

Abstract
Analogues of the P2X7 receptor antagonist KN-62, modified at the piperazine and arylsulfonyl groups, were synthesized and assayed at the human P2X7 receptor for inhibition of BzATP-induced effects, that is, uptake of a fluorescent dye (ethidium bromide) in stably transfected HEK293 cells and IL-1β release in differentiated THP-1 cells. Substitution of the arylsulfonyl moiety with a nitro group increased antagonistic potency relative to methyl substitution, such that compound 21 was slightly more potent than KN-62. Substitution with D-tyrosine in 36 and sterically bulky tyrosyl 2,6-dimethyl groups in 9 enhanced antagonistic potency.
Keywords: P2X7 receptor, Tyrosine-based antagonists, Ethidium bromide uptake, IL-1β release
The P2 purinergic receptor family is divided into P2X and P2Y subfamilies. The P2X receptors are ligandgated cation channels activated by extracellular ATP (adenosine-5-triphosphate). P2X receptors are further classified into seven receptor subtypes, P2X1–P2X7.1,2 In particular, the P2X7 receptor, formerly designated as the P2Z receptor,3,4 is highly expressed in cells of the immune system such as macrophages,5 lymphocytes,6 mast cells,7 and microglia.8 The P2X7 receptor, which displays 35–40% structural homology with other subtypes of P2X receptors, consists of two hydrophobic membrane-spanning domains (M1,M2), a large extracellular loop, a short intracellular N-terminus, and a long intracellular C-terminus.9 Interestingly, the C terminus of the P2X7 receptor is 239 amino acids longer (AA 352–595) than that of other P2X receptor subtypes. The long intracellular C-terminal domain of the P2X7 receptor modulates several functions, including signal transduction events and cytolytic pore formation.10–12 The activation of P2X7 receptors results in two distinct responses, depending on the exposure time to agonist. A brief stimulation of the receptor by extracellular ATP opens a membrane channel permeable to small cations (Na+, Ca2+, K+). However, repeated or sustained activation by ATP induces the formation of a pore permeable to large molecular weight molecules/ions up to 900 Da in mass, such as ethidium bromide (314 Da) and YO-Pro-1 (376 Da), leading to cellular death or cell lysis.13–15
The P2X7 receptor is implicated in the regulation of the expression and secretion of cytokines and inflammatory mediators including interleukin (IL)-1,16 IL-2,17 IL-18,18 and tumor necrosis factor (TNF)-α.19 Notably, the P2X7 receptor plays an important role in the processing and release of the proinflammatory cytokine IL-1β in the immune system by a complex sequence of events: Initially, the activated P2X7 receptor causes the depletion of K+ leading to the stimulation of IL-1β converting enzyme (caspase-1), which converts LPS-activated pro-IL-1β to mature IL-1β.16 Therefore, development of antagonists for inhibition of the P2X7 receptor could be a therapeutic strategy to treat inflammatory diseases (Fig. 1).
Figure 1.

Among potent and specific non-competitive antagonists at the human (h) P2X7 receptor developed so far20 is a tyrosyl derivative, the isoquinoline KN-62 (1-(N,O-bis(1,5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl)-4-phe-nylpiperazine) 1, first known as an inhibitor of Ca2+/calmodulin-dependent protein kinase II (CaMK II).21 Compound 2 (MRS2306), modified from KN-62, with the general structure R1-Tyr(OR2)-piperazinyl-R3, exhibited an IC50 value of 40 nM and was more potent than KN-62 in an ATP-induced K+ efflux assay in hP2X7-expressing HEK293 cells.22 Another series of conformationally constrained and N-arylpiperazinemodified analogues was reported.23,24 An N-p-fluorophenylpiperazine analogue, 3, displayed higher antagonistic potency than KN-62 in an ethidium bromide uptake assay and significantly inhibited ATP-stimulated secretion of IL-1β from human macrophages. Other P2X7 receptor antagonists, such as the thiazolidine-2,4-dione derivative 4, the 4,5-diarylimidazoline derivative 5, and the 1-benzyl-5-phenyltetrazole derivative 6, have been reported by several industrial pharmaceutical laboratories.25–27
In the present study, we report the structure–activity relationship (SAR) of a novel series of Cbz-substituted tyrosyl derivatives having substituted piperazine and arylsulfonyl groups. The compounds were evaluated in an assay of the uptake of a fluorescent dye ethidium bromide induced by 2′- and 3′-O-(4-benzoyl-benzoyl)-ATP (BzATP), which activates the hP2X7 receptor stably expressed in HEK293 cells, and in an ELISA for IL-1β release from differentiated THP-1 cells.
