

Abstract
The conformations of C8-dG adducts of 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) positioned in the C-X1-G, G-X2-C, and C-X3-C contexts in the C-G1-G2-C-G3-C-C recognition sequence of the NarI restriction enzyme were compared, using the oligodeoxynucleotides 5′-d(CTCXGCGCCATC)-3′·5′-d(GATGGCGCCGAG)-3′, 5′-d(CTCGXCGCCATC)-3′·5′-d(GATGGCGCCGAG)-3′, and 5′-d(CTCGGCXCCATC)-3′·5′-d(GATGGCGCCGAG)-3′ (X is the C8-dG adduct of IQ). These were the NarIIQ1, NarIIQ2, and NarIIQ3 duplexes, respectively. In each instance, the glycosyl torsion angle χ for the IQ-modified dG was in the syn conformation. The orientations of the IQ moieties were dependent upon the conformations of torsion angles α′ [N9–C8–N(IQ)–C2(IQ)] and β′ [C8–N(IQ)–C2(IQ)–N3(IQ)], which were monitored by the patterns of 1H NOEs between the IQ moieties and the DNA in the three sequence contexts. The conformational states of IQ torsion angles α′ and β′ were predicted from the refined structures of the three adducts obtained from restrained molecular dynamics calculations, utilizing simulated annealing protocols. For the NarIIQ1 and NarIIQ2 duplexes, the α′ torsion angles were predicted to be −176 ± 8° and −160 ± 8°, respectively, whereas for the NarIIQ3 duplex, torsion angle α′ was predicted to be 159 ± 7°. Likewise, for the NarIIQ1 and NarIIQ2 duplexes, the β′ torsion angles were predicted to be −152 ± 8° and −164 ± 7°, respectively, whereas for the NarIIQ3 duplex, torsion angle β′ was predicted to be −23 ± 8°. Consequently, the conformations of the IQ adduct in the NarIIQ1 and NarIIQ2 duplexes were similar, with the IQ methyl protons and IQ H4 and H5 protons facing outward in the minor groove, whereas in the NarIIQ3 duplex, the IQ methyl protons and the IQ H4 and H5 protons faced into the DNA duplex, facilitating the base-displaced intercalated orientation of the IQ moiety [Wang, F., Elmquist, C. E., Stover, J. S., Rizzo, C. J., and Stone, M. P. (2006) J. Am. Chem. Soc. 128, 10085−10095]. In contrast, for the NarIIQ1 and NarIIQ2 duplexes, the IQ moiety remained in the minor groove. These sequence-dependent differences suggest that base-displaced intercalation of the IQ adduct is favored when both the 5′- and 3′-flanking nucleotides in the complementary strand are guanines. These conformational differences may correlate with sequence-dependent differences in translesion replication.
The browning of protein-rich foods leads to the formation of heterocyclic amines (HCAs)1 such as 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) (1–4). Various HCAs, including IQ, have been identified in grilled foods at parts per billion levels (5, 6). Human intakes of HCAs, estimated to be ∼60 ng/day (7), are modest; however, exposures to these compounds, which have been isolated from human urine (8), are of concern. Human exposure to HCAs is associated with pancreatic (9), colon (10), prostate (11), and breast cancer (12, 13). Tumors in organs of rodents and in the livers of monkeys are induced by IQ (14–17). In mice, exposures lead to liver, forestomach, and lung tumors (18). In rats, exposures lead to cancers in the liver, intestine, zymbal gland, clitoral gland, skin (19), mammary glands, liver, and ear ducts (20). TD50 values in rats are 0.7 mg kg−1 day−1 and in mice are 14.7 mg kg−1 day−1 (21). In bacterial reversion assays (22–25), HCAs are active in point and frameshift tester strains (26).
IQ is one of the strongest chemical mutagens (27). It is less prevalent than 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) (28) but is 200-fold more mutagenic than the latter in Salmonella reversion assays (3). IQ is 1 order of magnitude more mutagenic than aflatoxin B1. In bacteria, mutations occur primarily at G·C base pairs (29, 30). It exhibits frameshift mutations in CG repeats. Similar levels of mutations are seen in mammalian hprt (31) and ef-2 (32) gene assays. In mammalian cells, point mutations are observed (33–36). Sister chromatid exchanges are observed in rodent cells (37–39).
IQ is activated primarily by the enzyme CYP P450 1A2 to an N-hydroxyl oxidation product (40–43). Extrahepatic CYP P450s oxidize HCAs with lower efficiencies (44). The N-hydroxyl oxidation product is acetylated by cellular N-acetyltransferases (NAT), particularly NAT2 (45–47). The resulting nitrenium ion is the ultimate reactive electrophile (36, 44). The NAT2 fast acetylator polymorphism is associated with an increased risk of colorectal cancer in humans (48, 49).
The C8-dG adducts of HCAs are observed in both rodents and primates, as measured by 32P postlabeling (35). The major adduct formed by IQ occurs by substitution at C8-dG (Chart 1); a minor N2-dG adduct is also formed (50). The structures of these adducts are established (51–53). The formation of the C8-dG adduct probably involves initial alkylation at N7-dG, followed by rearrangement (54). High-sensitivity liquid chromatography and electrospray ionization mass spectrometry (LC–ESI-MS) (55) have measured several adducts per 107 nucleotides in animal tissues (11, 56). The levels of C8 and N2-dG IQ adducts measured in tissues of rats and primates using mass spectrometry (57, 58) are in agreement with data obtained by 32P postlabeling methods.
Chart 1.

Metabolic Activation of IQ
The NarI sequence contains the 5′-d(CG1G2CG3CC)-3′ recognition site of the NarI restriction enzyme, in which the third guanine represents a hot spot for −2 bp frameshifts (Chart 2) (59–62). The NarI sequence contains the 5′-CG1G-3′, 5′-GG2C-3′, and 5′-CG3C-3′ sequence steps and offers a unique opportunity to study the effect of nearest-neighbor and next-nearest-neighbor sequences on the conformation of the C8-dG IQ adduct positioned opposite dC. A structural study of this adduct was conducted in 5′-d(C1T2C3G4G5C6-X7C8C9A10T11C12)-3′·5′-d(G13A14T15G16G17C18G19C20C21G22-A23G24)-3′, where X is 8-[(3-methyl-3H-imidazo[4,5-f]quinolin-2-yl)amino]-2′-deoxyguanosine, named the NarIIQ3 sequence. The results revealed a base-displaced intercalated structure (63). The adducted dG adopted the syn conformation about the glycosyl bond and was extruded into the major groove; the IQ moiety intercalated into the DNA, and the complementary dC was extruded from the helix.
Chart 2.
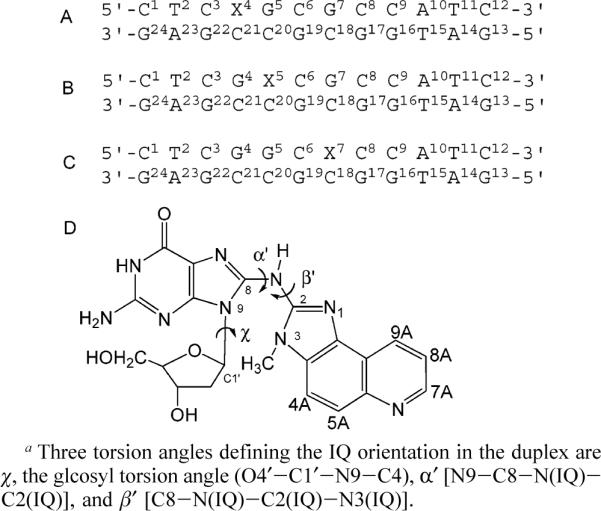
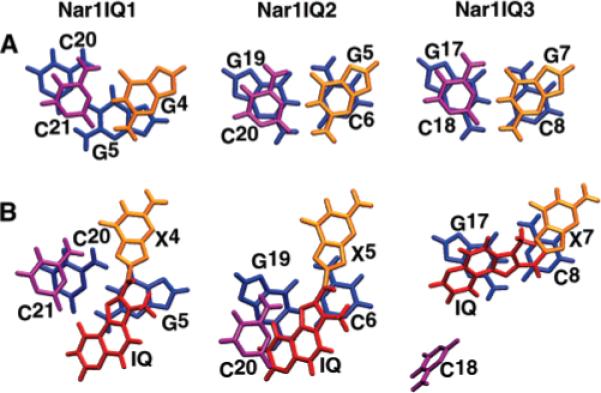
(A) C8-dG IQ Adducted NarIIQ1 Dodecamer, (B) C8-dG IQ Adducted NarIIQ2 Dodecamer, (C) C8-dG IQ Adducted NarIIQ3 Dodecamer, and (D) C8-dG IQ Adducta
A combination of ultraviolet spectroscopy and circular dichroism studies indicated that the conformation of the C8-dG IQ adduct positioned opposite dC differed in the 5′-GGCAX1G2TGGTG-3′ sequence found in codon 12 of the ras protooncogene, as compared to the NarIIQ3 sequence (64). It was concluded that the C8-dG adduct existed in a groove-bound conformation in the ras protoooncogene sequence (64). Subsequently, the C8-dG adduct was incorporated into the G1 and G2 positions of the NarI sequence (65). Analysis of the UV, CD, and NMR chemical shift data for the IQ protons was consistent with the C8-dG IQ adduct adopting a minor groove-bound conformation at the G1 and G2 positions of the NarI sequence, in contrast to the base-displaced intercalated conformation observed at the G3 position of the NarI sequence (65).
This NMR study compares the solution structures of C8-dG IQ adducts paired opposite dC in the duplexes 5′-d(CTCX4GCGCCATC)-3′·5′-d(GATGGCGCCGAG)-3′ and 5′-d(CTCGX5CGCCATC)-3′·5′-d(GATGGCGCCGAG)-3′ with the previously determined solution structure of the C8-dG IQ adduct in the duplex 5′-d(CTCGGCX7CCATC)-3′· 5′-d(GATGGCGCCGAG)-3′ (63), where X is the C8-dG adduct of IQ, where the X4 adduct is positioned at G1 of the NarI sequence (the NarIIQ1 duplex), the X5 adduct is positioned at G2 of the NarI sequence (the NarIIQ2 duplex), and the previously examined X7 adduct is positioned at G3 of the NarI sequence [the NarIIQ3 duplex (63)] (Chart 1). The NMR data reveal that the solution conformations of the C8-dG IQ adducts in the NarIIQ1 and NarIIQ2 sequences are similar, with the IQ moiety being oriented in the minor groove, in contrast to the base-displaced intercalated conformation of the C8-dG IQ adduct in the NarIIQ3 sequence (63), confirming the predictions based upon UV and CD spectroscopy (65). The minor groove versus base-displaced intercalation orientations of the IQ moieties in the NarIIQ1, NarIIQ2, and NarIIQ3 sequences are modulated by the torsion angles α′ [N9–C8–N(IQ)–C2(IQ)] and β′ [C8–N(IQ)–C2(IQ)–N3(IQ)]. For the NarIIQ1 and NarIIQ2 duplexes, the α′ torsion angles are predicted to be −176 ± 8° and −160 ± 8°, respectively, whereas for the NarIIQ3 duplex, torsion angle α′ is predicted to be 159 ± 7° (63). Likewise, for the NarIIQ1 and NarIIQ2 duplexes, the β′ torsion angles are predicted to be −152 ± 8° and −164 ± 7°, respectively, whereas for the NarIIQ3 duplex, torsion angle β′ is predicted to be −23 ± 8° (63). Consequently, in the NarIIQ1 and NarIIQ2 duplexes, the IQ methyl protons and IQ H4 and H5 protons face outward in the minor groove, whereas in the NarIIQ3 duplex, the IQ methyl protons and the IQ H4 and H5 protons face into the DNA duplex, facilitating the base-displaced intercalated orientation of the IQ moiety (63).
MATERIALS AND METHODS
Sample Preparation
The unmodified oligodeoxynucleotide 5′-d(GATGGCGCCGAG)-3′ was obtained from the Midland Certified Reagent Co. and was purified by anion exchange chromatography. The IQ-adducted oligodeoxy-nucleotides 5′-d(CTCXGCGCCATC)-3′ and 5′-d(CTCGXCGCCATC)-3′ were synthesized and purified as described previously (64). All oligodeoxynucleotides were characterized by MALDI-TOF mass spectrometry and enzymatic digestion, and their purities were also assessed by capillary zone electrophoresis (CZE). The NarIIQ1 and NarIIQ2 oligodeoxynucleotides were greater than 92% pure.
Oligodeoxynucleotide duplexes were annealed at 70 °C. The stoichiometry was established by monitoring the 1H NMR spectrum. The duplexes were dissolved in 0.250 mL of buffer solution containing 0.1 M NaCl, 10 mM NaH2-PO4, and 50 μM Na2EDTA (pH 7.0). The oligodeoxynucleotide concentrations were ∼0.7 mM using an extinction coefficient of 1.10 × 105 M−1 cm−1 at 260 nm (66).
NMR
1H NMR spectra were obtained at 500.13, 600.20, and 800.23 MHz. COSY spectra were collected at 15, 20, 25, 30, and 35 °C in 99.996% D2O. 1H NOESY experiments in D2O were conducted at 15 °C. To derive distance restraints, spectra were recorded consecutively at mixing times of 150, 200, and 250 ms, respectively, at the 1H NMR frequency of 800.23 MHz. The data were recorded with 1024 real data points in the t1 dimension and 2048 real points in the t2 dimension. The relaxation delay was 2 s. The data in the t1 dimension were zero-filled to give a matrix of 2K × 2K real points. NOESY spectra for the exchangeable protons were recorded at 5 °C, in a 90:10 H2O/D2O mixture, using a field gradient Watergate pulse sequence (67) for water suppression and a 250 ms mixing time at a 1H NMR frequency of 600.20 MHz. Chemical shifts of proton resonances were referenced to water. Double-quantum-filtered 1H correlation (DQF-COSY) (68, 69) and exclusive COSY (E-COSY) (70) spectra were collected at 25 °C and 500.13 MHz and zero-filled to give a matrix of 1024 × 2048 real points. A skewed sine-bell square apodization function with a 90° phase shift and a skew factor of 1.0 was used in both dimensions. 1H–31P HMBC spectra (71) were recorded at 30 °C. The data matrix was 256 (t1) × 2048 (t2) complex points. The data were Fourier-transformed after zero filling in the t1 dimension, resulting in a matrix size of 512 (D1) × 2048 (D2) real points. Trimethyl phosphate was used as an external standard. NMR data were processed using FELIX2000 (Accelrys, Inc., San Diego, CA) on Silicon Graphics (Mountain View, CA) Octane workstations.
Experimental Restraints
(a) Distance Restraints
Footprints were drawn around cross-peaks for the NOESY spectrum measured with a mixing time of 250 ms to define the size and shape of individual cross-peaks, using FELIX2000. Identical footprints were transferred and fit to the cross-peaks obtained at the other two mixing times. Cross-peak intensities were determined by volume integration of the areas under the footprints. The intensities were combined with intensities generated from complete relaxation matrix analysis of a starting DNA structure to generate a hybrid intensity matrix (72). MARDIGRAS (version 5.2) (73, 74) was used to refine the hybrid matrix by iteration to optimize the agreement between the calculated and experimental NOE intensities. The molecular motion was assumed to be isotropic. The noise level was set at half the intensity of the weakest cross-peak. Calculations were performed using the DNA starting models generated using INSIGHT II (Accelrys) and NOE intensities derived from experiments at three mixing times, and with three τc values (2, 3, and 4 ns), yielding 18 sets of distances. Analysis of these data yielded the experimental distance restraints and standard deviations for the distance restraints used in subsequent restrained molecular dynamics calculations. For partially overlapped cross-peaks, the lower and upper bounds on the distances were increased. The distance restraints were divided into four classes, reflecting the confidence level in the experimental data.
