and the on state is infinite). Although this ideal is never reached in biology, many systems have a sufficiently abrupt transition such that they function as true biological switches to drive discrete cell fate decisions.26,39,59,77-79
Digital Signaling
True switches are bistable, meaning they can switch between two distinct steady states but cannot rest in intermediate states.30-32 The low and high steady states represent ‘off’ and ‘on’ respectively. Biology is replete with instances where on/off decision-making is crucial, for example the decision to proliferate, differentiate or undergo programmed cell death. Much work has therefore been focused on biological mechanisms that can generate digital signal outputs. Under the appropriate conditions, bistability can arise from substrate inhibition, positive and double negative feedback, regulated protein translocation, or a combination of these mechanisms.28-33 In Xenopus oocytes, bistability is generated through a positive feedback loop combined with the switch-like kinetics of ERK activation caused by distributive phosphorylation.26 In addition, kinetic modeling has shown that bistability can arise solely from the multi-site distributive phosphorylation of MEK and ERK.34 The digital nature of MAP kinase signaling seems teleologically appropriate given how often MAPK modules are embedded in signaling pathways that regulate all-or-nothing cell fate decisions.
Analog Signaling
In contrast to digital circuits, analog circuits transmit continuous information that is directly proportional to the input stimulus (Fig. 2). Analog circuits have two main advantages over their digital counterparts. First, analogs circuits allow a range of different outputs from the same input source. This principle is strikingly illustrated by a morphogen that is produced at a localized source and forms a concentration gradient as it spreads through surrounding tissue.35 The graded signal acts directly on cells in a concentration-dependent manner to specify cell fate selection, thus functioning as an analog positional cue during development to organize patterns of cell differentiation.35 An example is the morphogen gradient along the dorsoventral axis of Drosophila embryos that specifies between four to seven thresholds of gene expression.36 Secondly, analog systems have the ability to condense data manipulation. For example a digital system has only two memory states, on or off, whereas an equivalent analog system can store a range of values to any arbitrarily high number.28 As the universe is inherently analog, and given the flexibility and data handling capabilities of analog systems, it has been convincingly argued that evolution has selected largely for analog based signaling networks.28,37 Indeed, several groups have shown that the MAP kinase pathway can generate a graded output in response to RTK activation.23,38-40 How signaling pathways in general, and the MAPK pathway in particular, are able to transmit graded information to control concentration-dependent cell functions has remained one of the outstanding questions in biology. Below, we explore how spatial regulation of MAPK signaling provides a simple and robust solution to this engineering conundrum.
Ras Proteins Generate Signaling Nanoclusters on the Plasma Membrane
The plasma membrane is a lipid barrier that separates cells from their external environment. Long thought to be disordered structure whose primary function was to act as a permeable boundary, it is now clear that the plasma membrane is a complex, dynamic structure that is compartmentalized on multiple lengths and time scales.41 Compartmentalization is driven by a variety of lipid-lipid, lipid-protein, protein-protein and actin cytoskeleton interactions, the net result of which is the non-random distribution of proteins and lipids across different types of transient nanoscale domains.41 An important role ascribed to these domains is that of selectively concentrating proteins to facilitate protein-protein interactions and the assembly of signaling complexes. Recent computer simulations of protein diffusion within two-dimensional membrane environments have provided novel general insights as to how this may occur.42,43
Nanodomain formation by the Ras proteins provides a specific example. Electron microscopy and single flourophore tracking reveals that Ras proteins operate in discrete nanoclusters on the inner plasma membrane.44-48 These nanoclusters contain ~7 Ras proteins, and have radii of 6–12 nm and short lifetimes (~0.4 s).46,47 The different Ras isoforms in their GTP and GDP loaded states occupy spatially distinct nanoclusters that have different structural requirements for actin, cholesterol and various scaffold proteins such as galectin-1, galectin-3 and sur-8.44,49-51 Only a fraction of any Ras isoform (~40%) is confined to nanoclusters at any given time point, with the remainder arrayed randomly over the cell surface.44 Current thinking is that Ras proteins actually drive the formation of the nanodomains in which they operate.48,52
For Ras nanoclusters to function as signaling platforms, they must be able to recruit downstream effectors. Single particle tracking of Ras and Raf interactions in live cells confirm that Raf is recruited from the cytosol by Ras to immobile Ras clusters.45,46 Raf proteins have three separate membrane binding motifs, a Ras binding domain (RBD), a cysteine rich domain (CRD) that binds to phosphatidyl serine, and a phosphatidic acid binding-motif.16 Although neither MEK nor ERK contain membrane-targeting sequences, MEK binds to both Raf and ERK, providing a direct method of recruitment of the entire MAP kinase cascade to Ras nanoclusters.53-55 In addition MAP kinase scaffold proteins act as docking platforms that bind to two or more components of the MAPK module to direct the MAP kinase module to membrane environments within cells (reviewed in ref. 3). KSR is the best characterized mammalian scaffold protein. KSR interacts with Raf, MEK and ERK and facilitates ERK activation at the plasma membrane.56,57 The localization of KSR is dynamic and regulated by RTK activation. In quiescent cells KSR is sequestered in the cytosol by interaction with 14-3-3 that masks the KSR membrane-targeting domain.58 RTK activation leads to dephosphorylation of one of the 14-3-3-binding sites, thus exposing the KSR membrane-targeting domain and allowing membrane recruitment.58
Building a High-fidelity Analog Circuit in Two Dimensions
Blocking the formation of Ras nanoclusters, by ablating critical scaffolds or perturbing the actin cytoskeleton, disrupts Ras-dependent MAPK signaling.44,47,48 These observations confirm that nanoclusters are critically important for MAPK activation, however they do not reveal what these signaling platforms actually do. To answer this question we used a combination of computational modeling and experiments to explore how Ras nanoclusters are used to build signaling circuitry in vivo.40 We first showed that MAPK signaling is under tight spatial regulation. Activation of the MAPK cascade by constitutively active Raf does not occur in the cytosol, but is actually confined to Ras nanoclusters on the plasma membrane (Fig. 3 and ref. 40).