Two variable positions of the skeleton, that is, the arylpiperazine and arylsulfonate moieties, were systematically modified through facile acylation and sulfonylation reactions as shown in Scheme 1 – Scheme 3. The starting materials, the compounds 7, 10, 11, and 12, were first reacted with a variably substituted arylpiperazine or Boc-piperazine to produce an amide in the presence of coupling reagents such as EDC·HCl or Bop-Cl. Then, the phenol was derivatized with various sulfonamide groups by the reaction of a para-substituted arylsulfonyl chloride in the presence of DMAP as catalyst. Compound 21 was used as the starting material in a synthetic route that led to the dimerized compound 40 (Scheme 3). This symmetric dimer was designed for the purpose of probing the proximity of adjacent binding sites on the receptor.22 The nitro group of compound 21 was reduced with NaBH4-copper (II) bisacetylacetonate to an amine, which was subsequently converted to the isothiocyanate in compound 38 by a reaction with thiophosgene. The isothiocyanate group in 38 was reacted with 1,4-diaminobutane to produce the primary amine congener 39. Finally, to make the dimer 40 a R5–R5 linker was created by a coupling reaction between 38 and 39.
Scheme 1.

Synthesis of a representative P2X7 receptor antagonist consisting of a 2,6-dimethyltyrosine derivative. Reagents: (a) benzoyl piperazine, HOBt, EDC.HCl, DMF, rt,8h; (b) p-methylbenzenesulfonyl chloride, DMAP, Et3N, CH2Cl2, rt, 12h
Scheme 3.

Dimerization of P2X7 receptor antagonists through formation of a thiourea linkage. Reagents and conditions: (a) NaBH4, Cu(acac)2, methanol, rt, 80–85%; (b) CSCl2, chloroform, water, sodium bicarbonate, 70–85%; (c) 1,4-diaminobutane, CH2Cl2, rt, 40–45%; (d) dry DMF, at 80 °C, 52%.
The SAR studies of tyrosine-based derivatives 9, 16–36, and 40, which have various substitutions at the toluenesulfonyl, substituted piperazine, and benzyloxycarbonyl (Cbz) groups, were assessed in the ethidium+ accumulation assay. Experiments were performed using HEK293 cells stably transfected with cDNA encoding the hP2X7 receptor. As shown in Table 1, compound 9 with 2,6-dimethyl groups at the phenyl ring of tyrosine displayed a 3-fold increase of the antagonistic activity compared with compound 16. In addition, when the methyl group was adjacent to the benzoyl piperazine group (e.g., 17 and 18), the antagonist effects were also increased by 2-fold compared with compound 16 regardless of the stereochemistry of the chiral carbon of the piperazine group. Thus, the steric effects near the sulfonyl ester of the phenyl group of tyrosine and the benzoyl group of piperazine ring were tolerated in the antagonism of the receptor. However, unlike compound 17 or 18, their regioisomer, 19, contained a methyl group adjacent to the α-carbonyl of the tyrosyl moiety, which reduced the antagonistic effect. In a comparison between compounds 20 and 16, the presence of a methyl group at the tyrosyl α-nitrogen decreased the antagonistic activity by 2-fold at the hP2X7 receptor, which is consistent with previously reported results.23 The 4-nitro group at the R5 position (compound 21) increased the antagonistic effect by 5-fold compared with the corresponding 4-methyl group of 16; compound 21 was slightly more potent than KN-62.
