

Abstract
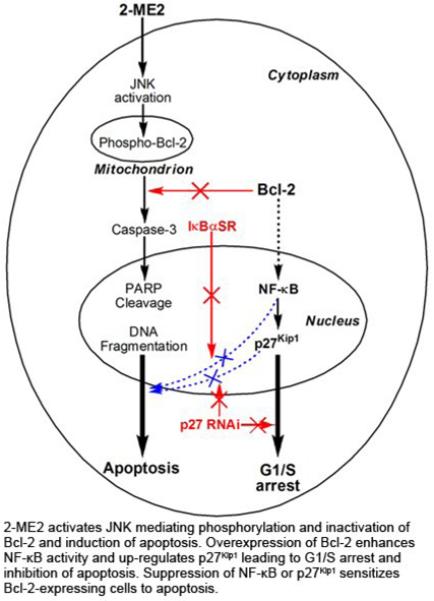
2-Methoxyestradiol (2-ME2) induces leukemia cells to undergo apoptosis in association with Bcl-2 inactivation but the mechanisms whereby Bcl-2 contributes to protection against programmed cell death in this context remain unclear. Here we showed that 2-ME2 inhibited the proliferation of Jurkat leukemia cells by markedly suppressing the levels of cyclins D3 and E, E2F1 and p21Cip1/Waf1 and up-regulating p16INK4A. Further, 2-ME2 induced apoptosis of Jurkat cells in association with downregulation and phosphorylation of Bcl-2 (as mediated by JNK), up-regulation of Bak, activation of caspases-9 and -3 and PARP-1 cleavage. To determine the importance and mechanistic role of Bcl-2 in this process we enforced its expression in Jurkat cells by retroviral transduction. Enforcing Bcl-2 expression in Jurkat cells abolished 2-ME2-induced apoptosis and instead produced a G1/S phase cell cycle arrest in association with markedly increased levels of p27Kip1. Bcl-2 and p27Kip1 were localized mainly in the nucleus in these apoptotic resistant cells. Interestingly NF-κB activity and p50 levels were increased by 2-ME2 and suppression of NF-κB signaling reduced p27Kip1 expression and sensitized cells to 2-ME2-induced apoptosis. Importantly, knocking-down p27Kip1 in Jurkat Bcl-2 cells sensitized them to spontaneous and 2-ME2-induced apoptosis. Thus, Bcl-2 prevented the 2-ME2-induced apoptotic response by orchestrating a p27Kip1-dependent G1/S phase arrest in conjunction with activating NF-κB. Thus, we achieved a much better understanding of the penetrance and mechanistic complexity of Bcl-2 dependent anti-apoptotic pathways in cancer cells and why Bcl-2 inactivation is so critical for the efficacy of apoptosis and anti-proliferative inducing drugs like 2-ME2.
Keywords: 2-Methoxyestradiol (2-ME2), Bcl-2, p27Kip1, NF-κB, cell cycle, apoptosis
1. Introduction
2-Methoxyestradiol (2-ME2), a catechol estrogen, is a natural metabolic byproduct of 17β-estradiol that acts independently of estrogen receptors to inhibit angiogenesis and tumor cell proliferation and to induce apoptosis in vitro and in vivo [1-8].
Multiple discrete mechanisms are involved in the specific anti-proliferative action of 2-ME2 in tumor cells including both G1/S and G2/M cell cycle phase arrest. 2-ME2 was shown to arrest the growth of many human cancer cell lines in vitro, including, Jurkat cells [9], multiple myeloma [10], epithelial [11-16], melanoma [17] and medulloblastoma cancer cells [18] and transformed fibroblasts [19] in G2/M phase. G2/M cell cycle arrest was characterized by the induction of cyclin B and Cdc2 kinase activity [9,11,13,17]. Others, however, showed that 2-ME2 inhibited the growth of pancreatic cancer cells by prolonging S-phase [20] or by inducing both G1/S and G2/M arrest of human osteosarcoma cells [21] or of pancreatic cell lines [22]. In contrast, 2-ME2 had no effect on the growth of normal cells [13,15,17,19,23] including lymphocytes [24]. The induction of apoptosis by 2-ME2 in tumor cells involves different molecular mechanisms. While several studies suggested that 2-ME2 can induce apoptosis both by p53-dependent and p53-independent mechanisms in various tumor cell types [8,13,15,17-19 ,23,25,26], scant evidence exists implicating NF-κB in 2-ME2-induced apoptosis [18,27]. While p38/JNK-dependent NF-κB activation was required for 2-ME2-induced apoptosis in prostate cancer cells [27], in contrast a reduction in NF-κB transcriptional and DNA binding activity was observed in 2-ME2-induced apoptosis of medulloblastoma cells [18].