Figure 3.

Cytosolic activation of the MAP kinase pathway is suppressed in vivo. Constitutively active human C-Raf mutants (C-Raf with all four activating phosphorylation sites mutated to acidic residues [S338D/Y341D/T491E/ S494D = Raf*]80 a generous gift from Dr Kun-Liang Guan) were engineered to have different cellular distributions in vivo. Raf* exists in a dynamic equilibrium between the plasma membrane and cytosol due to its membrane-targeting motifs. Raf*ΔM is exclusively cytosolic due to point mutations within each Raf membrane-targeting motifs that abrogates motif-substrate binding (R89L/C165S,C168S/R398A,K399A,R401A).81-83 This mutant was targeted constitutively to the plasma membrane by addition of the C-terminal membrane-targeting motif of K-Ras to generate Raf*ΔM-tK.84 (A) The schematic diagram shows the cellular distribution expected for each of the mutant proteins. (B) HEK-293 cells transiently transfected and expressing the mutant Raf proteins were harvested, separated into crude membrane and cytosol fractions, and each fraction immunoblotted for C-Raf and ERK2 (=input control). Non-transfected and empty vector transfections are also shown as control. Each of the mutant Raf proteins displays the expected cellular distribution. (C) HEK-293 cells transiently transfected with the mutant Raf proteins were serum starved overnight, harvested and whole cell lyates immunoblotted for C-Raf, ppERK and ERK2. Strikingly Raf*ΔM, which does not have access to the plasma membrane, was a poor activator of the MAP kinase pathway in vivo. We conclude from these results and others40 that activation of the MAP kinase pathway by Raf is suppressed within the cytosol.
We then assessed the signal output from individual nanoclusters and found that each nanocluster responds maximally to very low inputs (input = Raf kinase activity).40 Two possible mechanisms could account for this response. The first is that each nanocluster is a bistable switch with a very low threshold of activation as the system fires maximally over a 2-log range of inputs in vivo.40 The alternative mechanism is that each nanocluster is not bistable but is highly sensitive to, and rapidly saturated by low Raf input. Each nanocluster therefore functions as a high gain amplifier. Modeling and experimental data support the latter mechanism (Fig. 6 and refs. 40,53 and 59). Operationally either mechanism generates a maximal ERKpp burst during the short lifetime of the nanocluster. This is because the dominant factor within the system is the transient nature of individual nanoclusters. These concepts are captured by the term nanoswitch, reflecting both the spatiotemporal scale and the digital output characteristics of the Ras signaling nanocluster.
Figure 6.

Ras nanoclusters amplify low MEK activity in vivo. COS cells were transfected with constitutively active Raf proteins targeted to the plasma membrane (Raf*-tK) with either full activity [Raf*-tK], catalytically inactive [Raf*-tK(KD)], intermediate catalytic activity (Raf*-tK(S508N)], or very low catalytic activity (Raf*-tK(S508D)], serum starved for 18 hours then whole cell lysates immunoblotted for C-Raf, ppMEK, MEK1, ppERK and ERK2. Raf*-tK activated both MEK and ERK in vivo, whereas the kinase dead control did not. Raf*-tK(S508N) activated MEK equivalent to the fully active control, confirming that nanoclusters amplify low Raf activity in vivo. Raf*-tK(S508D), the Raf protein with the lowest catalytic activity, poorly activated MEK. Nevertheless, this low MEK activity was sufficient to fully activate ERK in vivo. Thus both in silico and in vivo experiments support the idea that Ras nanoclusters amplify both Raf and MEK activity, functioning as high-gain amplifiers for the MAP kinase module in vivo.
Next we asked how nanoswitches are utilized to build biological circuits in response to EGF receptor activation. We found that the number of Ras nanoswitches generated at the plasma membrane is a linear function of EGF concentration.40 Digitization is the process of approximating a continuous range of values using a finite set of discrete values or quanta. The plasma membrane therefore digitizes the EGF analog input, with individual nanoswitches functioning as the discrete quanta. Thus the plasma membrane functions as a linear analog-to-digital converter (ADC) by activating a discrete number of nanoswitches in direct proportion to the analog EGF input (Fig. 4). Each nanoswitch dumps the same quanta of active ERK into the cytoplasm, allowing the cells to convert the digital pulses from individual Ras nanoclusters into a final analog output by summing the total ppERK output from the total number of nanoclusters. In effect the cytoplasm functions as a linear digital-to-analog converter (DAC), reversing the analog-to-digital conversion performed by the plasma membrane (Fig. 4). This analog-digital-analog circuit relay generates high-fidelity signal transmission for both low and high EGF inputs in vivo.40
Figure 4.