Table 1.
Activities of synthesized tyrosine-based derivatives on ethidium accumulation in hP2X7-expressing HEK293 cells28
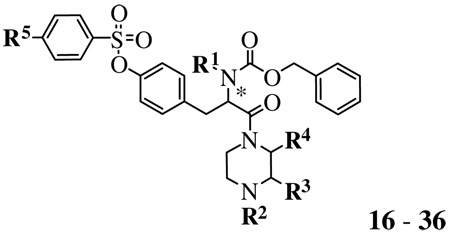 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R/S | R1 | R2 | R3 | R4 | R5 | IC50a ± SE (nM) |
| KN-62 (positive control) | 175 ± 35 | ||||||
| 9b | (S) | H | Ph-CO | H | H | CH3 | 219 ± 69 |
| 16 | (S) | H | Ph-CO | H | H | CH3 | 599 ± 22 |
| 17 | (S) | H | Ph-CO | (S)CH3 | H | CH3 | 260 ± 77 |
| 18 | (S) | H | Ph-CO | (R)CH3 | H | CH3 | 256 ± 96 |
| 19 | (S) | H | Ph-CO | H | (R)CH3 | CH3 | 504 ±136 |
| 20 | (S) | CH3 | Ph-CO | H | H | CH3 | 1360 ± 580 |
| 21 | (S) | H | Ph-CO | H | H | NO2 | 117 ± 44 |
| 22 | (S) | H | p-(CH3)2N-Ph-CO | (S)CH3 | H | CN | 539 ± 90 |
| 23 | (S) | H | p-(CH3)2N-Ph-CO | (S)CH3 | H | I | 55 ± 5%c |
| 24 | (S) | H | p-(CH3)2N-Ph-CO | (S)CH3 | H | CH3 | 54 ± 3%c |
| 25 | (S) | H | p-(CH3)2N-Ph-CO | H | H | CH3 | 2010 ± 340 |
| 26 | (S) | H | p-CF3-Ph-CO | H | H | CH3 | 724 ± 171 |
| 27 | (S) | H | p-F-Ph-CO | H | H | CH3 | 319 ± 65 |
| 28 | (S) | H | p-Br-Ph-CO | H | H | CH3 | 481 ± 41 |
| 29 | (S) | H | Boc | H | H | F | 5880 ± 3280 |
| 30 | (S) | H | Boc | H | H | Br | 517 ± 18 |
| 31 | (S) | H | Boc | H | H | I | 3540 ± 907 |
| 32 | (S) | H | Boc | H | H | CN | 780 ± 107 |
| 33 | (S) | H | Boc | H | H | OCF3 | 725 ±118 |
| 34 | (S) | H | Boc | H | H | CH2Br | 2460 ± 470 |
| 35 | (S) | H | Boc | H | H | CH3 | 1540 ± 102 |
| 36 | (R) | H | Boc | H | H | CH3 | 861 ± 123 |
| 40d | 28 ± 1%c | ||||||
IC50 = 50% inhibitory concentrations were obtained from concentration–response curves. Data values are expressed as means ± SD. All experiments were repeated at least 3–11 times.
This compound has 2,6-dimethyl groups at the phenyl ring of the tyrosyl moiety (see Scheme 1).
The compounds 23, 24, and 40 represented the % inhibition at 10 µM concentration level.
This compound is a dimer linked through formation of a thiourea linkage (see Scheme 3). 16, MRS2427; 21, MRS2447; 27, MRS2544.