Further studies have implicated the anti-apoptotic members of the Bcl-2 family in 2-ME2-induced apoptosis [15,27-30]. The Bcl-2 family comprises two mutually opposing groups of proteins including: anti-apoptotic Bcl-2 and Bcl-XL and pro-apoptotic Bak and Bax. While several models have ben proposed to explain the mechanism by which Bcl-2 family members regulate apoptosis, the ratio of anti-apoptotic:pro-apoptotic Bcl-2 family members is one key factor dictating the relative sensitivity or resistance of cells to a wide variety of apoptotic stimuli [31-33]. In addition, Bcl-2 and Bcl-XL are regulated by phosphorylation in their flexible loop between the BH4 and BH3 domains, which determines their cytoprotective function in response to cellular stresses as well as growth and survival factors [34]. Bcl-2 phosphorylation by ERK1/2 and PKCα kinases, either at the unique Ser70 residue or at multiple Thr69, Ser70, and Ser89 sites, positively regulates Bcl-2 anti-apoptotic function [35]. However, c-jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK)-mediated phosphorylation of Bcl-2 at multiple-sites hinders Bcl-2 survival function in paclitaxel-induced apoptosis [36,37]. Thus, the type of stimulus, the regulatory pathways involved, and the degree and duration of phosphorylation at specific Bcl-2 residues produce different outcomes.
In response to 2-ME2, both Bcl-2 [15,27-29] and Bcl-XL [22,30] are inactivated by phosphorylation at Ser70 and Ser62, respectively, mediated by JNK but not ERK1/2 [27,29,30,37]. Whether Bcl-2 phosphorylation induced by microtubule destabilizing agents such as taxol [37-39] or 2-ME2 [15,27-29] interferes with the heterodimerization of Bcl-2 to Bax remains elusive [37-39]. However, JNK-mediated phosphorylation of Bcl-2 leading to its inactivation in response to 2-ME2 will allow the pro-apoptotic members of the Bcl-2 family, to drive the cell towards death [31-33]. 2-ME2-induced phosphorylation of Bcl-2, mediated by JNK/SAPK, has been correlated with apoptosis of prostate [15,27] and leukemia cells [29]. Activation of JNK by 2-ME2 appears to be due to its ability to potently inhibit superoxide dismutase [40] resulting in enhanced formation of ROS [7,24-26,40] and Akt inhibition [26] to selectively kill tumor cells [24-26,40].
Although available data point to Bcl-2 phosphorylation as a key executing signal for 2-ME2-induced apoptosis, Bcl-2’s mechanisms of action in this context and its ability to protect cells from 2-ME2-induced apoptosis both remain undefined. We show here that 2-ME2 treatment of leukemia cells promoted a p53-independent apoptotic response characterized by Bcl-2 downregulation and phosphorylation mediated by JNK/SAPK, Bak up-regulation, proteolytic cleavages of caspases-9, -3 and PARP-1. Moreover, ectopic over-expression of Bcl-2 in leukemia cells prevented all of these aspects of the 2-ME2-induced apoptotic response by orchestrating a p27Kip1-dependent G1/S phase arrest in conjunction with activating NF-κB.
2. Materials and methods
2.1. Cell culture
Human Jurkat T lymphoma cells (clone E6-1) were cultured in RPMI-1640 and amphotropic Phoenix cells in DMEM supplemented with 10% FCS, (all from Biochrom AG, Germany), 2 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin (PAA Laboratories, Germany) at 37°C, 5% CO2.