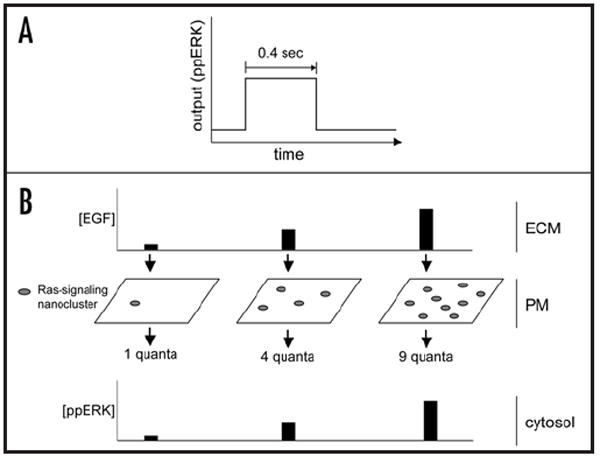
EGF signal transmission is digitised across the plasma membrane. (A) Ras nanoclusters have an avarage radius of 6–12 nanometers and short lifetimes (~4 s).45-47 During their brief existence, Ras nanoclusters generate maximal ppERK output, functioning as switches (termed nanoswitches to capture their size and digital signal output). (B) EGF is the analog input present in the extracellular matrix (ECM). Activated EGF receptors trigger the formation of Ras nanoswitches at the plasma membrane in direct proportion to the external EGF input through an unknown mechanism.40 The plasma membrane thus digitizes analog EGF inputs using individual Ras nanoswitches as discrete quanta, functioning as a linear analog-to-digital converter (ADC). As the output of each nanocluster is dumped into the cytoplasm, the digital pulses from individual nanoclusters are summed to give a final analog ppERK output. In this way the cytoplasm reverses the digital conversion that occurred at the plasma membrane and functions as a digital-to-analog converter (DAC). This biological circuit generates the high fidelity signal transmission across the plasma membrane that occurs in vivo.40
Why Does the Nanoswitch System Deliver High-Fidelity Signaling?
The fidelity of ADCs is a function of two factors: the sampling rate that controls how many samples are taken per second; and the sampling precision which controls how many graduations (quantization levels) lie between the minimum and maximum outputs. As the sampling rate and sampling precision increases, so too does the fidelity (fidelity = the similarity between the input and output signals) (Fig. 5). Ras nanoswitches are highly dynamic because each nanocluster turns over on average every 0.4 s: this means that the cell could sample the external environment over 100 times per minute if a ‘single’ nanocluster was activated. As more nanoclusters are activated, the sampling rate increases proportionally. Mammalian fibroblasts can generate ~50,000 Ras.GTP nanoclusters on the plasma membrane.40 This large number of signaling units affords the nanocluster system an extraordinarily high resolution of signal transfer as the signal between zero and maximum is effectively divided into 50,000 increments. The combination of a fast sampling rate and large sampling precision endows the nanocluster system with the high fidelity of signal transfer we observed in silico and in vivo.
Figure 5.

Fast sampling rates and large sampling precision generates high fidelity signal transmission. The fidelity of signal transmission in a linear ADC is controlled by two variables: the sampling rate, which determines how many samples are taken over time; and the sampling precision, which determines how many quantization levels are between zero and maximum. Figure 5 shows how increasing both these variables increases signal fidelity during signal transmission. The red line represents the analog input signal that changes in amplitude over time. In the upper left panel, both the sampling rate and the sampling precision of the ADC are low (5 and 4 respectively). When the DAC recreates the analog input curve from these numbers it generates the blue line shown in the upper right panel. It is clear from this graph that the output curve is a poor representation on the analog input and much data has been lost during signal transmission. If both the sampling rate and sampling precision of the ADC is increased (bottom panels), then the output curve generated from the DAC more precisely matches the input, generating an equivalent improvement in signal fidelity.
It might be theoretically possible to further increase the resolution of the system if each nanocluster generated an analog rather than digital output. We tested this hypothesis in silico by modifying key parameters within the MAPK module to realize an analog output from each nanocluster. Importantly, we found that only a digital nanocluster output returned a high-fidelity linear output. All nanocluster outputs between digital and linear generated nonlinear low-fidelity system outputs, with the degree of non-linearity being proportional to the degree of graded output from the nanoclusters.40 High-fidelity signal transmission therefore results directly from the digital nanocluster output combined with the simple first-order relationship between EGF input and the number of nanoclusters.