The rank order of potency according to IC50 values of para-substituents at the benzoyl group was F (27) > Br (28) > CF3 (26) > (CH3)2N (25). Comparing compounds 17 versus 24 and 16 versus 25 show that a 4-dimethylamino group significantly reduced the antagonistic activity. Thus, basic or bulky groups seem to be inappropriate recognition motifs for the antagonism. Compounds that were Boc-substituted at the R2 position showed different antagonistic effects depending on phenyl sulfonyl ring para-substitution (i.e., R5-substituent) with the following order of potency: Br (30) > OCF3 (33) > CN (32) >> CH3 (35) > CH2Br (34) > I (31) > F (29).
In the study of the effect of chirality of the tyrosine by comparing R- and S-isomers 35 and 36, the R-(d)-isomer was the more potent P2X7 receptor antagonist. The substitution with a d-tyrosyl residue might add stability to the compound in biological systems. The dimeric compound 40 showed only marginal antagonism at a 10 µM concentration.
The functional antagonism of the tyrosine-based derivatives (9, 16–36, and 40) was evaluated with an ELISA for the detection of BzATP-activated IL-1β release in LPS/ IFNγ-differentiated human THP-1 cells (Table 2). Most of the compounds with low submicromolar IC50 values in the ethidium accumulation assay (e.g., 9, 17, 18, 27, 28, etc.) except for 21, resulted in the parallel correlation with the inhibitory activity against the release of IL-1β by BzATP. However, compounds 24 and 31, which had very low antagonistic activity in the ethidium accumulation assay, showed a relatively high inhibitory effect on the release of IL-1β. The methyl group at the R4 position of 19 and the methyl group on the α-nitrogen of 20 seem to affect the inhibitory effect on the release of IL-1β as a comparison between them and compound 16 shows.
Table 2.
Antagonistic effects of tyrosyl derivatives against BzATP-stimulated IL-1β release in LPS/IFNγ-differentiated human THP-1 cells29
| Compound | % inhibitiona | Compound | % inhibition |
|---|---|---|---|
| KN-62 | 83 ± 9 | 26 | 65 ± 6 |
| 9 | 87 ± 8 | 27 | 68 ± 10 |
| 16 | 70 ± 14 | 28 | 78 ± 6 |
| 17 | 69 ± 12 | 29 | 56 ± 13 |
| 18 | 65 ± 3 | 30 | 68 ± 12 |
| 19 | 45 ± 4 | 31 | 72 ± 4 |
| 20 | 47 ± 12 | 32 | 60 ± 8 |
| 21 | 48 ± 7 | 33 | 46 ± 9 |
| 22 | 59 ± 11 | 34 | 52 ± 5 |
| 23 | 61 ± 6 | 35 | 72 ± 15 |
| 24 | 68 ± 7 | 36 | 56 ± 5 |
| 25 | 65 ± 12 | 40 | 37 ± 20 |
The values represent the % inhibition at a 1 µM concentration expressed as means ± SD against 1 mM Bz-ATP (n = 3).
Compounds 9 and 28, which showed inhibitory effects similar to KN-62 against the release of IL-1β by BzATP, were further evaluated with full concentration–response curves to compare IC50 values (Fig. 2). The IC50 values of compounds 9 (481 nM) and 27 (881 nM) were 2- and 4-fold higher than that of KN-62 (188 nM).
Figure 2.

Concentration-dependent inhibition of BzATP-stimulated IL-1β release in LPS/IFNγ-differentiated human THP-1 cells by compounds 9, 28, and KN-62. Data points represent means ± SD of values obtained (n = 3).
In conclusion, a novel series of tyrosine-based P2X7 receptor antagonists with modifications at the piperazine and arylsulfonyl groups were synthesized and evaluated in ethidium accumulation assays and ELISAs of the release of IL-1β. Substitution of the arylsulfonyl moiety with a nitro group, in comparison to a methyl group, increased antagonistic potency in compound 21, which was slightly more potent than KN-62. Several derivatives, including the 2,6-dimethylphenylsulfonate analog 9 and the methylpiperazine analogs 17 and 18, were nearly equipotent to the parent compound KN-62 and their functional inhibitory activity against the release of the inflammatory cytokine, IL-1β, was confirmed. Substitution with d-tyrosine in 36 and sterically bulky tyrosyl 2,6-dimethyl groups in 9 favored P2X7 receptor antagonistic potency.