(b) Torsion Angle Restraints
Pseudorotational angles (P) of the deoxyribose rings were estimated graphically by monitoring the 3JHH couplings of sugar protons (75). The JH1′–H2′ and JH1′–H2″ couplings were measured from the E-COSY experiment (70), while the intensities of JH2″–H3′ and JH3′–H4′ couplings were determined from the DQF-COSY experiment. The data were fit to curves relating the coupling constants to the deoxyribose pseudorotation (P), sugar pucker amplitude (ϕ), and the percentage S-type conformation. The pseudorotation and amplitude ranges were converted to the five dihedral angles, ν0–ν4.
Restrained Molecular Dynamics
Restrained molecular dynamics calculations using a simulated annealing protocol were performed in vacuo using X-PLOR (76). The force field was derived from CHARMM (77) and adapted for restrained molecular dynamics (rMD) calculations of nucleic acids. The empirical energy function consisted of terms for bonds, bond angles, torsion angles, tetrahedral and planar geometry, hydrogen bonding, and nonbonded interactions, including van der Waals and electrostatic forces. It treated hydrogens explicitly. The van der Waals energy term used the Lennard-Jones potential energy function. The electrostatic term used the Coulomb function, based on a full set of partial charges (−1 per residue) and a distance-dependent dielectric constant of 4r. The nonbonded pair list was updated if any atom moved more than 0.5 Å, and the cutoff radius for nonbonded interactions was 11 Å. The effective energy function included terms describing distance and dihedral restraints, in the form of square-well potentials. Sets of rMD calculations for NarIIQ1 and NarIIQ2 and different starting structures of NarIIQ1 and NarIIQ2 with IQ located in the minor groove (syn), major groove (anti), and intercalated position (syn) were considered. These were generated using INSIGHT II through modification at G4 C8 or G5 C8, followed by energy minimization using X-PLOR. Partial charges and atom types for IQ used for X-PLOR calculations were those obtained by Wu et al. (78). Restrained molecular dynamics calculations used the same protocol that we used in the previous study (63). Final structures were analyzed using X-PLOR to measure the root-mean-square deviation (rmsd) between an averaged structure and the converged structures. Back-calculation of theoretical NMR intensities from the emergent structures was performed using CORMA (version 5.2) (72). Helicoidal parameters were examined using 3DNA (79).
RESULTS
The C8-dG IQ adduct was site-specifically incorporated into the NarIIQ1 oligodeoxynucleotide 5′-d(C1T2C3X4G5C6-G7C8C9A10T11C12)-3′·5′-d(G13A14T15G16G17C18G19C20C21G22-A23G24)-3′ and NarIIQ2 oligodeoxynucleotide 5′-d(C1T2C3-G4X5C6G7C8C9A10T11C12)-3′·5′-d(G13A14T15G16G17C18G19C20-C21G22A23G24)-3′ containing the NarI restriction sequence (G1G2CG3CC) as previously described (64). The previously studied NarIIQ3 [oligodeoxynucleotide 5′-d(C1T2C3G4G5C6-X7C8C9A10T11C12)-3′·5′-d(G13A14T15G16G17C18G19C20C21G22-A23G24)-3′] sequence (63) is considered a “hot spot” for arylamine modification and is prone to −2 frameshift mutations (80). Both NarIIQ1 and NarIIQ2 duplexes yielded excellent NMR spectra at temperatures between 5 and 30 °C.
NMR Spectroscopy
(a) DNA Nonexchangeable Protons of the Nar IIQ1 Duplex
The NMR spectrum was well-resolved and yielded resonances with narrow line widths, indicative of a stable and ordered conformation. Panels A and B of Figure 1 detail the sequential NOE connectivity for the NarIIQ1 duplex (81, 82). The sequential NOE connectivity was interrupted. The absence of an imidazole proton in the C8-dG adduct precluded observation of the C3 H1′ → X4 H8 and X4 H8 → G5 H1′ sequential NOEs, as well as the intranucleotide X4 H8 → X4 H1′ NOE. The X4 H1′ → G5 H8 NOE was of normal intensity (see the START peak in Figure 1A). In the complementary strand, the C20 H1′ → C21 H6 NOE was missing. The C21 H1′ → G22 H8 sequential NOE was weak. C21 is the nucleotide opposite to X4 in the complementary strand of the duplex. The deoxyribose sugar proton resonances were assigned from DQF-COSY spectra (Figure S1 of the Supporting Information). A complete 1H assignment was achieved, with the exception of several of the H5′ and H5″ protons. The X4 H2′ resonance shifted downfield to 3.59 ppm. The 1H NMR assignments are listed in Table S1 of the Supporting Information.
Figure 1.
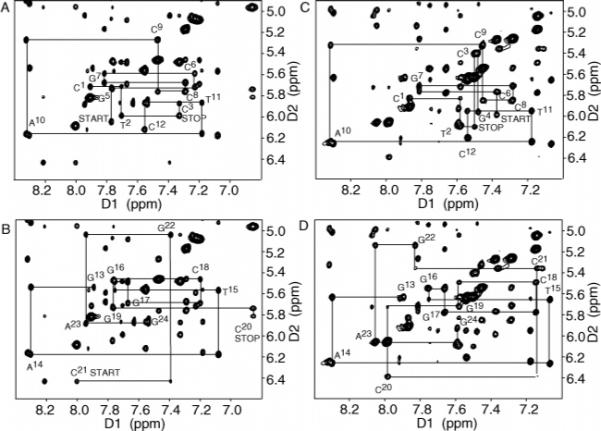
Expanded plots from the aromatic proton–anomeric proton region of the 800.13 MHz NOESY spectrum for the modified NarIIQ1 and NarIIQ2 duplexes at 15 °C using a mixing time of 250 ms, showing sequential NOE connectivity. (A) Nucleotides C1–C12 of the NarIIQ1 duplex. (B) Nucleotides G13–G24 of the NarIIQ1 duplex. (C) Nucleotides C1–C12 of the modified strand in the NarIIQ2 duplex. (D) Nucleotides G13–G24 of the complementary strand in the NarIIQ2 duplex.
(b) DNA Nonexchangeable Protons of the Nar IIQ2 Duplex
The NMR spectrum was well-resolved and yielded resonances with narrow line widths, indicative of a stable and ordered conformation (Figure 1C,D). A similar pattern of sequential NOEs was observed. The absence of a purine imidazole proton in the C8-dG adduct precluded observation of the G4 H1′ → X5 H8 and X5 H8 → C6 H1′ sequential NOEs, as well as the intranucleotide X5 H8 → X5 H1′ NOE. The X5 H1′ → C6 H6 NOE was of normal intensity (see the START peak in Figure 1C). In the complementary strand, the G19 H1′ → C20 H6 and C20 H1′ → C21 H6 sequential NOEs were weak. C20 is the nucleotide opposite to X5 in the complementary strand of the duplex. The deoxyribose sugar proton resonances were assigned from DQF-COSY spectra (Figure S1 of the Supporting Information). A complete 1H assignment was achieved, with the exception of several of the H5′ and H5″ protons. The X5 H2′ resonance shifted downfield to 3.57 ppm. The 1H NMR assignments are listed in Table S1 of the Supporting Information.
(c) DNA Exchangeable Protons of the NarI IQ1 Duplex
The guanine imino protons were assigned from sequential NOEs between adjacent base pairs and NOEs to their corresponding base-paired amino protons (83). The imino proton resonances arising from each of the three A·T base pairs, and from six of the nine G·C base pairs, were identified (Figure 2A). The resonance arising from X4 N1H was not observed, presumably due to fast exchange with water. Consequently, the sequential NOEs between Watson–Crick hydrogen-bonded amino and imino protons were interrupted between the C3·G22 and X4·C21 base pairs, and the X4·C21 and G5·C20 base pairs. The imino resonances from the terminal C1·G24 and C12·G13 base pairs were also not observed, due to rapid exchange with water. Four imino proton resonances, from the C6·G19,G7·C18,C8·G17, and C9·G16 base pairs, were located at approximately 13 ppm. Two resonances, arising from the C3·G22 and G5·C20 base pairs, were further upfield and observed at 12.6 and 11.6 ppm, respectively. At the 5′ neighbor C3·G22 base pair, G22 N1H exhibited NOEs to the C3 NH2 protons and to C3 H5. At the 3′ neighbor G5·C20 base pair, G5 N1H exhibited NOEs to the C20 NH2 protons and to C20 H5. The anticipated T2 N3H → A23 H2 and T11 N3H → A10 H2 NOEs were detected.
Figure 2.
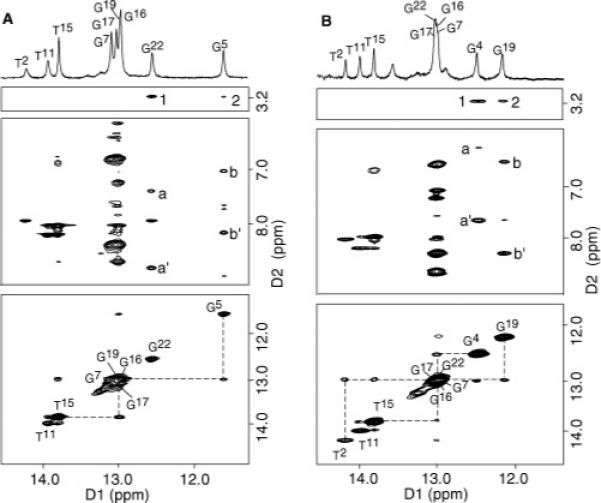
Comparison of expanded plots of the imino proton region of the 1H NOESY spectra for (A) the NarIIQ1 and (B) NarIIQ2 duplexes. In the bottom panels are expanded plots showing sequential NOE connectivity for the imino protons of base pairs T2·A23 to T11·A14 at 15 °C. The labels represent the imino proton of the designated base. In the middle panels are NOE connectivities between the imino protons and the base amino protons. The NOE cross-peaks involving the imino protons are labeled in the figure as follows: (A) (a′ and a) G22 N1H → C3 NH2-4b,e and (b′ and b) G5 N1H → C20 NH2-4b,e and (B) (a′ and a) G4 N1H → C21 NH2-4b,e and (b′ and b) G19 N1H → C6 NH2-4b,e. In the top panels are NOE connectivities between the imino protons and the IQ methyl protons. The IQ-DNA cross-peaks are labeled as follows: (A) 1, G22 N1H → X4 CH3; and 2, G5 N1H → X4 CH3; and (B) 1, G4 N1H → X5 CH3; and 2, G19 N1H → X5 CH3.
(d) DNA Exchangeable Protons of the NarIIQ2 Duplex
The guanine imino protons were assigned from sequential NOEs between adjacent base pairs and NOEs to their corresponding base-paired amino protons (83). The imino proton resonances arising from each of the three A·T base pairs, and from six of the nine G·C base pairs, were identified (Figure 2B). The resonance arising from X5 N1H was not observed, presumably due to fast exchange with water. Consequently, interruptions in the sequential NOEs between Watson-Crick hydrogen-bonded amino and imino protons occurred between the G4·C21 and X5·C20 base pairs, and the X5·C20 and C6·G19 base pairs. Two additional broad resonances were not assigned but presumably arose from the terminal C1·G24 and C12·G13 base pairs. Four imino proton resonances, from the C3·G22,G7·C18,C8·C17, andC9·G16 base pairs, were located at approximately 13 ppm. Two resonances, arising from the G4·C21 and C6·G19 base pairs, were further upfield and observed at 12.5 and 12.2 ppm, respectively. At the 5′-adjacent G4·C21 base pair, G4 N1H exhibited NOEs to the C21 NH2 protons and to C21 H5. At the 3′-adjacent C6·G19 base pair, G19 N1H exhibited NOEs to the C6 NH protons and to C6 H5. The anticipated T2 N3H → A23 H2 and T11 N3H → A10 H2 NOEs were detected.
(e) IQ Protons in the NarIIQ1 Duplex
The resonance assignments of the IQ protons were achieved using a combination of magnitude COSY and NOESY spectra (Figure 3A,B). The IQ H4A proton was assigned to 7.22 ppm on the basis of a cross-peak to the IQ methyl protons in the NOESY spectrum (Figure 3B). The IQ H5A proton resonance was assigned to 7.14 ppm on the basis of its scalar coupling to H4A. The IQ H7A, H8A, and H9A proton resonances were located at 8.60, 7.10, and 8.12 ppm, respectively. The H7A resonance exhibited broadening attributed to the adjacent pyridinyl nitrogen. It showed a strong NOE to the H8A proton and a weaker NOE to the H9A proton. The small scalar coupling between the IQ H7A and H8A protons was not observed at temperatures below 25 °C, presumably due to spectral line broadening at the lower temperatures. The COSY cross-peaks between the IQ protons also broadened above 35 °C, presumably due to thermal melting of the duplex as the temperature was increased.
Figure 3.
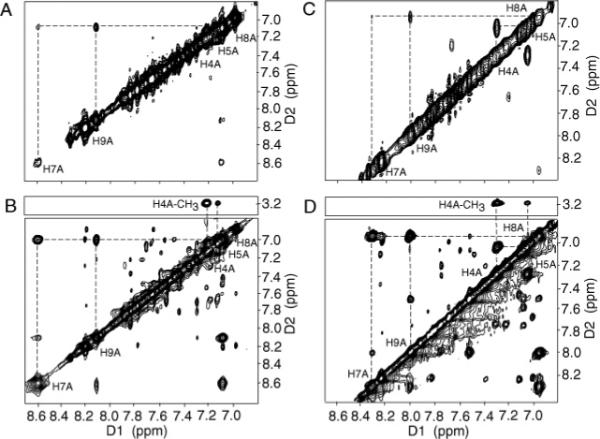
Assignments of the IQ proton resonances. Expanded plots from (A) the COSY spectrum and (B) the aromatic–aromatic region of the NOESY spectrum at 15 °C for the IQ-adducted NarIIQ1 duplex. Expanded plots from (C) the COSY spectrum and (D) the aromatic–aromatic region of the NOESY spectrum at 15 °C for the IQ-adducted NarIIQ2 duplex.
(f) IQ Protons in the NarIIQ2 Duplex
The resonance assignments of the IQ protons were achieved using a combination of magnitude COSY and NOESY spectra (Figure 3C,D). The IQ H4A proton was assigned to 7.30 ppm on the basis of a cross-peak to the IQ methyl protons in the NOESY spectrum. The IQ H5A proton resonance was assigned to 7.05 ppm on the basis of its scalar coupling to H4A. The IQ H7A, H8A, and H9A proton resonances were located at 8.32, 6.96, and 8.00 ppm, respectively. The H7A resonance exhibited broadening attributed to the adjacent pyridinyl nitrogen. It showed a strong NOE to the H8A proton and a weaker NOE to the H9A proton. The small scalar coupling between the IQ H8A and H7A protons was not observed at temperatures below 25 °C, presumably due to spectral line broadening at the lower temperatures. The COSY cross-peaks between the IQ protons also broadened above 35 °C, presumably due to thermal melting of the duplex as the temperature was increased.