2.2. Retroviral vectors and generation of infected Jurkat cells
The retroviral vectors pBabe-Puro and pBabe-Puro/Bcl-2 carrying human Bcl-2 cDNA [41] pSR-Puro (referred to as Vector thereafter) and pSR-Puro carrying p27Kip1 shRNA (referred to as p27KD, thereafter) [42] have been described elsewhere. IBIN, was a retroviral vector carrying a trans-dominant IκBα (S32A/S36A) super repressor (IκBαSR), with serines 32 and 36 mutated to alanines, in addition to Neo as a selection marker was described previously [43].
Jurkat cells were infected overnight in suspension with high titers of retroviruses generated using amphotropic phoenix cells. The infected Jurkat cells were centrifuged, the virus-containing supernatant was aspirated and the cell pellets were washed once in serum-free RPMI-1640 and incubated in complete RPMI-1640 growth medium for 48 h before being subjected to selection with 1 μg/ml puromycin (Sigma Co., Germany) for two weeks or to 0.5-1.0 mg/ml G418 (Geneticin) (Gibco/Invitrogen, UK) for three weeks.
2.3. Treatment of Jurkat cells with 2-methoxyestradiol
All Jurkat cells plated at 1.5 × 106 cells per 10 cm dish were treated with 0.5 -10 μM 2-ME2 (Sigma Co., Germany), dissolved in ethanol or with ethanol vehicle as control for different periods of time. Treated and untreated cells were harvested and DNA and proteins were extracted and analyzed as described below.
2.4. Growth of uninfected and retrovirus-infected Jurkat cells
Cells were plated at a density of 1.0 × 105 cells per well in 24-multiwell plates in complete growth medium in the presence or absence of 1.0 μM 2-ME2 or ethanol. Cell growth was monitored over a period of 9 days. The experiment was repeated three times and growth curves were constructed.
2.5. Flow cytometric analysis
1.5 × 106 exponentially growing Jurkat cells were incubated in complete growth medium in the presence or absence of 0.5 μM and 1.0 μM 2-ME2 or ethanol for 12 h and 24 h. The cells were collected by centrifugation, and processed for flow cytometric analysis on a Becton Dickinson FACScan flow cytometer as described previously using a CycleTEST PLUS DNA kit (Becton Dickinson, CA, USA), according to manufacturer’s instructions.
2.6. DNA fragmentation assay
1.5 × 106 Jurkat cells grown in complete growth medium were treated with increasing concentrations of 2-ME2 or ethanol for different periods of time. Low molecular weight DNA was extracted and analyzed as described previously [44].
2.7. Quantification of apoptosis using annexin V-PI
Simultaneous flow cytometric quantification of apoptotic and viable cells was performed with an annexin V/propidium iodide kit (Annexin V-FITC/PI) (Assay Designs, Ann Arbor, Michigan, USA). Untreated and 2-ME2-treated Jurkat cells were collected by centrifugation, resuspended in 1X binding buffer, incubated with annexin-FITC and propidium iodide and analyzed by flow cytometry (CyFlow ML, Partec, Germany) according to the manufacturer’s instructions.
2.8. Preparation of cytoplasmic and nuclear extracts and isolation of nuclei
Cytoplasmic and nuclear extracts were prepared essentially as described previously [45]. Nuclei were isolated through sucrose gradients essentially as described previously [46]. Protein concentration was determined using a BioRad protein assay reagent. The extracts were stored at -80°C or used immediately on western blots.