Scheme 2.

Synthesis of various P2X7 receptor antagonists consisting of tyrosyl derivatives. Reagents and conditions: (a) substituted arylpiperazine or Boc-piperazine, HOBt, EDC·HCl, DMF, rt, 93– 98%; (b) substituted benzenesulfonyl chloride, DMAP, CH2Cl2, rt, 70– 90%.
Acknowledgments
This work was supported by the Korea Research Foundation Grant (KRF-2004-042-E00169) and by the Intramural Program of NIDDK, NIH for Dr. K.A. Jacobson.
References and notes
- 1.Fredholm BB, Abbracchio MP, Burnstock G, Dubyak GR, Harden TK, Jacobson KA, Schwabe U, Williams M. Trends Pharmacol. Sci. 1997;18:79. doi: 10.1016/s0165-6147(96)01038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ralevic V, Burnstock G. Pharmacol. Rev. 1998;50:413. [PubMed] [Google Scholar]
- 3.Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. Science. 1996;272:735. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- 4.Di Virgilio F. Immunol. Today. 1995;16:524. doi: 10.1016/0167-5699(95)80045-X. [DOI] [PubMed] [Google Scholar]
- 5.Sikora A, Liu J, Brosnan C, Buell G, Chessel I, Bloom BR. J. Immunol. 1999;163:558. [PubMed] [Google Scholar]
- 6.Sluyter R, Barden JA, Wiley JS. Cell. Tissue Res. 2001;304:231. doi: 10.1007/s004410100372. [DOI] [PubMed] [Google Scholar]
- 7.Schulman ES, Glaum MC, Post T, Wang Y, Raible DG, Mohanty J, Butterfield JH, Pelleg A. Am. J. Respir. Cell Mol. Biol. 1999;20:530. doi: 10.1165/ajrcmb.20.3.3387. [DOI] [PubMed] [Google Scholar]
- 8.Di Virgilio F, Sanz JM, Chiozzi P, Falzoni S. Prog. Brain Res. 1999;120:355. doi: 10.1016/s0079-6123(08)63569-4. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson KA, Jarvis MF, Williams M. J. Med. Chem. 2002;45:4057. doi: 10.1021/jm020046y. [DOI] [PubMed] [Google Scholar]
- 10.Smart ML, Gu B, Panchal RG, Wiley J, Cromer B, Williams DA, Petrou S. J. Biol. Chem. 2003;278:8853. doi: 10.1074/jbc.M211094200. [DOI] [PubMed] [Google Scholar]
- 11.Denlinger LC, Fisette PL, Sommer JA, Watters JJ, Prabhu U, Dubyak GR, Proctor RA, Bertics PJ. J. Immunol. 2001;167:1871. doi: 10.4049/jimmunol.167.4.1871. [DOI] [PubMed] [Google Scholar]
- 12.Kim M, Jiang LH, Wilson HL, North RA, Surprenant A. EMBO J. 2001;20:6347. doi: 10.1093/emboj/20.22.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Virginio C, MacKenzie A, North RA, Surprenant A. J. Physiol. 1999;519(Pt 2):335. doi: 10.1111/j.1469-7793.1999.0335m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michel AD, Kaur R, Chessell IP, Humphrey PP. Br. J. Pharmacol. 2000;130:513. doi: 10.1038/sj.bjp.0703368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.North RA. Physiol. Rev. 2002;82:1013. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. J. Immunol. 2006;176:3877. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 17.Loomis WH, Namiki S, Ostrom RS, Insel PA, Junger WG. J. Biol. Chem. 2003;278:4590. doi: 10.1074/jbc.M207868200. [DOI] [PubMed] [Google Scholar]