(g) IQ-DNA NOEs in the NarIIQ1 Duplex
There were 16 NOEs observed between the IQ moiety and DNA protons. These involved nucleotides X4 and G5 in the modified strand and nucleotides C20, C21, and G22 in the complementary strand. The IQ methyl protons exhibited NOEs only to the modified strand. Strong NOEs were observed to X4 H1′, G5 H1′, and G5 H5′ of the modified strand (Figure 4A) and to G22 N1H, the Watson–Crick hydrogen-bonded imino proton at the 5′ neighbor base pair (Figure 4A). A weak NOE was observed to G5 N1H, the Watson–Crick hydrogen-bonded imino proton at the 3′ neighbor base pair (Figure 4A). The IQ H4A and H5A protons exhibited weak NOEs to G22 H1′ of the complementary strand, located in the 5′-direction in the minor groove (Figure 4A). The H7A and H8A protons did not exhibit NOEs to the DNA. The IQ H9A proton exhibited a strong NOE to C20 H2′, and medium-strength NOEs to C20 H2″ and C21 H1′. Weak NOEs were observed between the IQ H9A proton and C20 H1′, H3′, and H6, C21 H4′, and G22 H8 and H5′ located in the complementary strand (Figure 4A). The IQ-DNA distances estimated from the volume integrals of the NOE cross-peaks in the NarIIQ1-modified dodecamer are listed in Table S3 of the Supporting Information.
Figure 4.
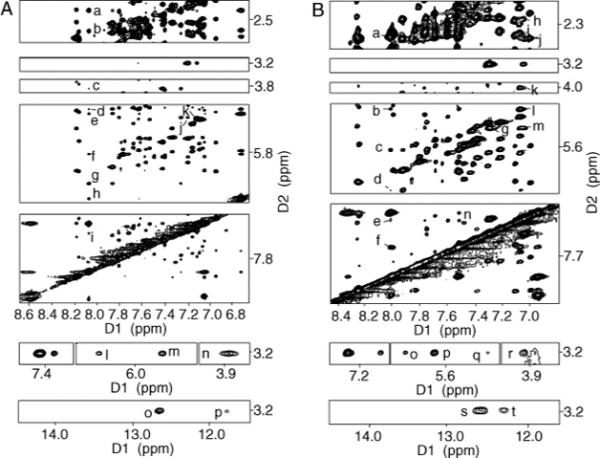
Tile plots showing NOE cross-peaks between nonexchangeable protons of DNA and IQ protons in the NarIIQ1 and NarIIQ2 duplexes. (A) NOE cross-peaks for the NarIIQ1 duplex: (a) C20 H2′ → IQ H9A, (b) C20 H2″ → IQ H9A, (c) G22 H5′ → IQ H9A, (d) C20 H3′ → IQ H9A, (e) C21 H4′ → IQ H9A, (f) C21 H1′ → IQ H9A, (g) C20 H1′ → IQ H9A, (h) C20 H8 → IQ H9A, (i) G17 H8 → IQ H9A, (j) G22 H1′ → IQ H4A, (k) G22 H1′ → IQ H5A, (l) X4 H1′ → IQ CH3, (m) G5 H1′ → IQ CH , (n) G5 H5′ → IQ CH3, (o) G22 N1H → IQ CH3, and (p) G5 N1H → IQ CH3. (B) NOE cross-peaks for the NarIIQ2 duplex: (a) G19 H2″ → IQ H9A, (b) G19 H3′ → IQ H9A, (c) G19 H1′ → IQ H9A, (d) C20 H1′ → IQ H9A, (e) C21 H6 → IQ H9A, (f) G19 H8 → IQ H9A, (g) C21 H1′ → IQ H4A, (h) C20 H2′ → IQ H5A, (i) C20 H2″ → IQ H5A, (j) G19 H2″ → IQ H8A, (k) C21 H4′ → IQ H5A, (l) C21 H5 → IQ H5A, (m) C21 H1′ → IQ H5A, (n) G19 H8 → IQ H8A, (o) X5 H1′ → IQ CH3, (p) C6 H1′ → IQ CH3, (q) C21 H1′ → IQ CH3, (r) C6 H4′ → IQ CH3, (s) G4 N1H → IQ CH3, and (t) G19 N1H → IQ CH3.
(h) IQ–DNA NOEs in the NarIIQ2 Duplex
There were 20 NOEs observed between the IQ moiety and DNA protons. These involved nucleotides G4, X5, and C6 in the modified strand and nucleotides G19, C20, and C21 in the complementary strand. The IQ methyl protons exhibited strong NOEs to the modified strand. These were observed between the IQ methyl protons and C6 H1′, C6 H4′, and G4 N1H, the Watson-Crick hydrogen-bonded imino proton of the 5′ neighbor base pair. A medium-intensity NOE was observed to X5 H1′. Weak NOEs were observed between the IQ methyl protons and G19 N1H, the Watson-Crick hydrogen-bonded imino proton of the 3′ neighbor base pair, and C21 H1′, in the complementary strand (Figure 4B). The IQ H4A proton exhibited a strong NOE to C21 H1′ in the complementary strand (Figure 4B). The IQ H5A proton exhibited medium-strength NOEs to C20 H2′ and H2″, C21 H1′, and C21 H4′, in the complementary strand. A weak NOE was observed to C21 H5 in the complementary strand. No NOEs were observed between the IQ H7A proton and the DNA. The IQ H8A proton exhibited a medium-intensity NOE to G19 H8 and a weak NOE to G19 H2″, located in the complementary strand. A weak NOE between IQ H8A and G19 H1′ could be observed only in the spectrum collected with a mixing time of 250 ms. The IQ H9A proton exhibited medium-intensity NOEs to G19 H1′, H2″, and H8 in the complementary strand and weak NOEs to G19 H3′, C20 H1′, and C21 H6, located in the complementary strand. The IQ-DNA distances estimated from the volume integrals of the NOE cross-peaks in the NarIIQ2-modified dodecamer are listed in Table S3 of the Supporting Information.
(i) Torsion Angle Analysis in the NarIIQ1 Duplex
The intensity of the X4 H8 → X4 H1′ NOE normally would have been utilized to assess the conformation of the glycosyl torsion angle χ. The absence of this proton in the adducted oligodeoxynucleotide necessitated the evaluation of chemical shift data at the deoxyribose H2′ and H2″ protons (84, 85). The X4 H2′ resonance shifted downfield to 3.59 ppm (Figure 5A; expanded DQF-COSY plots identifying scalar couplings between deoxyribose H1′ and H2′ and H2″ protons in the NarIIQ1 duplex are shown in Figure S1 of the Supporting Information). This was characteristic of the syn dG orientation at the modified position (84, 85). In the corresponding unmodified duplex, the G4 H8 → G4 H1′ NOE was of normal intensity and all scalar cross-peaks between deoxyribose H1′ and H2′ and H2″ protons were located in the anticipated chemical shift range of 1.6−3.0 ppm (63).
Figure 5.
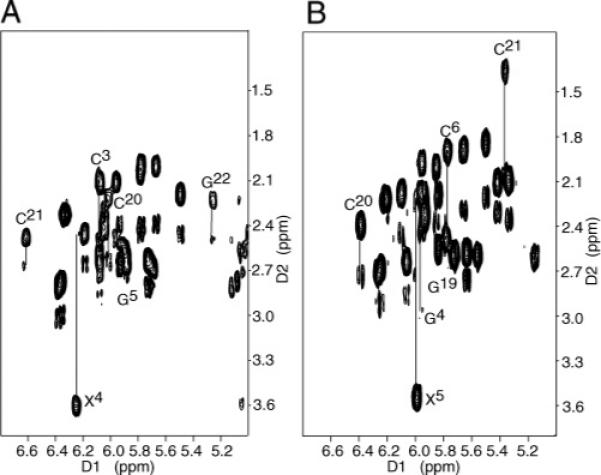
Expanded COSY contour plot at 15 °C establishing the connectivity between the H1′ and H2′ and H2″ protons. (A) NarIIQ1 duplex. The H2′ and H2″ protons of nucleotides C3, X4, G5, C20, C21, and G22 adjacent to the lesion site are connected by lines and labeled. (B) NarIIQ2 duplex. The H2′ and H2″ protons of nucleotides G4, X5, C6, G19, C20, and C21 adjacent to the lesion site are connected by lines and labeled.
Analysis of DQF-COSY and E-COSY spectra suggested that all of the pyrimidine pseudorotation values (P) were in the C1′-exo range of 126 ± 18°, and all of the purines in the center 10 bp had pseudorotation values in the C2′-endo range of 162 ± 18°. The sugar pucker of X4 was in the C2′-endo range. The C8-dG IQ adduct at X4 resulted in a dispersion of four 31P resonances, with the greatest change occurring at P4, the phosphodiester 3′ to X4 in the modified strand (∼0.8 ppm). The smaller differences, 0.35 and 0.25 ppm observed for 31P chemical shifts for P20 and P21, respectively, suggested that the phosphodiester linkages opposite to X in the complementary strand were less perturbed. Figure S1 of the Supporting Information shows the DQF-COSY and E-COSY spectral data. Figure S2 of the Supporting Information shows the 31P HMBC correlation spectra and the counterpart spectrum for the unmodified duplex.
(j) Torsion Angle Analysis in the NarIIQ2 Duplex
The intensity of the X5 H8 → X5 H1′ NOE normally would have been utilized to assess the conformation of the glycosyl torsion angle χ. The absence of this proton in the adducted oligodeoxynucleotide necessitated the evaluation of chemical shift data at the deoxyribose H2′ and H2″ protons (84, 85). The X5 H2′ resonance shifted downfield to 3.57 ppm (Figure 5B; expanded DQF-COSY plots identifying scalar couplings between deoxyribose H1′ and H2′ and H2″ protons in the NarIIQ2 duplex are shown in Figure S1 of the Supporting Information). This was characteristic of the syn dG orientation at the modified position (84, 85). In the corresponding unmodified duplex, the G5 H8 → G5 H1′ NOE was of normal intensity and all scalar cross-peaks between deoxyribose H1′ and H2′ and H2″ protons were located in the anticipated chemical shift range of 1.6−3.0 ppm (63), except C21 H2′.
Analysis of DQF-COSY and E-COSY spectra suggested that all of the pyrimidine pseudorotation values (P) were in the C1′-exo range of 126 ± 18°, except C21 was in the O1′-endo range, and all of the purines in the center 10 bp had pseudorotation values in the C2′-endo range of 162 ± 18°. The sugar pucker of X5 was in the C2′-endo range. The C8-dG IQ adduct at X5 resulted in a dispersion of four 31P resonances, with the most significant change occurring at P4, the phosphodiester 5′ to X5 in the modified strand (∼ 1.1 ppm). The downfield 31P chemical shift at P4 presumably reflected conformational perturbations associated with the P4 phosphodiester (86). The small differences, −0.1 and 0.25 ppm, observed for 31P chemical shifts for P19 and P20, respectively, suggested that the phosphodiester linkages opposite to X in the complementary strand in the modified duplex were less perturbed. Figure S1 of the Supporting Information shows the DQF-COSY and E-COSY spectral data. Figure S2 of the Supporting Information shows the 31P HMBC correlation spectrum and its counterpart for the unmodified duplex.
(k) Chemical Shift Perturbations of the NarIIQ1 Duplex
The 1H NMR chemical shifts of the NarIIQ1 dodecamer were compared with those of the unmodified dodecamer (Figure 6). The largest chemical shift perturbations were observed for the aromatic and anomeric protons of C21 in the complementary strand, opposite to the modified position. Smaller chemical shift perturbations were also observed for the G22 H8 and H1′, C3 H1′ and H6, and X4 H1′ and C20 H6 resonances. Significant downfield shifts were observed for H7A, H8A, and H9A for NarIIQ1, compared to those of NarIIQ3 (63) (Table 1).
Figure 6.
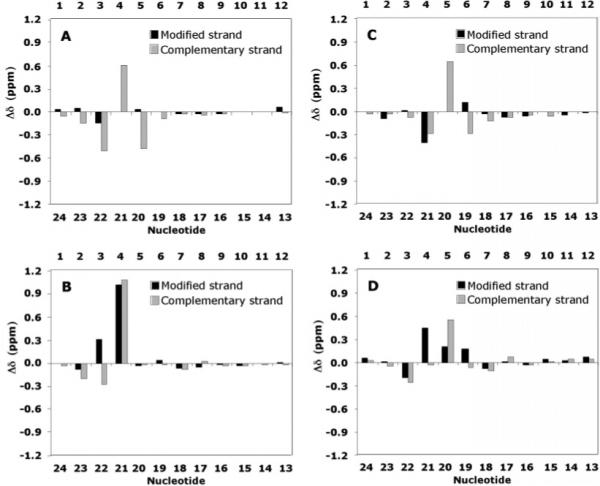
Chemical shift changes of (A) aromatic protons H6 and H8 and (B) anomeric H1′ protons of the NarIIQ1 duplex and (C) aromatic protons H6 and H8 and (D) anomeric H1′ protons of the NarIIQ2 duplex, relative to the unmodified duplex, where Δδ = δmodified oligodeoxynucleotide – δunmodified oligodeoxynucleotide (parts per million).
Table 1.
Chemical Shifts of the IQ Protons in the NarIIQ1, NarIIQ2, NarIIQ3, and RasIQ5 Sequences
| H4A | H5A | H7A | H8A | H9A | |
|---|---|---|---|---|---|
| NarIIQ1 | 7.218 | 7.141 | 8.596 | 7.100 | 8.121 |
| NarIIQ2 | 7.296 | 7.053 | 8.316 | 6.959 | 8.004 |
| NarIIQ3 | 7.269 | 7.105 | 8.067 | 6.702 | 7.798 |
| RasIQ5 | 7.340 | 7.249 | 8.580 | 7.252 | 8.309 |
(l) Chemical Shift Perturbations of the NarIIQ2 Duplex
The 1H NMR chemical shifts of the NarIIQ2 dodecamer were compared with those of the unmodified dodecamer (Figure 6). The largest chemical shift perturbations were observed for the aromatic and anomeric protons of C20 in the complementary strand, opposite to the modified position. Smaller chemical shift perturbations were also observed for the G22 H1′, C3 H1′, C21 H6, G4 H1′ and H8, X5 H1′, C6 H1′ and H6, and G19 H8 resonances. The aromatic protons of the flanking bases around the adduct site were all in some way perturbed in NarIIQ2, similar to what was observed in NarIIQ3 (63). Significant downfield shifts were observed for H7A, H8A, and H9A for NarIIQ2, compared those for NarIIQ3 (63) (Table 1).
Structural Refinement
(a) NarIIQ1 Duplex
The DNA starting conformation for structural refinement utilized the syn glycosyl torsion angle at X4. To determine the starting conformation for the IQ adduct, a searching strategy similar to that employed by Mao et al. (87) was guided by the intermolecular IQ–DNA restraints listed in Table S3 of the Supporting Information. The IQ–DNA orientation space was searched with 16 potential energy minimization trials in which linkage torsion angles α′ and β′ (Chart 2) were started at 0°, 90°, 180°, and 270° in all combinations, which generated four low-energy conformations in which the glycosyl bond remained in the syn conformation and in which IQ torsion angles α′ and β′ were either approximately 0° or 180°. Of these four structures, the potential energy-minimized structure starting from an α′ of 180° and a β′ of 180° exhibited the best fit to the 1H NOE data of the adducted NarIIQ1 dodecamer and was therefore selected as the starting conformation for subsequent restrained molecular dynamics calculations.