Ras Nanoclusters are Essential for Signal Transmission
A striking result to emerge from our analyses is that Ras nanoclusters are absolutely required for Ras signal transduction. As either the number or size of Ras nanoclusters decreases in silico, the system response to EGF progressively fails.40 When Ras nanoclusters disappear altogether and Ras proteins function as individual molecules, signal output decreases to 3% of the maximal ERKpp achievable with nanoclusters intact.40 Our computer simulations precisely match biological experiments, which show that Ras signal output is progressively lost as Ras clustering decreases.40,47 These combined data reveal that the cell cannot be activated without functional nanoclusters at the plasma membrane. Our in silico analyses provides insight into why this occurs. As nanoclusters are lost the target area of the plasma membrane available for Raf and KSR-MEK-ERK recruitment is progressively diminished, and thus the probability of a successful recruitment and activation event falls. An intriguing and previously unexplored mechanism to emerge from these observations is that cells may modulate responsiveness to EGF and other ligands by regulating the degree of Ras clustering. Ectopic expression of galectins enhances EGF-stimulated ERK activation:51 our results suggest that this is due to galectin expression increasing the target area of the plasma membrane for Raf and KSR-MEK-ERK recruitment by increasing the extent of Ras nanoclustering. Galectin expression is carefully regulated throughout development and during different physiological and pathological conditions,60 consistent with our hypothesis that galectin levels may modulate cellular responsiveness to EGF and other ligands. Galectin regulation of Ras nanoclusters may also have been hijacked in cancers that show increased galectin expression.61,62 Modulating cellular responsiveness to mitogens by regulating Ras nanoclustering appears crucial for both normal growth and development and may be relevant in oncogenic transformation.
The Ras Nanocluster System is Biologically Robust
Robustness is a fundamental characteristic of all biological systems and is defined as ‘a property that allows a system to maintain its functions against internal and external perturbations’.63 As the function of the Ras nanoswitch system is high-fidelity signal transmission, we asked whether the nanoswitch circuit confers robustness with respect to signal transmission in vivo. All biological systems are subject to a variety of random disturbances or variations, collectively referred to as noise. Biological systems suffer from both extrinsic noise (that is noise generated external to the system), and intrinsic noise (noise inherent to the system itself). Analog systems are particularly vulnerable to degradation by noise when they are copied or amplified (as occurs when transmitting the EGF signal across the plasma membrane), since small fluctuations can result in a significant change in the output signal. Because of the small numbers of reactive species within a single nanocluster, nanoclusters are susceptible to intrinsic noise.64,65 Despite this, the nanoswitch system is highly robust to intrinsic noise.40 This is because by spatially dividing the system into transient modules (i.e. nanoswitches), any perturbations caused by intrinsic noise are contained within the individual modules. Moreover, the large number of modules dampens the effects of intrinsic noise simply by averaging out the effects of noise within the total system.
Activation of the MAP kinase module exclusively at the plasma membrane renders the pathway resistant to pharmacological inhibitors of MEK activation,59 showing that the Ras nanoswitch system is robust to perturbations in MEK activity in vivo. In silico simulations suggest that Ras nanoclusters function as a high-gain amplifiers for low MEK catalytic activity (Tian and Hancock, unpubl. data). To test this prediction experimentally, we expressed a Raf mutant (RafS508D) that activates MEK very poorly even when targeted to the plasma membrane (Fig. 6 and ref. 53), and found that ERK activation was equivalent to the wild-type control in vivo (Fig. 6). Taken together these data confirm that Ras nanoclusters function as high-gain amplifiers for MEK activity in vivo, and this amplification renders Ras nanoclusters robust to perturbations in both Raf and MEK catalytic activity. Thus the Ras nanocluster system generates high fidelity data transmission for both low-strength and high-strength signals across the plasma membrane that is biologically robust, making it an ideal circuit configuration for signal transduction in vivo.
These data may also explain why tumor cells harboring oncogenic Ras are resistant to MEK inhibition, whereas tumor cells with oncogenic B-Raf are exquisitely sensitive.66 When the pathway is activated at the plasma membrane by oncogenic Ras, the robust Ras nanoswitch system generates high ERK output despite MEK inhibition. In contrast, when oncogenic B-Raf activates the pathway from the cytosol, where high-gain amplification is absent, the pathway is now extremely sensitive to MEK inhibition.66 In addition, our results potentially rationalize why Raf activation is so complex. It is now clear that Raf activation does not have to be anywhere near maximal to fully activate the MAP kinase pathway from Ras nanoclusters.40,53,59 We therefore suggest that the multiple Raf activation mechanisms are not necessarily required in combination, but instead provide redundancy to ensure Raf activation occurs on a sufficiently rapid time-scale to match Ras nanocluster lifetime. Indeed, the high basal catalytic activity of the B-Raf isoform means that membrane recruitment of B-Raf alone may be sufficient to activate the nanoswitch system.
In summary, we have shown that Ras nanoclusters function as digital nanoswitches in vivo. Cells use Ras nanoswitches to digitize the analog EGF input and build a linear ADC that transmits analog signals across the plasma membrane with high fidelity. This circuit configuration is biologically robust and absolutely necessary for Ras signal transduction. As many other signaling pathways are spatially organized within lipid rafts and plasma membrane nanoclusters, we speculate that the use of nanoclusters may represent a basic systems-level design principle for the generation of robust, high fidelity signal transduction circuits in vivo.