- 18.Mehta VB, Hart J, Wewers MD. J. Biol. Chem. 2001;276:3820. doi: 10.1074/jbc.M006814200. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y. J. Neurosci. 2004;24:1. doi: 10.1523/JNEUROSCI.3792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baraldi PG, Di Virgilio F, Romagnoli R. Curr. Top. Med. Chem. 2004;4:1707. doi: 10.2174/1568026043387223. [DOI] [PubMed] [Google Scholar]
- 21.Clyne CD, Nguyen A, Rainey WE. Endocr. Res. 1995;21:259. doi: 10.3109/07435809509030441. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Ravi RG, Kertesy SB, Dubyak GR, Jacobson KA. Bioconjug. Chem. 2002;13:1100. doi: 10.1021/bc020025i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baraldi PG, Romagnoli R, Tabrizi MA, Falzoni S, Di Virgilio F. Bioorg. Med. Chem. Lett. 2000;10:681. doi: 10.1016/s0960-894x(00)00083-4. [DOI] [PubMed] [Google Scholar]
- 24.Baraldi PG, del Carmen Nunez M, Morelli A, Falzoni S, Di Virgilio F, Romagnoli R. J. Med. Chem. 2003;46:1318. doi: 10.1021/jm021049d. [DOI] [PubMed] [Google Scholar]
- 25.Alcaraz L, Baxter A, Bent J, Bowers K, Braddock M, Cladingboel D, Donald D, Fagura M, Furber M, Laurent C, Lawson M, Mortimore M, McCormick M, Roberts N, Robertson M. Bioorg. Med. Chem. Lett. 2003;13:4043. doi: 10.1016/j.bmcl.2003.08.033. [DOI] [PubMed] [Google Scholar]
- 26.Merriman GH, Ma L, Shum P, McGarry D, Volz F, Sabol JS, Gross A, Zhao Z, Rampe D, Wang L, Wirtz-Brugger F, Harris BA, Macdonald D. Bioorg. Med. Chem. Lett. 2005;15:435. doi: 10.1016/j.bmcl.2004.10.052. [DOI] [PubMed] [Google Scholar]
- 27.Nelson DW, Gregg RJ, Kort ME, Perez-Medrano A, Voight EA, Wang Y, Grayson G, Namovic MT, Donnelly-Roberts DL, Niforatos W, Honore P, Jarvis MF, Faltynek CR, Carroll WA. J. Med. Chem. 2006;49:3659. doi: 10.1021/jm051202e. [DOI] [PubMed] [Google Scholar]
- 28.Ethidium accumulation in hP2X7-expressing HEK 293 cells. All experiments were performed using adherent HEK293 cells stably transfected with cDNA encoding the human P2X7 receptor. Synthesized tyrosine-based derivatives was added to each well of 96-well plate (black, clear bottom).hP2X7-expressing HEK293 cells were then re-suspended at 2.5 × 106 cells/ml in Hepes-buffered salt solution that comprised (in mM): ethidium bromide 0.1, ethylene diamine tetraacetic acid (EDTA) 1, glucose 5, Hepes 20, and potassium chloride 140 (pH 7.4). The cell suspension was treated to the wells of 96-well plate followed by addition of BzATP. The plates were incubated at 37°Cfor 120 min, and cellular accumulation of ethidium+ was determined by measuring fluorescence with a fluorescent plate reader (excitation filter of 530/20 and emission filter of 590/20).
- 29.Inhibition effects of BzATP-mediated IL-1β release by ELISA measurements. IL-1β release was measured in differentiated THP-1 cells primed for 3 h with 25 ng/ml LPS and 10 ng/ml IFNγ, and then was stimulated with 1 mM BzATP for 30 min. Synthesized tyrosine-based derivatives at 1 µM were treated for 30 min prior to BzATP. Supernatants were collected by centrifugation at 1000 rpm for 5 min and assayed for the presence of mature human IL-1β using an ELISA kit.