The restrained molecular dynamics calculation employed a simulated annealing protocol. A total of 504 NOE-based distance restraints were included, consisting of 139 inter-residue and 365 intra-residue distances. They included 16 DNA-IQ distances. Ten distance restraints were utilized so that IQ H8A, H4A, and methyl protons would be required to be more than 5 Å from specific DNA protons to which no NOEs were observed. The pyrimidine pseudorotation values (P) were constrained in the C1′-exo range of 126 ± 18°, and the purines in the center 10 bp were constrained with pseudorotation values in the C2′-endo range of 162 ± 18°. No backbone torsion angle constraints were used at the lesion site in the modified strand. Elsewhere, backbone angles α, β, and ξ were constrained to −60 ± 30°, 180 ± 30°, and −90 ± 30°, respectively, to allow both A- and B-like geometry (88). No empirical base pairing restraints were used at the lesion site. Elsewhere, empirical base pair planarity restraints and Watson–Crick hydrogen bonding restraints were used. These were consistent with crystal-lographic data (89). Their inclusion was based on NMR data that showed the modified DNA maintained Watson–Crick base pairing.
The structural statistics arising from the calculations are listed in Table 2. An ensemble of 10 convergent structures was obtained from randomly seeded calculations. The precision of the rMD-calculated structures was determined by pairwise rmsd measurement. Figure 7A shows a stereoview of 10 superimposed structures emergent from the rMD calculations. These structures exhibited a maximum pairwise rmsd of 0.83 Å, which suggested that excellent convergence had been achieved.
Table 2.
Analysis of the rMD-Generated Structures of the NarIIQ1 and NarIIQ2 Duplexesa
| NarIIQ1 | NarIIQ2 | |
|---|---|---|
| NMR restraints | ||
| total no. of distance restraints | 504 | 450 |
| no. of inter-residue distance restraints | 139 | 141 |
| no. of intra-residue distance restraints | 365 | 309 |
| no. of DNA–IQ distance restraints | 16 | 20 |
| no. of IQ–Q distance restraints | 6 | 6 |
| no. of H-bonding restraints | 30 | 30 |
| no. of dihedral planarity restraints | 22 | 22 |
| no. of sugar pucker restraints | 120 | 120 |
| no. of backbone torsion angle restraints | 78 | 78 |
| structural statistics | ||
| NMR R-factor (R1x)b | ||
| 〈rMDRi〉 | 0.0891 ± 0.0004 | 0.0913 ± 0.0005 |
| rmsd of NOE violations (Å) | 0.00778 ± 0.00002 | 0.00783 ± 0.00002 |
| no. of NOE violations >0.2 Å | 0 | 0 |
| root-mean-square deviations from ideal geometry | ||
| bond lengths (Å) | 0.02824 ± 0.00005 | 0.02875 ± 0.00006 |
| bond angles (deg) | 2.726 ± 0.007 | 2.734 ± 0.006 |
| improper angles (deg) | 0.78 ± 0.02 | 0.81 ± 0.02 |
| pairwise rmsd (Å) over all atoms | ||
| 〈rMDRi〉 vs 〈rMDav〉c | 0.83 ± 0.01 | 0.87 ± 0.02 |
The mixing time was 250 ms.
R1x = Σ|(ao)i1/6 – (ac)i1/6|/Σ|(ao)i1/6|, where ao and ac are the intensities of observed (non-zero) and calculated NOE cross-peaks, respectively.
〈rMDRi〉, 10 converged structures starting from randomly seeded calculations; 〈rMDav〉, average of 10 converged structures.
Figure 7.
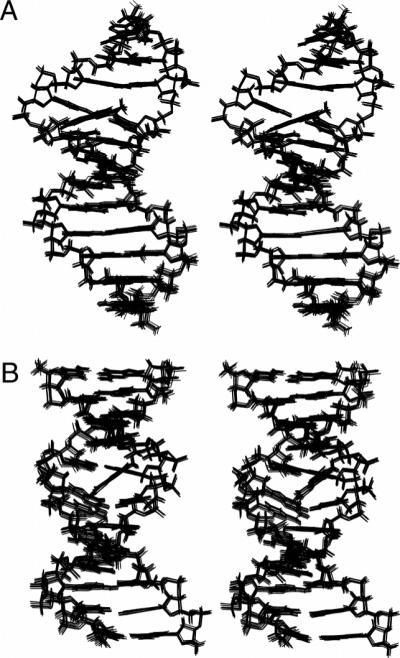
Stereoviews of 10 randomly seeded superimposed structures of the (A) NarIIQ1 and (B) NarIIQ2 duplexes emergent from rMD simulated annealing calculations, looking into the minor groove at the lesion site.
The accuracy of the rMD-calculated structures was determined by back-calculation of theoretical NMR intensities from the emergent structures using CORMA (version 5.2) (72). For the structures shown in Figure 8, the overall value of the sixth-root residual R1x was 8.9 × 10−2. Both inter-residue and intra-residue R1x values were consistently on the order of 10% (Figure 8). At the adduct site, the R1x residuals were 6.7, 10.2, 11.7, and 6.8 × 10−2 for G5, C20, C21, and G22, respectively.
Figure 8.
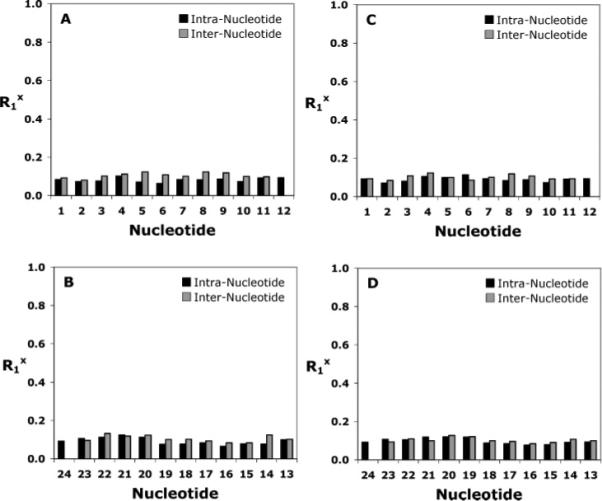
Distribution of Rx1 values calculated using CORMA (72). (A) Nucleotides C1–C12 of the modified NarIIQ1 duplex. (B) Nucleotides G13–G24 of the modified NarIIQ1 duplex. (C) Nucleotides C1–C12 of the modified NarIIQ2 duplex. (D) Nucleotides G13–G24 of the modified NarIIQ2 duplex. The black bars represent intranucleotide values and the gray bars internucleotide values.
(b) NarIIQ2 Duplex
The DNA starting conformation for structural refinement utilized the syn glycosyl torsion angle at X5. To determine the starting conformation for the IQ adduct, a searching strategy similar to that employed by Mao et al. (87) was guided by the intermolecular IQ–DNA restraints listed in Table S3 of the Supporting Information. The IQ–DNA orientation space was searched with 16 potential energy minimization trials in which linkage torsion angles α′ and β′ (Chart 2) were started at 0°, 90°, 180°, and 270° in all combinations, which generated four low-energy conformations in which the glycosyl bond remained in the syn conformation and in which IQ torsion angles α′ and β′ were either approximately 0° or 180°. Of these four structures, the potential energy-minimized structure starting from an α′ of 180° and a β′ of 180° exhibited the best fit to the 1H NOE data of the adducted NarIIQ2 dodecamer and was therefore selected as the starting conformation for subsequent restrained molecular dynamics calculations.
The restrained molecular dynamics calculation employed a simulated annealing protocol. A total of 450 NOE-based distance restraints were included, consisting of 141 inter-residue and 309 intra-residue distances. They included 20 DNA-IQ distances. Thirteen distance restraints were utilized so that IQ H8A, H9A, H5A and methyl protons would be required to be more than 5 Å from specific DNA protons to which no NOEs were observed. The pyrimidine pseudorotation values (P) were constrained in the C1′-exo range of 126 ± 18°, and the purines in the center 10 bp were constrained with pseudorotation values in the C2′-endo range of 162 ± 18°. No backbone torsion angle constraints were used at the lesion site in the modified strand. Elsewhere, backbone angles α, β, and ξ were constrained to −60 ± 30°, 180 ± 30°, and −90 ± 30°, respectively, to allow both A- and B-like geometry (88). No empirical base pairing restraints were used at the lesion site. Elsewhere, empirical base pair planarity restraints and Watson-Crick hydrogen bonding restraints were used. These were consistent with crystallographic data (89). Their inclusion was based on NMR data that showed the modified DNA maintained Watson–Crick base pairing.
The structural statistics arising from the calculations are listed in Table 2. An ensemble of 10 convergent structures was obtained from randomly seeded calculations. The precision of the rMD-calculated structures was determined by pairwise rmsd measurement. Figure 8B shows a stereoview of 10 superimposed structures emergent from the rMD calculations. These structures exhibited a maximum pairwise rmsd of 0.87 Å, suggesting that excellent convergence had been achieved.
The accuracy of the rMD-calculated structures was determined by back-calculation of theoretical NMR intensities from the emergent structures using CORMA (version 5.2) (72). For the structures shown in Figure 7, the overall value of the sixth-root residual R1x was 9.1 × 10−2. Both inter-residue and intra-residue R1x values were consistently on the order of 10% (Figure 8). At the adduct site, the R1x residuals were 12.0, 10.3, 13.1, 10.2, and 11.3 × 10−2 for nucleotides G4, C6, G19, C20, and C21, respectively.
Solution Conformation
(a) The NarIIQ1 Duplex
A view normal to the helix axis and looking into the minor groove of the 5′-d(C3X4G5)-3′·5′-d(C20C21G22)-3′ segment of the representative structure from the ensemble of 10 refined structures is shown in Figure 9A. At the adduct site, the glycosyl bond of the modified nucleotide X4 was in the syn conformation. The glycosyl torsion angle χ was predicted from the calculations to be ∼96°. The IQ moiety was oriented into the minor groove, with the methyl group and the H4A, H5A, and H7A protons facing out. This oriented the IQ H8A and H9A protons toward G21 and C20 in the complementary strand. The X4 methyl group was oriented close to the modified strand. The orientation of the IQ ring with respect to the complementary strand resulted in the buckling of complementary nucleotide C21. The complementary nucleotide C21 was slightly displaced toward the major groove but remained intrahelical. The rMD calculations suggested that the IQ ring was tilted with respect to the C3·G22 and G5·C20 base pairs. This tilt was defined by the IQ torsion angle β′, which was measured from the refined structures as 149°. IQ torsion angle α′ was calculated to be −115°. The orientation of the IQ ring in NarIIQ1 was different from that of NarIIQ3 (Figure 10).
Figure 9.
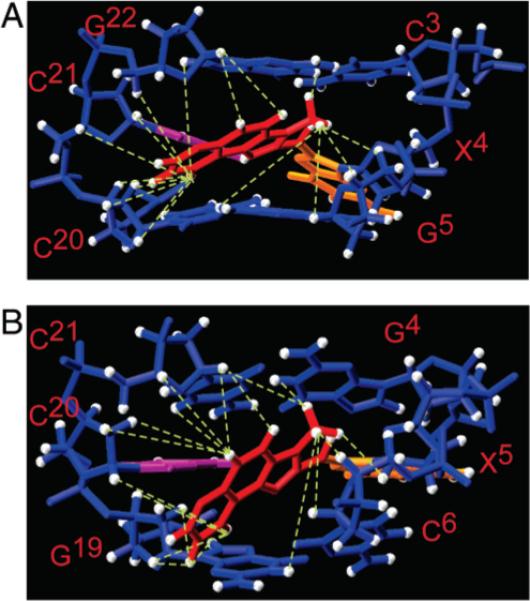
Comparison of averaged refined structures, looking into the minor groove, and normal to the helix axis of the central segment: (A) NarIIQ1 duplex and (B) NarIIQ2 duplex. The NOEs defining the IQ orientation are indicated by the green dashed lines.
Figure 10.
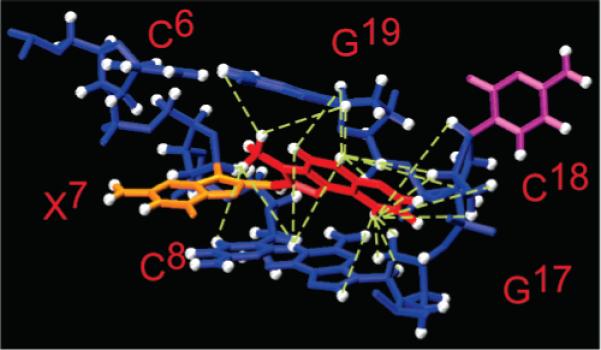
Average refined structure of the NarIIQ3 duplex, looking into the major groove, and normal to the helix axis of the central segment (63). The NOEs defining the IQ orientation are indicated by the green dashed lines.
Views looking down the helix axis of the 5′-d(C3X4G5)-3′·5′-d(C20C21G22)-3′ segment are shown in Figures 11 and 12. These views monitor overlap geometry between the IQ ring and the flanking Watson–Crick base pairs, as compared to that of the corresponding unmodified duplexes. The syn conformation of the glycosyl bond at X4 allowed the guanine ring of the modified nucleotide to stack with C3, the 5′-flanking nucleotide (Figure 11). However, it disrupted the stacking of the modified nucleotide with the 3′-flanking nucleotide G5, and the stacking geometry at the G5·C20 base pair was perturbed (Figure 12). The duplex was underwound at base pair step C3 → X4 as measured by the helicoidal twist angle of −26°. The rMD calculations predicted an adduct-induced bend in the duplex of 16 ± 5°. This was consistent with 31P chemical shift perturbations at phosphodiester linkages P4, P20, and P21. The calculated glycosyl torsion angles and sugar pseudorotation P angles are listed in Table S4 of the Supporting Information.
Figure 11.
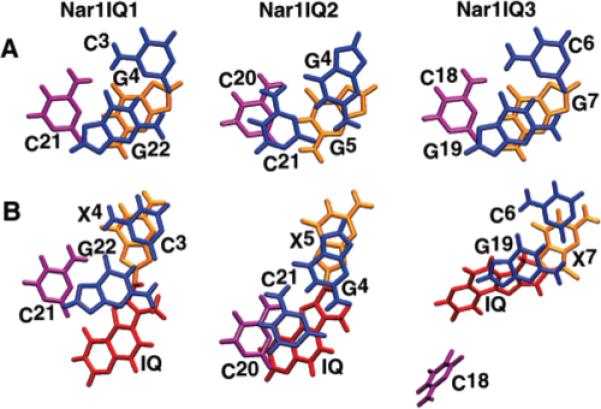
Base stacking orientations of the NarI, NarIIQ1, NarIIQ2, and NarIIQ3 (63) duplexes. (A) Unmodified duplex detailing base stacking corresponding to the modified duplexes. (B) NarIIQ1, NarIIQ2, and NarIIQ3 (63) duplexes detailing base stacking between the base pair at the modified position and its 5′ neighboring base pair, with the 5′ neighboring base pair aligned as in the unmodified duplex.
Figure 12.
Base stacking orientations of the NarI, NarIIQ1, NarIIQ2, and NarIIQ3 (63) duplexes. (A) Unmodified duplex detailing base stacking corresponding to the modified duplexes. (B) NarIIQ1, NarIIQ2, and NarIIQ3 (63) duplexes detailing base stacking between the base pair at the modified position and its 3′ neighboring base pair, with the 3′ neighboring base pair aligned as in the unmodified duplex.