2.9. Isolation of total proteins and western blot analysis
1.5 × 106 Jurkat cells grown in complete growth medium were treated with 0.5 μM and 1.0 μM 2-ME2 or ethanol for 24 h. The cells were collected by centrifugation and total proteins were extracted and analyzed by western immunoblotting, as described previously [44]. Antibodies used were: Mouse monoclonals to Bcl-2 (sc-509), cyclin D3 (sc-6283), E2F1 (sc-251), pRb (sc-102), p16INK4A (sc-1661) (Santa Cruz Biotech, Germany), p21Cip1/Waf1 (Ab-1, OP64), p27Kip1 (NCL-p27; Novacatsra Ltd or M-7203; DakoCytomation, Denmark), caspase-8 (sc-5263), PARP-1 (CII-10; BD Transduction Laboratories), caspase-3 (sc-7272) and β-actin (A5441; Sigma Co, Germany), rabbit polyclonals to JNK/SAPK (#9252) and phospho-JNK/SAPK(Thr182/Tyr185) (#9251) (Cell Signaling, USA), Bak (A3538; Dako), cyclin E (sc-481), caspase-9 (sc-8355), NF-κB p65 (sc-372) and lamin B and goat polyclonals to NF-κB p50 (sc-1190) and Pim-2 (ab13616; Abcam Ltd, UK), followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Antibody binding was detected using an ECL detection kit (GE HealthCare, Bucks, UK).
3. Results
3.1. Induction of Jurkat cell apoptosis by 2-ME2
Actively proliferating Jurkat cells were treated with different concentrations of 2-ME2 ranging from 0.5 μM to 10 μM or ethanol as a negative control for 24 and 48 h and low molecular weight DNA was extracted and analyzed by agarose gel electrophoresis (Fig. 1A). Whereas ethanol-treated cells did not undergo apoptosis, 2-ME2-treated Jurkat cells exhibited DNA fragmentation, producing a DNA ladder characteristic of cells undergoing apoptosis even with 2-ME2 as low as 0.5-1.0 μM. Based on the densities of DNA fragmentation patterns, the extent of apoptosis appeared to be higher in cells treated with 2-ME2 for 48 h vs. 24 h. (Fig. 1A). Moreover, Jurkat cells were treated with 0.5 μM and 1.0 μM 2-ME2 or ethanol for 0 - 24 h exhibited a marked induction of apoptosis at 8 h, which became more pronounced at 12 and 24 h at both concentrations (Fig. 1B).
Fig. 1.

Induction of apoptosis of Jurkat cells by 2-ME2. (A) Dose-dependent induction of apoptosis of Jurkat cells. 1.5 × 106 cells were treated with increasing concentrations of 2-ME2 for 24 and 48 h as indicated or ethanol (Et) as control and low molecular weight DNA was extracted and analyzed by agarose gel electrophoresis. (B) Time-dependent apoptosis of Jurkat cells. 1.5 × 106 cells were treated with 0.5 and 1.0 μM of 2-ME2 or Et for different periods of time as indicated and low molecular weight DNA was extracted and analyzed by agarose gel electrophoresis. (C) Analysis of apoptosis of asynchronous cultures of Jurkat cells treated with 2-ME2 as determined by flow cytometry. Cells treated with 0.5 and 1.0 μM of 2-ME2 or Et for 12 and 24 h were subjected to flow cytometric analysis as described under materials and methods. Results were reproduced in two independent experiments.
Apoptosis of Jurkat cells treated with 2-ME2 was further evaluated by flow cytometry (Fig. 1C). Jurkat cells treated with 2-ME2 for 12 h or 24 h exhibited a decrease in the G1 population due to the appearance of a considerable fraction of 2-ME2-treated cells as a sub-G1 population indicative of 2-ME2-induced apoptosis. The sub-G1 cell population undergoing apoptosis increased from 2.2% in the ethanol-treated cells to 24% and 26.3% following treatment of the cells with 0.5 μM and 1.0 μM 2-ME2 for 12 h, respectively (Fig. 1C). 2-ME2 treatment at 0.5 μM and 1.0 μM respectively for 24 h produced much higher proportions of 39.6% and 42.2% of sub-G1 apoptotic cells vs only 5% for ethanol treated controls. Collectively, these data show that 2-ME2 induced apoptosis of Jurkat cells in a time- and dose-dependent manner.
3.2. Enforced Bcl-2 expression suppressed 2-ME2-induced apoptosis
Next we directly investigated the effects of Bcl-2 in this response by comparing the properties of Jurkat cells stably transduced with a human Bcl-2 retroviral vector vs. a puromycin (Puro) empty vector control. Western blot analysis showed that the Bcl-2-transduced cells expressed the 26 kDa human Bcl-2 protein at much higher levels than parental Jurkat or Jurkat Puro control cells (Fig. 2A). Jurkat Puro and Jurkat Bcl-2 cell populations were treated with 2-ME2 or ethanol for 24 h and their genomic DNA was subjected to agarose gel electrophoresis (Fig. 2B). Whereas 2-ME2 induced apoptosis in a dose-dependent manner in the control cells, enforced Bcl-2 expression blocked this apoptotic response.