The Role of RKIP
A critical question posed by our study is why constitutively active Raf does not activate MEK in the cytosol. Our recent experiments provide some new insights here. Raf kinase Inhibitor Protein (RKIP) can bind to both Raf and MEK, but this binding is mutually exclusive because the binding sites for Raf and MEK overlap within the RKIP molecule.67,68 One function of RKIP is to block Raf signaling and this is achieved via two mechanisms: RKIP prevents a direct interaction between Raf and MEK68 and inhibits the phosphorylation of activating residues on C-Raf.69 Protein kinase C (PKC) phosphorylates RKIP on S153 causing dissociation of RKIP from Raf and subsequent activation of the MAPK pathway.70 Given that RKIP is predominantly localized to the cytosol, we hypothesized that RKIP mediates the cytosolic inhibition of the MAP kinase pathway we observed in vivo. A prediction of this model is that membrane localization should protect Raf from RKIP inhibition. We tested this experimentally by co-expressing RKIP with constitutively active Raf either localized to the cytosol or targeted to the plasma membrane. We found that Raf targeted to the plasma membrane could activate ERK despite the presence of RKIP and drive cell proliferation, whereas activation of the MAPK pathway by cytosolic Raf was efficiently inhibited by RKIP co-expression (Fig. 7). We conclude that RKIP suppresses activation of the MAP kinase cascade in the cytosol and suggest that an additional role Raf membrane recruitment is to remove the inhibitory effect of RKIP.
Figure 7.

RKIP inhibits cytosolic but not plasma membrane activation of the MAPK module. (A) COS cells transfected with constitutively active Raf-1 (Raf*) or constitutively active Raf targeted to the plasma membrane (Raf*-tK) with or without RKIP were serum starved, fractionated and the membrane and cytosolic fractions immunoblotted for C-Raf. The cytosolic fractions were immunoblotted for ppERK, ERK2 and RKIP. RKIP expression efficiently inhibited ERK activation by Raf*, whereas RKIP did not inhibit ERK activation by membrane targeted Raf*-tK. (B) Identical aliquots of COS cells were co-transfected with 5 μg of either Raf* or Raf*-tK plasmid, and a range of RKIP plasmid concentrations (0–20 μg). After 48 hours cells were harvested and proliferation quantified by counting cells. Figure 6B shows that even at high levels of RKIP expression, cells expressing membrane targeted Raf*-tK continued to proliferate. In contrast, low levels of RKIP expression inhibited proliferation of cells expressing cytosolic Raf* (The RKIP expression vector was a generous gift from Dr. Walter Kolch68).
Using Three Dimensions to Generate Multiple Outputs
Rat adrenal pheochromocytoma cells (PC-12) cells use the MAPK module to generate two opposing cell fate decisions. Activation of the MAPK pathway by the EGF drives proliferation, whereas activation of the MAPK pathway by NGF through activation of the receptor tyrosine kinase TrkA leads to differentiation.71 The duration of ERK activation has long been thought to generate these opposing biological outcomes.72 EGF stimulates a rapid and transient activation of ERK that is required for proliferation. In contrast, NGF stimulates the sustained activation of ERK that is required for differentiation.72 Immediate early gene (IEG) products are stabilized by ERK phosphorylation during prolonged ERK activation, providing a mechanism for cells to interpret and respond differently to transient and sustained ERK activation.73 However recent results show that EGF and NGF treatment produce qualitatively different signal outputs from the MAP kinase module in PC-12 cells, revealing additional layers of MAPK network control over cell fate decisions.39
Working from the hypothesis that different ERK activation dynamics are ultimately controlled by different network configurations, Santos et al showed that EGF generates an analog ERK activation in PC-12 cells, whereas NGF treatment produced a digital ERK output.39 Guided by modular response analysis (MRA) results and historic modeling predictions, Santos et al made the striking observation that the EGF-stimulated MAPK network could be rewired by the concomitant activation of protein kinase C (PKC) to generate digital ERK activation and PC-12 differentiation. The converse also held true, with NGF-stimulated MAPK activation rewired to generate an EGF-like output by inhibition of PKC. As PKC modulates ERK activity through RKIP, the authors next investigated the role of RKIP in MAPK network configuration. Downregulation of RKIP protein levels by RNAi rewired the EGF response to give digital ERK activation and PC-12 differentiation. In combination these results place PKC upstream of the MAPK module, and suggest that PKC phosphorylation of RKIP reconfigures the MAPK module from an analog system to digital one, thereby directing opposing cell fate decisions.39
In combination with our results, the two studies reveal how spatial regulation can be used to rewire network configurations and give different outputs. By suppressing cytosolic signaling with RKIP, activation of the MAPK module is restricted to the plasma membrane; in this case the system generates a high-fidelity analog output.40 However, if the MAPK pathway is activated in conjunction with protein kinase C, RKIP is inactivated and cytosolic activation of the pathway also occurs.39 In this case the bistable nature of the module is dominant and the system output is digital. Thus spatial regulation provides cells with a simple yet robust mechanism to generate discrete biological outputs using the same fundamental network components.
Conclusions
It has been known for some time that MAP kinase modules generate fundamentally different types of output in vivo, although the underlying mechanisms allowing multiple outputs from a single pathway have remained obscure. In this review we have proposed a spatial model whereby membrane nanocluster environments are used to re-wire signaling networks to produce distinct signal outputs and biological outcomes. A particularly intriguing discovery of the nanocluster system is the use of both digital and analog subsystems to create a high-fidelity analog circuit. Characterizing the input/output characteristics of the various components that make up signaling cascades is critical to understand how these pathways are used as control systems during normal growth and development. This approach has important therapeutic applications in identifying pathway components that are robust to perturbations in activity, as is the case for the MAPK module, versus those components that are non-robust and therefore viable targets for therapeutic intervention. Finally, as many core-signaling pathways are spatially organized within lipid rafts and plasma membrane nanoclusters, it is imperative we continue to develop a systems level understanding of the contribution of membrane environments to normal and pathological signal transduction.