(b) NarIIQ2 Duplex
A view normal to the helix axis and looking into the minor groove of the 5′-d(G4X5C6)-3′·5′-d(G19C20C21)-3′ segment of the representative structure from the ensemble of 10 refined structures is shown in Figure 9B. At the adduct site, the glycosyl bond of the modified nucleotide X5 was in the syn conformation. The glycosyl torsion angle χ was predicted from the calculations to be ∼118°. The IQ moiety was oriented into the minor groove, with the methyl group and the H4A, H5A, and H7A protons facing out. This oriented the IQ H8A and H9A protons toward C20 and G19 in the complementary strand. The X5 methyl group was oriented close to the modified strand. The complementary nucleotide C21 remained intrahelical. The 5′ neighbor G4·C21 base pair remained planar, but the 3′ neighbor C6·G19 base pair buckled away from the IQ moiety. The rMD calculations suggested that the IQ ring was tilted with respect to the C4·G21 and C6·G19 base pairs. This tilt was defined by the IQ torsion angle β′, which was measured from the refined structures to be 168°. IQ torsion angle α′ was calculated to be −124°.
Views looking down the helix axis of the 5′-d(G4X5C6)-3′·5′-d(G19C20C21)-3′ segment are shown in Figures 11 and 12. These views monitor overlap geometry between the IQ ring and the flanking Watson–Crick base pairs, as compared to that of the corresponding unmodified duplexes. The syn conformation of the glycosyl bond at X5 allowed the guanine ring of the modified nucleotide to stack with G4, the 5′-flanking nucleotide (Figure 11). However, it disrupted the stacking of the modified nucleotide with the 3′-flanking nucleotide C6, and the stacking geometry at C6·G19 was perturbed (Figure 12). The rMD calculations predicted an adduct-induced bend in the duplex of 20 ± 5°. This was consistent with 31P chemical shift perturbations at phosphodiester linkages P4, P20, and P19. The calculated glycosyl torsion angles and sugar pseudorotation P angles are listed in Table S4 of the Supporting Information.
DISCUSSION
The site-specific synthesis of oligodeoxynucleotides containing the C8-dG IQ adduct (64, 90) has enabled high-resolution structural studies of this food-borne genotoxin in the NarIIQ1, NarIIQ2, and NarIIQ3 duplexes. Minor groove, major groove, and base-displaced insertion (64, 78) conformations have been proposed for the C8-dG IQ adduct in duplex DNA. On the basis of molecular modeling, the energetic differences between these proposed conformations were predicted to be modest (78). Our study of the C8-dG IQ adduct in the NarIIQ3 duplex (63) showed that it preferred to adopt a base-displaced intercalated conformation. In contrast, ultraviolet spectroscopy and circular dichroism studies were consistent with the C8-dG IQ adduct adopting a minor groove-bound conformation at the G1 and G2 positions of the NarI sequence (65). These NMR studies confirm this prediction.
Orientation of the IQ Moiety in the NarIIQ1 and NarIIQ2 Duplexes
(a) NarIIQ1 Duplex
The C8-dG IQ adduct is located at position G1 of the 5′-d(CG1G2CG3CC)-3′ recognition site of the NarI restriction enzyme. The X4 glycosyl torsion angle of the modified dG exists in the syn conformation. The key evidence supporting this conclusion is the downfield chemical shift for the X4 H2′ resonance, observed at ∼3.60 ppm (Figure 5 and Figure S1 of the Supporting Information). This downfield shift of the H2′ resonance represents a characteristic marker of the syn conformation of dG in modified duplexes (84, 85). This corroborated the results of earlier work at the nucleoside level showing that the dG-C8 IQ adduct was in the syn conformation (53).
Rotation of the glycosyl bond into the syn conformation at X4 places the Watson–Crick hydrogen bonding edge of the modified dG in the major groove. The X4 imino and amino protons are exposed to solvent. Displacement of the modified dG into the major groove is consistent with the disappearance of spectral resonances from the X4 imino protons, due to the rapid exchange rate with solvent. This displacement of the modified dG might cause the local electrostatic potential at phosphodiester linkage P4 to be perturbed. This notion is supported by the 31P chemical shift perturbation observed at P4 (Figure S2 of the Supporting Information). A strong X4 H1′ → G5 H8 NOE for the X4 → G5 base step (Figure 1A) is consistent with a separation between these protons of 3.0 ± 0.4 Å, as measured in the intensity-refined structures of the two modified duplexes.
The IQ moiety is located in the minor groove. There is no stacking between the IQ moiety and C3, G5, C20, or G22 (Figures 11 and 12). The NOEs (Table S3 of the Supporting Information) are consistent with the edge of the ring containing the IQ H9A proton being directed toward C20 and C21 in the complementary strand (Figure 9). The IQ methyl protons are closer to the G22 N1H imino proton than to the G5 N1H imino proton (Figure 9 and Table S3 of the Supporting Information), and this is also consistent with the NOEs (Figure 2).
The displacement of C21 results in a break in the 1H sequential NOE connectivity at the C20 → C21 step (Figure 1B). This distance is predicted to be 6.4 ± 0.4 Å in the refined structures. The amino proton resonances arising from the complementary dC are not observed. This is consistent with the disruption of Watson–Crick hydrogen bonding and the buckling of the complementary cytosine. These proton resonances are presumably broadened due to an intermediate rate of rotation about the C4–N4 bond and exchange with solvent.
(b) NarIIQ2 Duplex
The C8-dG IQ adduct is located at position G2 of the 5′-d(CG1G2CG3CC)-3′ recognition site of the NarI restriction enzyme. The downfield chemical shift for the X5 H2′ resonance, observed at ∼3.60 ppm (Figure 5 and Figure S1 of the Supporting Information), indicates that X5 glycosyl torsion angle χ of the modified dG exists in the syn conformation. This places the Watson–Crick hydrogen bonding edge of the modified dG in the major groove. The X5 imino and amino protons are exposed to solvent. This is consistent with the disappearance of the spectral resonance for the X5 imino proton, presumably due to rapid exchange with solvent. The 31P chemical shift perturbation observed at P4 (Figure S2 of the Supporting Information) suggests that the local electrostatic potential at phosphodiester linkage P4 is perturbed. A strong X5 H1′ → C6 H6 NOE for the X5 → C6 base step (Figure 1C) is consistent with a separation between these protons as measured in the intensity-refined structures of the two modified duplexes of 2.8 ± 0.4 Å.
The minor groove conformation of the IQ moiety in the NarIIQ2 duplex is similar to that observed in the NarIIQ1 duplex. The IQ moiety is wedged into the duplex somewhat more than for the NarIIQ1 duplex (Figures 11 and 12). The NOEs (Table S3 of the Supporting Information) are consistent with the edge of the IQ moiety containing the H4A and H5A protons being directed toward C20 and C21 in the complementary strand, while the edge of the IQ moiety containing the H8A and H9A protons is directed toward G19 and C20 in the complementary strand (Figure 9). The IQ methyl protons are closer to the G4 N1H imino proton than to the G19 N1H imino proton (Figure 9 and Table S3 of the Supporting Information). This is also consistent with the NOEs (Figure 2). The absence of NOEs between the IQ amine and methyl protons is attributed to exchange of the amine proton with solvent.
The exocyclic amino proton resonances of the complementary nucleotide C20 are not observed. This is consistent with the disruption of Watson–Crick hydrogen bonding at the modified X5·C20 base pair. These resonances are presumably broadened due to an intermediate rate of rotation about the C4–N4 bond and exchange with solvent. The weak sequential NOE at the G19 → C20 base step is consistent with the positioning of the IQ moiety between nucleotides G19 and C20 in the complementary strand, thus increasing the distance between G19 H8 and C20 H1′. Likewise, the weak sequential NOE at the C20 → C21 base step reflects the disruption of Watson–Crick base pairing at the modified X5·C20 base pair, thus increasing the distance between C20 H6 and C21 H1′.
Sequence-Dependent Conformation of C8-dG IQ Adducts in the NarI Sequence
The C8-dG IQ adduct adopts a minor groove conformation in the NarIIQ1 and NarIIQ2 duplexes, whereas in the NarIIQ3 duplex, it assumes a base-displaced intercalation conformation. In the NarIIQ1 5′-d(CX1G)-3′· 5′-d(CCG)-3′ sequence context, the C8-dG IQ adduct is flanked by dC in the 5′ direction and by dG in the 3′ direction in the complementary strand. In the NarIIQ2 5′-d(GX2C)-3′·5′-d(GCC)-3′ sequence context, the C8-dG IQ adduct is flanked by dG in the 5′ direction and by dC in the 3′ direction in the complementary strand. In contrast, the base-displaced intercalated structure was favored in the NarIIQ3 5′-d(CX3C)-3′·5′-d(GCG)-3′ sequence context, in which the C8-dG IQ adduct was flanked by dG in both the 5′ and 3′ directions in the complementary strand. In the NarIIQ3 duplex, it seems possible that the stacking of both G17 and G19 on the IQ moiety (Figures 11 and 12) stabilizes the base-displaced intercalation structure. It seems possible that the IQ ring might stack more favorably with flanking dG than flanking dC in the complementary strand, which is the case for the NarIIQ1 and NarIIQ2 duplexes, which consequently favor the minor groove conformation of the IQ moiety.
Roles of the α′ and β′ Torsion Angles in Modulating Base-Displaced Intercalation versus Minor Groove Conformations for the C8-dG IQ Adducts
Significantly, the orientation of the IQ moiety also differs between the NarIIQ1 and NarIIQ2 duplexes and the NarIIQ3 duplex. The IQ group in the minor groove-bound conformations (NarIIQ1 and NarIIQ2) is flipped nearly 180° relative to the base-displaced inserted structure (NarIIQ3). These data lead to the conclusion that the base-displaced intercalation conformation of the C8-dG IQ adduct requires that the α′ and β′ torsion angles be approximately 180° and 0°, respectively. This orients the IQ methyl group into the helix and toward the major groove, introducing a distortion of the duplex (Figure 10) and the displacement of the complementary cytosine nucleotide into the major grove (Figures 11 and 12), which presumably causes a significant energy penalty. The stacking of both G17 and G19 on the IQ moiety in the NarIIQ3 duplex (Figures 11 and 12) appears to stabilize the base-displaced intercalation structure. In contrast, the IQ methyl group is oriented toward the minor groove for the NarIIQ1 and NarIIQ2 duplexes.
Chemical Shifts of the IQ Protons
The initial conclusions regarding the conformations of the C8-dG IQ adducts in the NarIIQ1, NarIIQ2, and NarIIQ3 duplexes were based on chemical shift analyses of the IQ protons (Table 1), as well as UV and CD spectroscopy (65). The relative chemical shifts of the IQ moiety are indicative of conformation (base-displaced inserted or groove-bound) and consistent with the full structural analyses. The chemical shifts of the H4A and H5A proton resonances were similar for the NarIIQ1 and NarIQ2 duplexes, which was indicative of a similar chemical environment. These protons are positioned in the minor groove for the NarIIQ1 and NarIQ2 duplexes, whereas they were positioned in the major groove for the NarIIQ3 duplex. Greater chemical shift differences were observed for the H7A, H8A, and H9A protons. The pyridine ring of the IQ moiety is stacked between the neighboring dG residues of the complementary strand in the NarIIQ3 duplex. Consequently, the IQ H7A, H8A, and H9A protons are the most shielded in the NarIIQ3 duplex. The same protons are the most exposed in the NarIIQ1 duplex, which is reflected in greater downfield chemical shifts, between 0.32 and 0.53 ppm versus NarIIQ3. The chemical shifts for the IQ protons for the NarIIQ2 duplex fell between those of the NarIIQ1 and NarIIQ3 duplexes. We considered the possibility that for the NarIIQ2 duplex the IQ adduct underwent conformational exchange between groove-bound and base-displaced inserted conformations; however, this was ruled out on the basis of the sharp spectral line shapes that were consistent with a single conformation. Formation of the base-displaced intercalation conformation from the minor groove conformation requires major shifts of the α′ and β′ torsion angles of IQ and is not simply a matter of moving the IQ moiety from the minor groove into the helix. The final structure of the NarIIQ2 duplex shows that the IQ ring is partially inserted into the DNA base stack with the H4A, H5A, and N6 positions exposed to the minor groove. The H7A, H8A, and H9A protons are closer to the base stack, and consequently, these resonances experience a shielding effect compared to the NarIIQ1 duplex.
Conformational Prediction
We examined the properties of the C8-dG IQ adduct located in the 5′-d(GGCAXGTGGTG)-3′·5′-d(CACCACCTGCC)-3′ duplex (named the RasIQ5 sequence). Our NMR data (Table 1), based on the comparison of the chemical shifts of IQ aromatic protons of RasIQ5 to those of NarIIQ1 (minor groove bound) and NarIIQ3 (63) (base-displaced intercalated), predict that in the 5′-d(AXG)-3′·5′-d(CCT)-3′ sequence the C8-dG IQ adduct adopts a minor groove-bound conformation. The modified dG in this sequence context adopts the syn orientation about the glycosyl bond, as evidenced by a characteristic downfield shift of the X H2′ resonance. Utilizing a combination of thermal UV melting studies, UV spectroscopy, and circular dichroism, Elmquist et al. (64) concluded that the C8-dG IQ adduct adopted a minor groove-bound conformation in the ras12 sequence, similar to that predicted by Wu et al. (78). Figure S3 of the Supporting Information shows magnitude COSY contour plots of the RasIQ5 duplex. Figure S4 of the Supporting Information shows the DQF-COSY data for the C8-dG IQ adduct in the RasIQ5 duplex.
(a) Comparison with the C8-dG PhIP Adduct
The solution structure of the C8-dG PhIP adduct was reported in 5′-d(CCATCXCTACC)-3′·5′-d(GGTAGCGATGG)-3′ (91). The PhIP-modified duplex with 5′-d(CXC)-3′ sequence adopted a conformation similar to that of the C8-dG IQ adduct in the NarIIQ3 sequence. The C8-dG PhIP adduct existed with the modified dG in the syn conformation and displaced into the major groove. The complementary dC was displaced into the major groove. The imidazo[4,5-b]pyridine (IP) ring system inserted into the duplex, stacking with the flanking G18 purine and the C5 and G16 rings. However, the out-of-plane geometry of the C6-phenyl group with respect to the IP ring in the PhIP adduct contributed to a greater unwinding and twisting of the helix as compared to those of the C8-dG IQ adduct in the NarIIQ3 sequence. The PhIP phenyl ring was inclined out-of-plane relative to the IP ring, rotating rapidly, precluding stacking with the flanking bases. Additionally, the PhIP methyl group was positioned toward the modified strand, directed toward the minor groove edge of the DNA, whereas in the NarIIQ3 duplex, the IQ methyl group was stacked between the flanking bases. Corresponding to the differences described above, the PhIP-dG linkage site was defined by torsion angles α′ and β′ of 221.3 ± 3.0° and 132.5 ± 8.0°, respectively.