Acknowledgments
This work was supported by grants from the Australian Research Council (ARC), the Queensland Cancer Fund, the National Health and Medical Research Council (Australia) and the National Institutes of Health. IMB is a Special Research Centre of the ARC.
Abbreviations
- ADC
analog to digital converter
- CRD
cysteine rich domain
- DAC
digital to analog converter
- EGF
epithelial growth factor
- EGFR
epithelial growth factor receptor
- IEG
immediate early gene
- KSR
kinase suppressor of Ras
- MRA
modular response analysis
- NGF
neuronal growth factor
- PA
phosphatidic acid
- PC-12
rat adrenal pheochromocytoma cells
- PH
pleckstrin homology
- PKC
protein kinase C
- PLD
phospholipase D
- PTB
phosphotyrosine binding
- REM
Ras exchanger motif
- MAPK
mitogen activated protein kinase
- RBD
Ras binding domain
- RKIP
Raf kinase inhibitor protein
- RTK
receptor tyrosine kinase
- SH2
Src homology domain
- SOS
son of sevenless
Footnotes
Previously published online as a Cell Cycle E-publication:
References
- 1.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 2.Pawson T. Regulation and targets of receptor tyrosine kinases. Eur J Cancer. 2002;38:3–10. doi: 10.1016/s0959-8049(02)80597-4. [DOI] [PubMed] [Google Scholar]
- 3.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–21. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 4.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–77. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 5.Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63:133–9. doi: 10.1016/0092-8674(90)90294-o. [DOI] [PubMed] [Google Scholar]
- 6.Quilliam LA, Khosravi-Far R, Huff SY, Der CJ. Guanine nucleotide exchange factors: activators of the Ras superfamily of proteins. Bioessays. 1995;17:395–404. doi: 10.1002/bies.950170507. [DOI] [PubMed] [Google Scholar]
- 7.Baass PC, Di Guglielmo GM, Authier F, Posner BI, Bergeron JJ. Compartmentalized signal transduction by receptor tyrosine kinases. Trends Cell Biol. 1995;5:465–70. doi: 10.1016/s0962-8924(00)89116-3. [DOI] [PubMed] [Google Scholar]
- 8.Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol. 2007;9:706–12. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- 9.Mor A, Campi G, Du G, Zheng Y, Foster DA, Dustin ML, Philips MR. The lymphocyte function-associated antigen-1 receptor costimulates plasma membrane Ras via phospholipase D2. Nat Cell Biol. 2007;9:713–9. doi: 10.1038/ncb1592. [DOI] [PubMed] [Google Scholar]
- 10.Hancock JF. PA promoted to manager. Nat Cell Biol. 2007;9:615–7. doi: 10.1038/ncb0607-615. [DOI] [PubMed] [Google Scholar]
- 11.Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119:393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-Sagi D, Marqusee S, Kuriyan J. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc Natl Acad Sci USA. 2006;103:16692–7. doi: 10.1073/pnas.0608127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boykevisch S, Zhao C, Sondermann H, Philippidou P, Halegoua S, Kuriyan J, Bar-Sagi D. Regulation of ras signaling dynamics by Sos-mediated positive feedback. Curr Biol. 2006;16:2173–9. doi: 10.1016/j.cub.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 14.Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 15.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 16.Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol. 1997;9:174–9. doi: 10.1016/s0955-0674(97)80060-9. [DOI] [PubMed] [Google Scholar]
- 17.Dhillon AS, Kolch W. Untying the regulation of the Raf-1 kinase. Arch Biochem Biophys. 2002;404:3–9. doi: 10.1016/s0003-9861(02)00244-8. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda M, Gotoh I, Adachi M, Gotoh Y, Nishida E. A novel regulatory mechanism in the mitogen-activated protein (MAP) kinase cascade. Role of nuclear export signal of MAP kinase kinase. J Biol Chem. 1997;272:32642–8. doi: 10.1074/jbc.272.51.32642. [DOI] [PubMed] [Google Scholar]
- 19.Fukuda M, Gotoh Y, Nishida E. Interaction of MAP kinase with MAP kinase kinase: its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J. 1997;16:1901–8. doi: 10.1093/emboj/16.8.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrell JE, Jr, Bhatt RR. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J Biol Chem. 1997;272:19008–16. doi: 10.1074/jbc.272.30.19008. [DOI] [PubMed] [Google Scholar]
- 21.Burack WR, Sturgill TW. The activating dual phosphorylation of MAPK by MEK is non-processive. Biochemistry. 1997;36:5929–33. doi: 10.1021/bi970535d. [DOI] [PubMed] [Google Scholar]
- 22.Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 23.Mackeigan JP, Murphy LO, Dimitri CA, Blenis J. Graded mitogen-activated protein kinase activity precedes switch-like c-Fos induction in mammalian cells. Mol Cell Biol. 2005;25:4676–82. doi: 10.1128/MCB.25.11.4676-4682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrell JE., Jr Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996;21:460–6. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]
- 25.Huang CY, Ferrell JE., Jr Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1996;93:10078–83. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrell JE, Jr, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280:895–8. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 27.Kholodenko BN. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur J Biochem. 2000;267:1583–8. doi: 10.1046/j.1432-1327.2000.01197.x. [DOI] [PubMed] [Google Scholar]