(b) Comparison with Aminofluorene and Acetylaminofluorene C8-dG Adducts
The AF- and AAF-derived C8-dG adducts have been studied with respect to structure in various DNA sequences (92) and in complex with the T7 DNA polymerase (93). The C8-dG AAF adduct exhibited a base-displaced inserted structure when placed opposite dC in the 5′-d(CXC)-3′ context, with the modified dG in the syn conformation about the glycosyl bond (94). The C8-dG AF adduct consistently yielded NMR data indicating conformational heterogeneity, in a variety of sequence contexts (87, 95–100). Of particular interest with regard to this work are structural data for the C8-dG AF adduct obtained at each of the three deoxyguanosines in the NarI sequence (87) and characterized by a mixture of base-displaced intercalated (87) and major groove external conformations (101) in each instance. The ratio of base-displaced intercalated to external conformers was 30:70, 10:90, and 50:50, in the NarIIQ1, NarIIQ2, and NarIIQ3 sequences, respectively (87). In the base-displaced intercalated conformation, the C8-dG AF adduct was in the syn conformation about the glycosyl bond, whereas in the major groove external conformation, it was in the anti conformation about the glycosyl bond (101). An AF-intercalated conformer with the modified dG in the syn conformation and displaced with the 5′-flanking dC residue in the major groove was reported for the C8-dG AF adduct opposite a −2 base deletion in the NarI sequence (102). The C8-dG AF adduct was also examined in a model primer–template duplex. The C8-dG AF adduct was in the syn conformation about the glycosyl torsion angle, and the modified dG was displaced into the major groove. The complementary dC was displaced into the minor groove. This was accompanied by stacking of the fluorene ring into the duplex (103). Crystallographic analyses of the C8-dG AAF and C8-dG AAF adducts complexed with the bacteriophage T7 DNA polymerase are now available. The crystallographic data with the C8-dG- AF adduct in the templating position of the polymerase showed that the modified dG was in the syn conformation with the fluorene ring inserted into a hydrophobic pocket on the surface of the fingers subdomain of the protein, locking the fingers in an open, inactive conformation (93). Two crystal structures with the C8-dG AF adduct in the templating position of the enzyme were not well defined by the electron density, consistent with weak binding to the polymerase and, again, suggesting heterogeneity between the syn and anti conformations of the modified dG (93).
(c) Comparison with C8-dG Aminobiphenyl and Aminopyrene Adducts
The C8-dG aminobiphenyl (ABP) adduct is similar to the C8-dG AF adduct but lacks the methylene bridge between the two phenyl rings. This allows the two phenyl rings of the ABP adduct to twist relative to each other. A structural study revealed that the major conformation of the ABP adduct was one in which the ABP moiety was oriented in the major groove (104), similar to the major groove external conformation of the C8-dG AF adduct (101). The C8-dG aminopyrene (AP) adduct has also been examined with respect to structure in duplex DNA. Its structure (105) was similar to that of the base-displaced intercalated C8-dG AF adduct structure (87) in the same sequence context.
Structure–Activity Relationships
Overall, these data enhance our understanding of the dual roles played by DNA sequence and adduct structure in determining the conformations of C8-dG arylamine DNA adducts. Early studies concluded that the model arylamine compound N-acetyl-2-aminofluorene (AAF) was a frameshift mutagen in Salmonella typhimurium (15, 106). The metabolism of 2-aminofluorene (AF) and AAF and consequent formation of specific DNA adducts have been characterized (44, 107). In Escherichia coli, the C8-dG AAF adduct gave frameshift mutations (84, 108), while the C8-dG AF adduct yielded base pair substitutions (109). Significantly, the guanine in the third position of the E. coli NarI restriction sequence was identified as a hot spot for frameshift mutations (84, 108, 110–112). In human cells, both the C8-dG AAF adduct and the C8-dG AF adduct yielded primarily base pair substitutions (113, 114).
The 5′-d(CG1G2CG3CC)-3′ NarI hot spot sequence in E. coli represents the strongest known hot spot for frameshift mutagenesis (60, 110). Within the NarI restriction sequence, the propensity for frameshift mutagenesis is sequence-dependent. These mutations occur following adduct formation at the G3 position but not the G1 or G2 position in the sequence (84, 108, 110–112). A single C8-dG acetylaminofluorene adduct located at position G3 in this sequence induced −2 bp frameshifts more than 107-fold over background mutagenesis in E. coli (115). The −2 bp frameshift mutations induced at position G3 in the NarI sequence by the aromatic amine AAF (116, 117) presumably arose via AAF-induced stabilization of a transient strand slippage intermediate during translesion replication (117–119), and it is thought that the −2 bp frameshifts induced by the C8-dG PhIP adduct arise via the same mechanism (120). Crystallographic analysis of the bypass polymerase Dpo4 from Sulfolobus solfataricus involving complexes with damaged DNA templates supports the notion that error-prone lesion bypass can involve the formation of transient slippage intermediates (121–123). It will now be of interest to examine the structure of the C8-dG IQ adducts in primer-template complexes with various human lesion bypass polymerases. Koffel-Schwartz and Fuchs (1995) demonstrated that the dinucleotide repeat GCGC was essential for the −2 bp frameshifts in the NarI sequence, while the flanking nucleotides NaGCGCNb, particularly Nb, modulated the relative mutagenic strength of the sequence (115). In the case of AAF, it is thought that the 3′ neighboring base Nb forms favorable stacking interactions with the fluorene ring that stabilize the transient two-base strand slippage intermediate (124).
The preference in adopting syn conformation of the three IQ-modified duplexes may play a role in modulating the repair of the C8-dG IQ adducts. Turesky et al. (125) proposed that differences in the accumulation and rates of removal of C8-dG IQ and N2-dG IQ adducts in rodents and non-human primates may be attributable to differences in conformation about the glycosyl bond in the two classes of adducts. Adducts in the syn conformation are proposed to create greater distortions of the DNA duplex; hence, they may be more easily recognized and excised. In contrast, adducts in the anti conformation are proposed to be more refractory toward repair. Turesky et al. (125) observed a preferential removal of the C8-dG IQ adduct, whereas the N2-dG IQ adduct was more persistent. The latter existed in the anti conformation about the glycosyl bond at the nucleoside level.
Supplementary Material
Table 3.
Torsion Angles (degrees) Defining the IQ Orientation in the rMD-Generated Structures of the NarIIQ1, NarIIQ2, and NarIIQ3 Duplexesa
| duplex | χ | α′ | β′ |
|---|---|---|---|
| NarIIQ1 | 96 ± 9 | −115 ± 8 | 149 ± 8 |
| NarIIQ2 | 118 ± 8 | −124 ± 8 | 168 ± 7 |
| NarIIQ3 | 85 ± 10 | 159 ± 7 | −23 ± 8 |
χ is the glycosyl torsion angle (O4′–C1′–N9–C4), α′ is the N9–C8–N(IQ)–C2(IQ) angle, and β′ is the C8–N(IQ)–C2(IQ)–N3(IQ) angle (see Chart 1).
ACKNOWLEDGMENT
We thank Ms. Albena Kozekova for assistance with the oligodeoxynucleotide synthesis and mass spectrometry and Mr. Markus Voehler for assistance with NMR experiments.
Footnotes
This work was supported by NIH Grant CA-55678 (M.P.S.). The Vanderbilt Center for Molecular Toxicology is funded by a center grant from the National Institute of Environmental Health Sciences (NIEHS) (ES-00267). J.S.S. was supported by NIEHS Predoctoral Traineeship ES-07028. Funding for the NMR spectrometers was supplied in part by NIH Grant RR-05805, the Vanderbilt Center in Molecular Toxicology, and Vanderbilt University. The Vanderbilt-Ingram Cancer Center is funded by a center grant from the National Center Institute (NCI) (CA-68485).
The PDB ID code for the NarIIQ1 duplex structure is 2Z2G and the PDB ID code for the NarIIQ2 duplex structure is 2Z2H.
Abbreviations: HCA, heterocyclic amine; EDTA, ethylenediaminetetraacetic acid; HPLC, high-pressure liquid chromatography; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser enhancement; DSS, sodium 4,4-dimethyl-4-silapentanesulfonate; TPPI, time-proportional phase increment; TOCSY, total homonuclear correlated spectroscopy; 1D, one-dimensional; 2D, two-dimensional. The oligodeoxynucleotides discussed in this paper do not have terminal phosphate groups; we abbreviate the nomenclature for oligodeoxynucleotides by leaving out the phosphodiester linkage. A, C, G, T, and X refer to mononucleotide units; X is the C8-dG IQ adduct. A right superscript refers to the numerical position in the oligodeoxynucleotide sequence starting from the 5′-terminus of chain A and proceeding to the 3′-terminus of chain A and then from the 5′-terminus of chain B to the 3′-terminus of chain B. H2, H5, H6, H8, H1′, H2′, H2″, etc., represent the protons attached to these carbons.
SUPPORTING INFORMATION AVAILABLE
Nonexchangeable proton chemical shifts of the modified NarIIQ1 and NarIIQ2 duplexes (Table S1), exchangeable proton chemical shifts for the modified NarIIQ1 and NarIIQ2 duplexes (Table S2), comparison of distance restraints for the NarIIQ1 and NarIIQ2 duplexes (Table S3), pseudorotation and glycosyl torsion angles for the NarIIQ1 and NarIIQ2 duplexes (Table S4), DQF-COSY contour plots of the NarIIQ1 and NarIIQ2 duplexes (Figure S1), nonselective excitation 31P-1H HMBC spectra for the NarIIQ1 and NarIIQ2 duplexes (Figure S2), COSY and NOESY spectra for the RasIQ5 duplex, showing assignments for the IQ protons (Figure S3), and COSY spectrum of the RasIQ5 duplex (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Wakabayashi K, Nagao M, Esumi H, Sugimura T. Food derived mutagens and carcinogens. Cancer Res. 1992;52:2092–2098. [PubMed] [Google Scholar]
- 2.Layton DW, Bogen KT, Knize MG, Hatch FT, Johnson VM, Felton JS. Cancer risk of heterocyclic amines in cooked foods: An analysis and implications for research. Carcinogenesis. 1995;16:39–52. doi: 10.1093/carcin/16.1.39. [DOI] [PubMed] [Google Scholar]
- 3.Sugimura T. Overview of carcinogenic heterocyclic amines. Mutat. Res. 1997;376:211–219. doi: 10.1016/s0027-5107(97)00045-6. [DOI] [PubMed] [Google Scholar]
- 4.Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 2004;95:290–299. doi: 10.1111/j.1349-7006.2004.tb03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kataoka H, Nishioka S, Kobayashi M, Hanaoka T, Tsugane S. Analysis of mutagenic heterocyclic amines in cooked food samples by gas chromatography with nitrogen-phosphorus detector. Bull. Environ. Contam. Toxicol. 2002;69:682–689. doi: 10.1007/s00128-002-0115-5. [DOI] [PubMed] [Google Scholar]
- 6.Felton JS, Knize MG, Salmon CP, Malfatti MA, Kulp KS. Human exposure to heterocyclic amine food mutagens/carcinogens: Relevance to breast cancer. Environ. Mol. Mutagen. 2002;39:112–118. doi: 10.1002/em.10070. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi M, Hanaoka T, Nishioka S, Kataoka H, Tsugane S. Estimation of dietary HCA intakes in a large-scale population-based prospective study in Japan. Mutat. Res. 2002;506−507:233–241. doi: 10.1016/s0027-5107(02)00170-7. [DOI] [PubMed] [Google Scholar]
- 8.Ushiyama H, Wakabayashi K, Hirose M, Itoh H, Sugimura T, Nagao M. Presence of carcinogenic heterocyclic amines in urine of healthy volunteers eating normal diet, but not of inpatients receiving parenteral alimentation. Carcinogenesis. 1991;12:1417–1422. doi: 10.1093/carcin/12.8.1417. [DOI] [PubMed] [Google Scholar]
- 9.Anderson KE, Hammons GJ, Kadlubar FF, Potter JD, Kaderlik KR, Ilett KF, Minchin RF, Teitel CH, Chou HC, Martin MV, Guengerich FP, Barone GW, Lang NP, Peterson LA. Metabolic activation of aromatic amines by human pancreas. Carcinogenesis. 1997;18:1085–1092. doi: 10.1093/carcin/18.5.1085. [DOI] [PubMed] [Google Scholar]
- 10.Lang NP, Butler MA, Massengill J, Lawson M, Stotts RC, Hauer-Jensen M, Kadlubar FF. Rapid metabolic phenotypes for acetyltransferase and cytochrome P4501A2 and putative exposure to food-borne heterocyclic amines increase the risk for colorectal cancer or polyps. Cancer Epidemiol., Biomarkers Prev. 1994;3:675–682. [PubMed] [Google Scholar]
- 11.Shirai T, Sano M, Tamano S, Takahashi S, Hirose M, Futakuchi M, Hasegawa R, Imaida K, Matsumoto K, Wakabayashi K, Sugimura T, Ito N. The prostate: A target for carcinogenicity of 2-amino-1-methyl-6-phenylimidazo-[4,5-b]pyridine (PhIP) derived from cooked foods. Cancer Res. 1997;57:195–198. [PubMed] [Google Scholar]
- 12.Snyderwine EG. Some perspectives on the nutritional aspects of breast cancer research. Food-derived heterocyclic amines as etiologic agents in human mammary cancer. Cancer. 1994;74:1070–1077. doi: 10.1002/1097-0142(19940801)74:3+<1070::aid-cncr2820741515>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Ronco A, De Stefani E, Mendilaharsu M, Deneo-Pellegrini H. Meat, fat and risk of breast cancer: A case-control study from Uruguay. Int. J. Cancer. 1996;65:328–331. doi: 10.1002/(SICI)1097-0215(19960126)65:3<328::AID-IJC9>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 14.Ohgaki H, Hasegawa H, Kato T, Suenaga M, Ubukata M, Sato S, Takayama S, Sugimura T. Carcinogenicity in mice and rats of heterocyclic amines in cooked foods. Environ. Health Perspect. 1986;67:129–134. doi: 10.1289/ehp.8667129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heflich RH, Neft RE. Genetic toxicity of 2-acetylaminofluorene, 2-aminofluorene and some of their metabolites and model metabolites. Mutat. Res. 1994;318:73–114. doi: 10.1016/0165-1110(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 16.Thorgeirsson UP, Snyderwine EG, Gomez DE, Adamson RH. Dietary heterocyclic amines as potential human carcinogens: Experimental data from nonhuman primates. In Vivo. 1996;10:145–152. [PubMed] [Google Scholar]