- 28.Sauro HM, Kholodenko BN. Quantitative analysis of signaling networks. Prog Biophys Mol Biol. 2004;86:5–43. doi: 10.1016/j.pbiomolbio.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol. 2006;7:165–76. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thron CD. A model for a bistable biochemical trigger of mitosis. Biophys Chem. 1996;57:239–51. doi: 10.1016/0301-4622(95)00075-5. [DOI] [PubMed] [Google Scholar]
- 31.Ferrell JE, Xiong W. Bistability in cell signaling: How to make continuous processes discontinuous, and reversible processes irreversible. Chaos. 2001;11:227–36. doi: 10.1063/1.1349894. [DOI] [PubMed] [Google Scholar]
- 32.Ferrell JE., Jr Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr Opin Cell Biol. 2002;14:140–8. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]
- 33.Ferrell JE., Jr How regulated protein translocation can produce switch-like responses. Trends Biochem Sci. 1998;23:461–5. doi: 10.1016/s0968-0004(98)01316-4. [DOI] [PubMed] [Google Scholar]
- 34.Markevich NI, Hoek JB, Kholodenko BN. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J Cell Biol. 2004;164:353–9. doi: 10.1083/jcb.200308060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashe HL, Briscoe J. The interpretation of morphogen gradients. Development. 2006;133:385–94. doi: 10.1242/dev.02238. [DOI] [PubMed] [Google Scholar]
- 36.Stathopoulos A, Levine M. Linear signaling in the Toll-Dorsal pathway of Drosophila: activated Pelle kinase specifies all threshold outputs of gene expression while the bHLH protein Twist specifies a subset. Development. 2002;129:3411–9. doi: 10.1242/dev.129.14.3411. [DOI] [PubMed] [Google Scholar]
- 37.Hazzalin CA, Mahadevan LC. MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol. 2002;3:30–40. doi: 10.1038/nrm715. [DOI] [PubMed] [Google Scholar]
- 38.Whitehurst A, Cobb MH, White MA. Stimulus-coupled spatial restriction of extracellular signal-regulated kinase 1/2 activity contributes to the specificity of signal-response pathways. Mol Cell Biol. 2004;24:10145–50. doi: 10.1128/MCB.24.23.10145-10150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos SD, Verveer PJ, Bastiaens PI. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol. 2007;9:324–30. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- 40.Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol. 2007;9:905–14. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 41.Hancock JF. Lipid rafts: contentious only from simplistic standpoints. Nat Rev Mol Cell Biol. 2006;7:456–62. doi: 10.1038/nrm1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicolau DV, Jr, Burrage K, Parton RG, Hancock JF. Identifying optimal lipid raft characteristics required to promote nanoscale protein-protein interactions on the plasma membrane. Mol Cell Biol. 2006;26:313–23. doi: 10.1128/MCB.26.1.313-323.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicolau DV, Jr, Hancock JF, Burrage K. Sources of anomalous diffusion on cell membranes: a Monte Carlo study. Biophys J. 2007;92:1975–87. doi: 10.1529/biophysj.105.076869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prior IA, Muncke C, Parton RG, Hancock JF. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–70. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hibino K, Watanabe TM, Kozuka J, Iwane AH, Okada T, Kataoka T, Yanagida T, Sako Y. Single-and multiple-molecule dynamics of the signaling from H-Ras to cRaf-1 visualized on the plasma membrane of living cells. Chemphyschem. 2003;4:748–53. doi: 10.1002/cphc.200300731. [DOI] [PubMed] [Google Scholar]
- 46.Murakoshi H, Iino R, Kobayashi T, Fujiwara T, Ohshima C, Yoshimura A, Kusumi A. Single-molecule imaging analysis of Ras activation in living cells. Proc Natl Acad Sci USA. 2004;101:7317–22. doi: 10.1073/pnas.0401354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plowman SJ, Muncke C, Parton RG, Hancock JF. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci USA. 2005;102:15500–5. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hancock JF, Parton RG. Ras plasma membrane signalling platforms. Biochem J. 2005;389:1–11. doi: 10.1042/BJ20050231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sieburth DS, Sun Q, Han M. SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell. 1998;94:119–30. doi: 10.1016/s0092-8674(00)81227-1. [DOI] [PubMed] [Google Scholar]
- 50.Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene. 2001;20:7486–93. doi: 10.1038/sj.onc.1204950. [DOI] [PubMed] [Google Scholar]
- 51.Elad-Sfadia G, Haklai R, Ballan E, Gabius HJ, Kloog Y. Galectin-1 augments Ras activation and diverts Ras signals to Raf-1 at the expense of phosphoinositide 3-kinase. J Biol Chem. 2002;277:37169–75. doi: 10.1074/jbc.M205698200. [DOI] [PubMed] [Google Scholar]
- 52.Abankwa D, Gorfe AA, Hancock JF. Ras nanoclusters: Molecular structure and assembly. Semin Cell Dev Biol. 2007 doi: 10.1016/j.semcdb.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harding A, Hsu V, Kornfeld K, Hancock JF. Identification of residues and domains of Raf important for function in vivo and in vitro. J Biol Chem. 2003;278:45519–27. doi: 10.1074/jbc.M303106200. [DOI] [PubMed] [Google Scholar]