- 17.Adamson RH, Thorgeirsson UP, Snyderwine EG, Thorgeirsson SS, Reeves J, Dalgard DW, Takayama S, Sugimura T. Carcinogenicity of 2-amino-3-methylimidazo-[4,5-f]quinoline in nonhuman primates: Induction of tumors in three macaques. Jpn. J. Cancer Res. 1990;81:10–14. doi: 10.1111/j.1349-7006.1990.tb02500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohgaki H, Kusama K, Matsukura N, Morino K, Hasegawa H, Sato S, Takayama S, Sugimura T. Carcinogenicity in mice of a mutagenic compound, 2-amino-3-methylimidazo[4,5-f]quinoline, from broiled sardine, cooked beef and beef extract. Carcinogenesis. 1984;5:921–924. doi: 10.1093/carcin/5.7.921. [DOI] [PubMed] [Google Scholar]
- 19.Takayama S, Nakatsuru Y, Masuda M, Ohgaki H, Sato S, Sugimura T. Demonstration of carcinogenicity in F344 rats of 2-amino-3-methyl-imidazo[4,5-f]quinoline from broiled sardine, fried beef and beef extract. Gann. 1984;75:467–470. [PubMed] [Google Scholar]
- 20.Tanaka T, Barnes WS, Williams GM, Weisburger JH. Multipotential carcinogenicity of the fried food mutagen 2-amino-3-methylimidazo[4,5-f]quinoline in rats. Jpn. J. Cancer Res. 1985;76:570–576. [PubMed] [Google Scholar]
- 21.Sugimura T, Nagao M, Wakabayashi K. Complex Factors Pertinent to Human Hazard and Risk. Wiley; New York: 2000. pp. 349–359. [Google Scholar]
- 22.Josephy PD, Evans DH, Parikh A, Guengerich FP. Metabolic activation of aromatic amine mutagens by simultaneous expression of human cytochrome P450 1A2, NADPH-cytochrome P450 reductase, and N-acetyltransferase in Escherichia coli. Chem. Res. Toxicol. 1998;11:70–74. doi: 10.1021/tx970171q. [DOI] [PubMed] [Google Scholar]
- 23.Josephy PD, Gruz P, Nohmi T. Recent advances in the construction of bacterial genotoxicity assays. Mutat. Res. 1997;386:1–23. doi: 10.1016/s1383-5742(96)00041-5. [DOI] [PubMed] [Google Scholar]
- 24.Oda Y, Yamazaki H, Watanabe M, Nohmi T, Shimada T. Development of high sensitive umu test system: Rapid detection of genotoxicity of promutagenic aromatic amines by Salmonella typhimurium strain NM2009 possessing high O-acetyltransferase activity. Mutat. Res. 1995;334:145–156. doi: 10.1016/0165-1161(95)90005-5. [DOI] [PubMed] [Google Scholar]
- 25.Oda Y, Aryal P, Terashita T, Gillam EM, Guengerich FP, Shimada T. Metabolic activation of heterocyclic amines and other procarcinogens in Salmonella typhimurium umu tester strains expressing human cytochrome P4501A1, 1A2, 1B1, 2C9, 2D6, 2E1, and 3A4 and human NADPH-P450 reductase and bacterial O-acetyltransferase. Mutat. Res. 2001;492:81–90. doi: 10.1016/s1383-5718(01)00154-1. [DOI] [PubMed] [Google Scholar]
- 26.Nagao M. In: Food-Borne Carcinogens: Heterocyclic Amines. Nagao M, Sugimura T, editors. Wiley; New York: 2000. pp. 31–71. [Google Scholar]
- 27.Sugimura T, Sato S. Mutagens: Carcinogens in foods. Cancer Res. 1983;43:2415s–2421s. [PubMed] [Google Scholar]
- 28.Hecht SS. Tobacco smoke carcinogens and breast cancer. Environ. Mol. Mutagen. 2002;39:119–126. doi: 10.1002/em.10071. [DOI] [PubMed] [Google Scholar]
- 29.Kosakarn P, Halliday JA, Glickman BW, Josephy PD. Mutational specificity of 2-nitro-3,4-dimethylimidazo-[4,5-f]quinoline in the lacI gene of Escherichia coli. Carcinogenesis. 1993;14:511–517. doi: 10.1093/carcin/14.3.511. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe M, Ohta T. Analysis of mutational specificity induced by heterocyclic amines in the lacZ gene of Escherichia coli. Carcinogenesis. 1993;14:1149–1153. doi: 10.1093/carcin/14.6.1149. [DOI] [PubMed] [Google Scholar]
- 31.Terada M, Nagao M, Nakayasu M, Sakamoto H, Nakasato F, Sugimura T. Mutagenic activities of heterocyclic amines in chinese hamster lung cells in culture. Environ. Health Perspect. 1986;67:117–119. doi: 10.1289/ehp.8667117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson LH, Tucker JD, Stewart SA, Christensen ML, Salazar EP, Carrano AV, Felton JS. Genotoxicity of compounds from cooked beef in repair-deficient CHO cells versus Salmonella mutagenicity. Mutagenesis. 1987;2:483–487. doi: 10.1093/mutage/2.6.483. [DOI] [PubMed] [Google Scholar]
- 33.Felton JS, Fultz E, Dolbeare FA, Knize MG. Effect of microwave pretreatment on heterocyclic aromatic amine mutagens/carcinogens in fried beef patties. Food Chem. Toxicol. 1994;32:897–903. doi: 10.1016/0278-6915(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 34.Felton JS, Knize MG, Dolbeare FA, Wu RW. Mutagenic activity of heterocyclic amines in cooked foods. Environ. Health Perspect. 1994;102:201–204. doi: 10.1289/ehp.94102s6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schut HA, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: Implications for mutagenesis and carcinogenesis. Carcinogenesis. 1999;20:353–368. doi: 10.1093/carcin/20.3.353. [DOI] [PubMed] [Google Scholar]
- 36.Turesky RJ. Heterocyclic aromatic amine metabolism, DNA adduct formation, mutagenesis, and carcinogenesis. Drug Metab. Rev. 2002;34:625–650. doi: 10.1081/dmr-120005665. [DOI] [PubMed] [Google Scholar]
- 37.Thompson LH, Carrano AV, Salazar E, Felton JS, Hatch FT. Comparative genotoxic effects of the cooked-food-related mutagens TRP-P-2 and IQ in bacteria and cultured mammalian cells. Mutat. Res. 1983;117:243–257. doi: 10.1016/0165-1218(83)90125-8. [DOI] [PubMed] [Google Scholar]
- 38.Aeschbacher HU, Ruch E. Effect of heterocyclic amines and beef extract on chromosome aberrations and sister chromatid exchanges in cultured human lymphocytes. Carcinogenesis. 1989;10:429–433. doi: 10.1093/carcin/10.3.429. [DOI] [PubMed] [Google Scholar]
- 39.Tohda H, Oikawa A, Kawachi T, Sugimura T. Induction of sister-chromatid exchanges by mutagens from amino acid and protein pyrolysates. Mutat. Res. 1980;77:65–69. doi: 10.1016/0165-1218(80)90121-4. [DOI] [PubMed] [Google Scholar]
- 40.Yamazoe Y, Shimada M, Kamataki T, Kato R. Microsomal activation of 2-amino-3-methylimidazo[4,5-f]quinoline, a pyrolysate of sardine and beef extracts, to a mutagenic intermediate. Cancer Res. 1983;43:5768–5774. [PubMed] [Google Scholar]
- 41.Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, Guengerich FP, Sutter TR. Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res. 1996;56:2979–2984. [PubMed] [Google Scholar]
- 42.Boobis AR, Lynch AM, Murray S, de la Torre R, Solans A, Farre M, Segura J, Gooderham NJ, Davies DS. CYP 1A2-catalyzed conversion of dietary heterocyclic amines to their proximate carcinogens is their major route of metabolism in humans. Cancer Res. 1994;54:89–94. [PubMed] [Google Scholar]
- 43.Hammons GJ, Milton D, Stepps K, Guengerich FP, Tukey RH, Kadlubar FF. Metabolism of carcinogenic heterocyclic and aromatic amines by recombinant human cytochrome P450 enzymes. Carcinogenesis. 1997;18:851–854. doi: 10.1093/carcin/18.4.851. [DOI] [PubMed] [Google Scholar]
- 44.Guengerich FP. N-hydroxyarylamines. Drug Metab. Rev. 2002;34:607–623. doi: 10.1081/dmr-120005663. [DOI] [PubMed] [Google Scholar]
- 45.Hickman D, Pope J, Patil SD, Fakis G, Smelt V, Stanley LA, Payton M, Unadkat JD, Sim E. Expression of arylamine N-acetyltransferase in human intestine. Gut. 1998;42:402–409. doi: 10.1136/gut.42.3.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hein DW, Doll MA, Rustan TD, Gray K, Feng Y, Ferguson RJ, Grant DM. Metabolic activation and deactivation of arylamine carcinogens by recombinant human NAT1 and polymorphic NAT2 acetyltransferases. Carcinogenesis. 1993;14:1633–1638. doi: 10.1093/carcin/14.8.1633. [DOI] [PubMed] [Google Scholar]
- 47.Minchin RF, Reeves PT, Teitel CH, McManus ME, Mojarrabi B, Ilett KF, Kadlubar FF. N- and O-acetylation of aromatic and heterocyclic amine carcinogens by human monomorphic and polymorphic acetyltransferases expressed in COS-1 cells. Biochem. Biophys. Res. Commun. 1992;185:839–844. doi: 10.1016/0006-291x(92)91703-s. [DOI] [PubMed] [Google Scholar]
- 48.Le Marchand L, Hankin JH, Pierce LM, Sinha R, Nerurkar PV, Franke AA, Wilkens LR, Kolonel LN, Donlon T, Seifried A, Custer LJ, Lum-Jones A, Chang W. Well-done red meat, metabolic phenotypes and colorectal cancer in hawaii. Mutat. Res. 2002;506−507:205–214. doi: 10.1016/s0027-5107(02)00167-7. [DOI] [PubMed] [Google Scholar]
- 49.Ishibe N, Sinha R, Hein DW, Kulldorff M, Strickland P, Fretland AJ, Chow WH, Kadlubar FF, Lang NP, Rothman N. Genetic polymorphisms in heterocyclic amine metabolism and risk of colorectal adenomas. Pharmacogenetics. 2002;12:145–150. doi: 10.1097/00008571-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 50.Zhou Y, Chladek S, Romano LJ. Synthesis of oligonucleotides containing site-specific carcinogen adducts. Preparation of the 2-cyanoethyl-N,N-diisopropylphosphoramidite of N-(2′-deoxyguanosin-8-yl)-2-(acetylamino)fluorene with FMOC as the base-protecting group. J. Org. Chem. 1994;59:556–563. [Google Scholar]
- 51.Snyderwine EG, Roller PP, Adamson RH, Sato S, Thorgeirsson SS. Reaction of N-hydroxylamine and N-acetoxy derivatives of 2-amino-3-methylimidazolo[4,5-f]quinoline with DNA. Synthesis and identification of N-(deoxyguanosin-8-yl)-IQ. Carcinogenesis. 1988;9:1061–1065. doi: 10.1093/carcin/9.6.1061. [DOI] [PubMed] [Google Scholar]
- 52.Nagaoka H, Wakabayashi K, Kim SB, Kim IS, Tanaka Y, Ochiai M, Tada A, Nukaya H, Sugimura T, Nagao M. Adduct formation at C-8 of guanine on in vitro reaction of the ultimate form of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine with 2′-deoxyguanosine and its phosphate esters. Jpn. J. Cancer Res. 1992;83:1025–1029. doi: 10.1111/j.1349-7006.1992.tb02716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turesky RJ, Rossi SC, Welti DH, Lay JO, Jr., Kadlubar FF. Characterization of DNA adducts formed in vitro by reaction of N-hydroxy-2-amino-3-methylimidazo[4,5-f]quinoline and N-hydroxy-2-amino-3,8-dimethylimidazo[4,5-f]-quinoxaline at the C-8 and N2 atoms of guanine. Chem. Res. Toxicol. 1992;5:479–490. doi: 10.1021/tx00028a005. [DOI] [PubMed] [Google Scholar]
- 54.Guengerich FP, Mundkowski RG, Voehler M, Kadlubar FF. Formation and reactions of N7-aminoguanosine and derivatives. Chem. Res. Toxicol. 1999;12:906–916. doi: 10.1021/tx990094u. [DOI] [PubMed] [Google Scholar]
- 55.Gorlewska-Roberts K, Green B, Fares M, Ambrosone CB, Kadlubar FF. Carcinogen-DNA adducts in human breast epithelial cells. Environ. Mol. Mutagen. 2002;39:184–192. doi: 10.1002/em.10060. [DOI] [PubMed] [Google Scholar]
- 56.Takayama K, Yamashita K, Wakabayashi K, Sugimura T, Nagao M. DNA modification by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in rats. Jpn. J. Cancer Res. 1989;80:1145–1148. doi: 10.1111/j.1349-7006.1989.tb01644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gangl ET, Turesky RJ, Vouros P. Detection of in vivo formed DNA adducts at the part-per-billion level by capillary liquid chromatography/microelectrospray mass spectrometry. Anal. Chem. 2001;73:2397–2404. doi: 10.1021/ac0100401. [DOI] [PubMed] [Google Scholar]
- 58.Soglia JR, Turesky RJ, Paehler A, Vouros P. Quantification of the heterocyclic aromatic amine DNA adduct N-(deoxyguanosin-8-yl)-2-amino-3-methylimidazo[4,5-f]quinoline in livers of rats using capillary liquid chromatography/microelectrospray mass spectrometry: A dose-response study. Anal. Chem. 2001;73:2819–2827. doi: 10.1021/ac010218j. [DOI] [PubMed] [Google Scholar]
- 59.Bichara M, Fuchs RPP. DNA binding and mutation spectra of the carcinogen N-2-aminofluorene in Escherichia coli. A correlation between the conformation of the premutagenic lesion and the mutation specificity. J. Mol. Biol. 1985;183:341–351. doi: 10.1016/0022-2836(85)90005-1. [DOI] [PubMed] [Google Scholar]
- 60.Burnouf D, Koehl P, Fuchs RPP. Single adduct mutagenesis: Strong effect of the position of a single acetylaminofluorene adduct within a mutation hot spot. Proc. Natl. Acad. Sci. U.S.A. 1989;86:4147–4151. doi: 10.1073/pnas.86.11.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Melchior WB, Jr., Marques MM, Beland FA. Mutations induced by aromatic amine DNA adducts in pBR322. Carcinogenesis. 1994;15:889–899. doi: 10.1093/carcin/15.5.889. [DOI] [PubMed] [Google Scholar]
- 62.Tebbs RS, Romano LJ. Mutagenesis at a site-specifically modified NarI sequence by acetylated and deacetylated aminofluorene adducts. Biochemistry. 1994;33:8998–9006. doi: 10.1021/bi00196a018. [DOI] [PubMed] [Google Scholar]
- 63.Wang F, Demuro NE, Elmquist CE, Stover JS, Rizzo CJ, Stone MP. Base-displaced intercalated structure of the food mutagen 2-amino-3-methylimidazo[4,5-f]quinoline in the recognition sequence of the NarI restriction enzyme, a hotspot for −2 bp deletions. J. Am. Chem. Soc. 2006;128:10085–10095. doi: 10.1021/ja062004v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elmquist CE, Stover JS, Wang Z, Rizzo CJ. Site-specific synthesis and properties of oligonucleotides containing C8-deoxyguanosine adducts of the dietary mutagen IQ. J. Am. Chem. Soc. 2004;126:11189–11201. doi: 10.1021/ja0487022. [DOI] [PubMed] [Google Scholar]
- 65.Elmquist CE, Wang F, Stover JS, Stone MP, Rizzo CJ. Conformational differences of the C8-deoxyguanosine adduct of 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) within the NarI recognition sequence. Chem. Res. Toxicol. 2007;20:445–454. doi: 10.1021/tx060229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cavaluzzi MJ, Borer PN. Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 2004;32:e13. doi: 10.1093/nar/gnh015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 68.Piantini U, Sorensen OW, Ernst RR. Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc. 1982;104:6800–6801. [Google Scholar]