- 54.Zhu J, Balan V, Bronisz A, Balan K, Sun H, Leicht DT, Luo Z, Qin J, Avruch J, Tzivion G. Identification of Raf-1 S471 as a novel phosphorylation site critical for Raf-1 and B-Raf kinase activities and for MEK binding. Mol Biol Cell. 2005;16:4733–44. doi: 10.1091/mbc.E05-02-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terai K, Matsuda M. Ras binding opens c-Raf to expose the docking site for mitogen-activated protein kinase kinase. EMBO Rep. 2005;6:251–5. doi: 10.1038/sj.embor.7400349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen A, Burack WR, Stock JL, Kortum R, Chaika OV, Afkarian M, Muller WJ, Murphy KM, Morrison DK, Lewis RE, McNeish J, Shaw AS. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol. 2002;22:3035–45. doi: 10.1128/MCB.22.9.3035-3045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ritt DA, Daar IO, Morrison DK. KSR Regulation of the Raf-MEK-ERK Cascade. Methods Enzymol. 2005;407:224–37. doi: 10.1016/S0076-6879(05)07019-9. [DOI] [PubMed] [Google Scholar]
- 58.Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol. 2003;13:1356–64. doi: 10.1016/s0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- 59.Harding A, Tian T, Westbury E, Frische E, Hancock JF. Subcellular localization determines MAP kinase signal output. Curr Biol. 2005;15:869–73. doi: 10.1016/j.cub.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 60.Chiariotti L, Salvatore P, Frunzio R, Bruni CB. Galectin genes: regulation of expression. Glycoconj J. 2004;19:441–9. doi: 10.1023/B:GLYC.0000014073.23096.3a. [DOI] [PubMed] [Google Scholar]
- 61.Nakahara S, Oka N, Raz A. On the role of galectin-3 in cancer apoptosis. Apoptosis. 2005;10:267–75. doi: 10.1007/s10495-005-0801-y. [DOI] [PubMed] [Google Scholar]
- 62.Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16:137R–57R. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- 63.Kitano H. Towards a theory of biological robustness. Mol Syst Biol. 2007;3:137. doi: 10.1038/msb4100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rao CV, Wolf DM, Arkin AP. Control, exploitation and tolerance of intracellular noise. Nature. 2002;420:231–7. doi: 10.1038/nature01258. [DOI] [PubMed] [Google Scholar]
- 65.Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–64. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 66.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, Rosen N. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 68.Yeung K, Janosch P, McFerran B, Rose DW, Mischak H, Sedivy JM, Kolch W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–85. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trakul N, Menard RE, Schade GR, Qian Z, Rosner MR. Raf kinase inhibitory protein regulates Raf-1 but not B-Raf kinase activation. J Biol Chem. 2005;280:24931–40. doi: 10.1074/jbc.M413929200. [DOI] [PubMed] [Google Scholar]
- 70.Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem. 2003;278:13061–8. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- 71.Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J. 1992;288:351–5. doi: 10.1042/bj2880351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–85. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 73.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–64. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 74.Matheny SA, White MA. Ras-Sensitive IMP Modulation of the Raf/MEK/ERK Cascade Through KSR1. Methods Enzymol. 2005;407:237–47. doi: 10.1016/S0076-6879(05)07020-5. [DOI] [PubMed] [Google Scholar]
- 75.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–24. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 76.Li C, Scott DA, Hatch E, Tian X, Mansour SL. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development. 2007;134:167–76. doi: 10.1242/dev.02701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bagowski CP, Besser J, Frey CR, Ferrell JE., Jr The JNK cascade as a biochemical switch in mammalian cells: ultrasensitive and all-or-none responses. Curr Biol. 2003;13:315–20. doi: 10.1016/s0960-9822(03)00083-6. [DOI] [PubMed] [Google Scholar]
- 78.Pomerening JR, Sontag ED, Ferrell JE., Jr Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat Cell Biol. 2003;5:346–51. doi: 10.1038/ncb954. [DOI] [PubMed] [Google Scholar]
- 79.Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005;3:e356. doi: 10.1371/journal.pbio.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chong H, Lee J, Guan KL. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001;20:3716–27. doi: 10.1093/emboj/20.14.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fabian JR, Vojtek AB, Cooper JA, Morrison DK. A single amino acid change in Raf-1 inhibits Ras binding and alters Raf-1 function. Proc Natl Acad Sci USA. 1994;91:5982–6. doi: 10.1073/pnas.91.13.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roy S, Lane A, Yan J, McPherson R, Hancock JF. Activity of plasma membrane-recruited Raf-1 is regulated by Ras via the Raf zinc finger. J Biol Chem. 1997;272:20139–45. doi: 10.1074/jbc.272.32.20139. [DOI] [PubMed] [Google Scholar]
- 83.Ghosh S, Moore S, Bell RM, Dush M. Functional analysis of a phosphatidic acid binding domain in human Raf-1 kinase: mutations in the phosphatidate binding domain lead to tail and trunk abnormalities in developing zebrafish embryos. J Biol Chem. 2003;278:45690–6. doi: 10.1074/jbc.M302933200. [DOI] [PubMed] [Google Scholar]
- 84.Hancock JF, Cadwallader K, Paterson H, Marshall CJ. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991;10:4033–9. doi: 10.1002/j.1460-2075.1991.tb04979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]