- 69.Rance M, Sorensen OW, Hodenhausen G, Wagner G, Ernst RR, Wuthrich K. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 1983;177:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 70.Griesinger G, Sorensen OW, Ernst RR. Two-dimensional correlation of connected NMR transitions. J. Am. Chem. Soc. 1985;107:6394–6396. [Google Scholar]
- 71.Sklenar V, Miyashiro H, Zon G, Miles HT, Bax A. Assignment of the 31P and 1H resonances in oligonucleotides by two-dimensional NMR spectroscopy. FEBS Lett. 1986;208:94–98. doi: 10.1016/0014-5793(86)81539-3. [DOI] [PubMed] [Google Scholar]
- 72.Keepers JW, James TL. A theoretical study of distance determination from NMR. Two-dimensional nuclear Overhauser effect spectra. J. Magn. Reson. 1984;57:404–426. [Google Scholar]
- 73.Borgias BA, James TL. MARDIGRAS: A procedure for matrix analysis of relaxation for discerning geometry of an aqueous structure. J. Magn. Reson. 1990;87:475–487. [Google Scholar]
- 74.Liu H, Tonelli M, James TL. Correcting NOESY cross-peak intensities for partial relaxation effects enabling accurate distance information. J. Magn. Reson., Ser. B. 1996;111:85–89. doi: 10.1006/jmrb.1996.0064. [DOI] [PubMed] [Google Scholar]
- 75.Salazar M, Fedoroff OY, Miller JM, Ribeiro NS, Reid BR. The DNA strand in DNA:RNA hybrid duplexes is neither B-form nor A-form in solution. Biochemistry. 1993;32:4207–4215. doi: 10.1021/bi00067a007. [DOI] [PubMed] [Google Scholar]
- 76.Brunger AT. X-PLOR. Version 3.1. A system for X-ray crystallography and nmr. Yale University Press; New Haven, CT: 1992. [Google Scholar]
- 77.Nilsson L, Karplus M. Empirical energy functions for energy minimization and dynamics of nucleic acids. J. Comput. Chem. 1986;7:591–616. [Google Scholar]
- 78.Wu X, Shapiro R, Broyde S. Conformational analysis of the major DNA adduct derived from the food mutagen 2-amino-3-methylimidazo[4,5-f]quinoline. Chem. Res. Toxicol. 1999;12:895–905. doi: 10.1021/tx990108w. [DOI] [PubMed] [Google Scholar]
- 79.Lu XJ, Olson WK. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoffmann GR, Fuchs RP. Mechanisms of frameshift mutations: Insight from aromatic amines. Chem. Res. Toxicol. 1997;10:347–359. doi: 10.1021/tx960128n. [DOI] [PubMed] [Google Scholar]
- 81.Reid BR. Sequence-specific assignments and their use in NMR studies of DNA structure. Q. Rev. Biophys. 1987;20:2–28. doi: 10.1017/s0033583500004212. [DOI] [PubMed] [Google Scholar]
- 82.Patel DJ, Shapiro L, Hare D. DNA and RNA: NMR studies of conformations and dynamics in solution. Q. Rev. Biophys. 1987;20:35–112. doi: 10.1017/s0033583500004224. [DOI] [PubMed] [Google Scholar]
- 83.Boelens R, Scheek RM, Dijkstra K, Kaptein R. Sequential assignment of imino- and amino-proton resonances in 1H NMR spectra of oligonucleotides by two-dimensional NMR spectroscopy. Application to a lac operator fragment. J. Magn. Reson. 1985;62:378–386. [Google Scholar]
- 84.Fuchs RP, Schwartz N, Daune MP. Hot spots of frameshift mutations induced by the ultimate carcinogen N-acetoxy-N-2-acetylaminofluorene. Nature. 1981;294:657–659. doi: 10.1038/294657a0. [DOI] [PubMed] [Google Scholar]
- 85.Norman D, Abuaf P, Hingerty BE, Live D, Grunberger D, Broyde S, Patel DJ. NMR and computational characterization of the N-(deoxyguanosin-8-yl)aminofluorene adduct [(AF)G] opposite adenosine in DNA: (AF)G[syn]:A[anti]-pair formation and its pH dependence. Biochemistry. 1989;28:7462–7476. doi: 10.1021/bi00444a046. [DOI] [PubMed] [Google Scholar]
- 86.Gorenstein DG. Phosphorus-31 chemical shifts and spin-spin coupling constant principles and empirical observations. In: Gorenstein DG, editor. Phosphorus-31 NMR Principles and Application. Academic Press; New York: 1984. pp. 7–56. [Google Scholar]
- 87.Mao B, Hingerty BE, Broyde S, Patel DJ. Solution structure of the aminofluorene [AF]-intercalated conformer of the syn-[AF]-C8-dG adduct opposite dC in a DNA duplex. Biochemistry. 1998;37:81–94. doi: 10.1021/bi972257o. [DOI] [PubMed] [Google Scholar]
- 88.Tjandra N, Tate S-I, Ono A, Kainosho M, Bax A. The NMR structure of a DNA dodecamer in an aqueous dilute liquid crystalline phase. J. Am. Chem. Soc. 2000;122:6190–6200. [Google Scholar]
- 89.Saenger W. Principles of Nucleic Acid Structure. Springer; New York: 1984. [Google Scholar]
- 90.Wang Z, Rizzo CJ. Synthesis of the C8-deoxyguanosine adduct of the food mutagen IQ. Org. Lett. 2001;3:565–568. doi: 10.1021/ol006968h. [DOI] [PubMed] [Google Scholar]
- 91.Brown K, Hingerty BE, Guenther EA, Krishnan VV, Broyde S, Turteltaub KW, Cosman M. Solution structure of the 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine C8-deoxyguanosine adduct in duplex DNA. Proc. Natl. Acad. Sci. U.S.A. 2001;98:8507–8512. doi: 10.1073/pnas.151251898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Patel DJ, Mao B, Gu Z, Hingerty BE, Gorin A, Basu AK, Broyde S. Nuclear magnetic resonance solution structures of covalent aromatic amine-DNA adducts and their mutagenic relevance. Chem. Res. Toxicol. 1998;11:391–407. doi: 10.1021/tx9702143. [DOI] [PubMed] [Google Scholar]
- 93.Dutta S, Li Y, Johnson D, Dzantiev L, Richardson CC, Romano LJ, Ellenberger T. Crystal structures of 2-acetylaminofluorene and 2-aminofluorene in complex with T7 DNA polymerase reveal mechanisms of mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 2004;101:16186–16191. doi: 10.1073/pnas.0406516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O'Handley SF, Sanford DG, Xu R, Lester CC, Hingerty BE, Broyde S, Krugh TR. Structural characterization of an N-acetyl-2-aminofluorene (AAF) modified DNA oligomer by NMR, energy minimization, and molecular dynamics. Biochemistry. 1993;32:2481–2497. doi: 10.1021/bi00061a005. [DOI] [PubMed] [Google Scholar]
- 95.Eckel LM, Krugh TR. 2-Aminofluorene modified DNA oligomer duplex exists in two interchangeable conformations. Nat. Struct. Biol. 1994;1:89–94. doi: 10.1038/nsb0294-89. [DOI] [PubMed] [Google Scholar]
- 96.Eckel LM, Krugh TR. Structural characterization of two interchangeable conformations of a 2-aminoflourene-modified DNA oligomer by NMR and energy minimization. Biochemistry. 1994;33:13611–13624. doi: 10.1021/bi00250a012. [DOI] [PubMed] [Google Scholar]
- 97.Cho BP, Beland FA, Marques MM. NMR structural studies of a 15-mer DNA duplex from ras protooncogene modified with the carcinogen 2-aminoflourene: Conformational heterogeneity. Biochemistry. 1994;33:1373–1384. doi: 10.1021/bi00172a013. [DOI] [PubMed] [Google Scholar]
- 98.Zhou L, Rajabzadeh M, Traficante DD, Cho BSP. Conformational heterogeneity of arylamine-modified DNA: F-19 NMR evidence. J. Am. Chem. Soc. 1997;119:5384–5389. [Google Scholar]
- 99.Cho BP, Zhou L. Probing the conformational heterogeneity of the acetylaminofluorene-modified 2′-deoxyguanosine and DNA by 19F NMR spectroscopy. Biochemistry. 1999;38:7572–7583. doi: 10.1021/bi990182d. [DOI] [PubMed] [Google Scholar]
- 100.Meneni S, Liang F, Cho BP. Examination of the long-range effects of aminofluorene-induced conformational heterogeneity and its relevance to the mechanism of translesional DNA synthesis. J. Mol. Biol. 2007;366:1387–1400. doi: 10.1016/j.jmb.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mao B, Hingerty BE, Broyde S, Patel DJ. Solution structure of the aminofluorene [AF]-external conformer of the anti-[AF]-C8-dG adduct opposite dC in a DNA duplex. Biochemistry. 1998;37:95–106. doi: 10.1021/bi972258g. [DOI] [PubMed] [Google Scholar]
- 102.Mao B, Gorin A, Gu Z, Hingerty BE, Broyde S, Patel DJ. Solution structure of the aminofluorene-intercalated conformer of the syn [AF]-C8-dG adduct opposite a–2 deletion site in the NarI hot spot sequence context. Biochemistry. 1997;36:14479–14490. doi: 10.1021/bi972205z. [DOI] [PubMed] [Google Scholar]
- 103.Mao B, Gu Z, Gorin A, Hingerty BE, Broyde S, Patel DJ. Solution structure of the aminofluorene-stacked conformer of the syn [AF]-C8-dG adduct positioned at a template-primer junction. Biochemistry. 1997;36:14491–14501. doi: 10.1021/bi972206r. [DOI] [PubMed] [Google Scholar]
- 104.Cho BP, Beland FA, Marques MM. NMR structural studies of a 15-mer DNA sequence from a ras protooncogene, modified at the first base of codon 61 with the carcinogen 4-aminobiphenyl. Biochemistry. 1992;31:9587–9602. doi: 10.1021/bi00155a011. [DOI] [PubMed] [Google Scholar]
- 105.Mao B, Vyas RR, Hingerty BE, Broyde S, Basu AK, Patel DJ. Solution conformation of the N-(deoxyguanosin-8-yl)-1-aminopyrene ([AP]dG) adduct opposite dC in a DNA duplex. Biochemistry. 1996;35:12659–12670. doi: 10.1021/bi961078o. [DOI] [PubMed] [Google Scholar]
- 106.Ames BN, Gurney EG, Miller JA, Bartsch H. Carcinogens as frameshift mutagens: Metabolites and derivatives of 2-acetylaminofluorene and other aromatic amine carcinogens. Proc. Natl. Acad. Sci. U.S.A. 1972;69:3128–3132. doi: 10.1073/pnas.69.11.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Beland FA, Kadlubar FF. Formation and persistence of arylamine DNA adducts in vivo. Environ. Health Perspect. 1985;62:19. doi: 10.1289/ehp.856219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Koffel-Schwartz N, Verdier JM, Bichara M, Freund AM, Daune MP, Fuchs RPP. Carcinogen-induced mutation spectrum in wild-type, uvra and umuc strains of Escherichia coli. J. Mol. Biol. 1984;177:33–51. doi: 10.1016/0022-2836(84)90056-1. [DOI] [PubMed] [Google Scholar]
- 109.Bichara M, Fuchs RP. DNA binding and mutation spectra of the carcinogen N-2-aminofluorene in Escherichia coli. A correlation between the conformation of the premutagenic lesion and the mutation specificity. J. Mol. Biol. 1985;183:341–351. doi: 10.1016/0022-2836(85)90005-1. [DOI] [PubMed] [Google Scholar]
- 110.Broschard TH, Koffel-Schwartz N, Fuchs RP. Sequence-dependent modulation of frameshift mutagenesis at narI-derived mutation hot spots. J. Mol. Biol. 1999;288:191–199. doi: 10.1006/jmbi.1999.2667. [DOI] [PubMed] [Google Scholar]
- 111.Burnouf DY, Miturski R, Fuchs RP. Sequence context modulation of translesion synthesis at a single N-2-acetylaminofluorene adduct located within a mutation hot spot. Chem. Res. Toxicol. 1999;12:144–150. doi: 10.1021/tx9801920. [DOI] [PubMed] [Google Scholar]
- 112.Tan X, Suzuki N, Grollman AP, Shibutani S. Mutagenic events in Escherichia coli and mammalian cells generated in response to acetylaminofluorene-derived DNA adducts positioned in the narI restriction enzyme site. Biochemistry. 2002;41:14255–14262. doi: 10.1021/bi0202878. [DOI] [PubMed] [Google Scholar]
- 113.Mah MC, Maher VM, Thomas H, Reid TM, King CM, McCormick JJ. Mutations induced by aminofluorene-DNA adducts during replication in human cells. Carcinogenesis. 1989;10:2321–2328. doi: 10.1093/carcin/10.12.2321. [DOI] [PubMed] [Google Scholar]
- 114.Mah MC, Boldt J, Culp SJ, Maher VM, McCormick JJ. Replication of acetylaminofluorene-adducted plasmids in human cells: Spectrum of base substitutions and evidence of excision repair. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10193–10197. doi: 10.1073/pnas.88.22.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Koffel-Schwartz N, Fuchs RP. Sequence determinants for –2 frameshift mutagenesis at NarI-derived hot spots. J. Mol. Biol. 1995;252:507–513. doi: 10.1006/jmbi.1995.0515. [DOI] [PubMed] [Google Scholar]
- 116.Lambert IB, Napolitano RL, Fuchs RPP. Carcinogen-induced frameshift mutagenesis in repetitive sequences. Proc. Natl. Acad. Sci. U.S.A. 1992;89:1310–1314. doi: 10.1073/pnas.89.4.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garcia A, Lambert IB, Fuchs RP. DNA adduct-induced stabilization of slipped frameshift intermediates within repetitive sequences: Implications for mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5989–5993. doi: 10.1073/pnas.90.13.5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Milhe C, Dhalluin C, Fuchs RP, Lefevre JF. NMR evidence of the stabilisation by the carcinogen N-2-acetylaminofluorene of a frameshift mutagenesis intermediate. Nucleic Acids Res. 1994;22:4646–4652. doi: 10.1093/nar/22.22.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Milhe C, Fuchs RP, Lefevre JF. NMR data show that the carcinogen N-2-acetylaminofluorene stabilises an intermediate of –2 frameshift mutagenesis in a region of high mutation frequency. Eur. J. Biochem. 1996;235:120–127. doi: 10.1111/j.1432-1033.1996.00120.x. [DOI] [PubMed] [Google Scholar]
- 120.Shibutani S, Fernandes A, Suzuki N, Zhou L, Johnson F, Grollman AP. Mutagenesis of the N-(deoxyguanosin-8-yl)-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine DNA adduct in mammalian cells. Sequence context effects. J. Biol. Chem. 1999;274:27433–27438. doi: 10.1074/jbc.274.39.27433. [DOI] [PubMed] [Google Scholar]
- 121.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: A mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 122.Ling H, Boudsocq F, Woodgate R, Yang W. Snapshots of replication through an abasic lesion: Structural basis for base substitutions and frameshifts. Mol. Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- 123.Zang H, Goodenough AK, Choi JY, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, Guengerich FP. DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4. Analysis and crystal structures of multiple base-pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J. Biol. Chem. 2005;280:29750–29764. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
- 124.Roy D, Hingerty BE, Shapiro R, Broyde S. A slipped replication intermediate model is stabilized by the syn orientation of N-2-aminofluorene- and N-2-(acetyl)aminofluorene-modified guanine at a mutational hotspot. Chem. Res. Toxicol. 1998;11:1301–1311. doi: 10.1021/tx980106w. [DOI] [PubMed] [Google Scholar]
- 125.Turesky RJ, Box RM, Markovic J, Gremaud E, Snyderwine EG. Formation and persistence of DNA adducts of 2-amino-3-methylimidazo[4,5-f]quinoline in the rat and nonhuman primates. Mutat. Res. 1997;376:235–241. doi: 10.1016/s0027-5107(97)00048-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.