Fig. 2.

Bcl-2 suppressed Jurkat cell apoptosis induced by 2-ME2 through the mitochondrial pathway. (A) Expression of Bcl-2 in retrovirally-transduced Jurkat cells. Total proteins extracted from Jurkat, Jurkat Puro and Jurkat Bcl-2 cells were analyzed by immunobloting for the expression of Bcl-2. (B) DNA fragmentation analysis of uninfected and retrovirus-infected Jurkat cells. DNA isolated from untreated and ethanol- or 2-ME2-treated Jurkat, Jurkat Puro and Jurkat Bcl-2 cells was analyzed by agarose gel electrophoresis. (C) Expression of Bcl-2 and Bak in control and Puro- or Bcl-2-expressing cells following treatment with 2-ME2. Total proteins isolated were analyzed by immunoblotting for the expression of Bcl-2- or Bak. In the Bcl-2 blot, the upper slower migrating band represents the phosphorylated form of Bcl-2. (D) Expression of total and phospho-JNK (Thr183/Tyr185) in control and Puro- or Bcl-2-expressing cells following treatment with 2-ME2. (E) Expression of caspases and PARP-1. Total proteins isolated as in (C) were analyzed for the expression of caspase-9, caspase-3 and PARP-1 or β-actin. All experiments were repeated 2-3 times with comparable results.
One of the likely mechanisms of 2-ME2-induced apoptosis involves the phosphorylation and inactivation of the anti-apoptotic Bcl-2 protein [15,27-29]. As shown in Fig. 2C, 2-ME2 downregulated and triggered the phosphorylation of Bcl-2 in Jurkat and Jurkat Puro cells, compared to their untreated or ethanol-treated controls, thus confirming these previous reports [15,27-29]. Although phosphorylated Bcl-2 protein was also present in 2-ME2-treated Jurkat Bcl-2 cells, its level of expression remained high following 2-ME2 exposure (Fig. 2C). Thus, because most of the overexpressed Bcl-2 was not phosphorylated, this could in part explain why the additional Bcl-2 was protective against apoptosis. Because Bcl-2 phosphorylation inactivates the protein and may interfere with its dimerization with pro-apoptotic partner proteins [36-38], we next investigated the expression of the pro-apoptotic Bcl-2 family member, Bak, which was previously shown to play a key role in Jurkat cell apoptosis [47,48]. Western blot analysis showed that while Bak expression was modestly induced in Jurkat and Jurkat Puro cells, it remained unaffected in Jurkat Bcl-2 cells (Fig. 2C). Thus the Bcl-2:Bak ratio was shifted towards the former in Jurkat Bcl-2 cells.
Because JNK/SAPK-mediated phosphorylation of Bcl-2 inactivates its anti-apoptotic function [27,29,30,36,37], we also investigated the expression of total and phospho-JNK/SAPK by immunoblotting (Fig. 2D). Indeed, 2-ME2 induced JNK activation, as revealed by enhanced amounts of phosphorylated p46 and p54 forms of JNK/SAPK in Jurkat and Jurkat Puro cells and phospho-p54 in Jurkat Bcl-2 cells. In response to 2-ME2, these latter activated JNKs correlated well with Bcl-2 phosphorylation (Fig. 2C), but only p54 phosphorylation was enhanced in 2-ME2 treated Jurkat Bcl-2 cells (Fig. 2D).
Next to further elaborate the mode of action of Bcl-2 in this context, we examined the status of caspases-9, -8 and -3 and PARP-1. Western blot analysis detected two bands of ~50 kDa and 35 kDa corresponding to caspase-9 (Fig. 2E). While treatment of Jurkat and Jurkat Puro cells with 2-ME2 resulted in a reduction in the levels of p35 compared to their untreated or ethanol-treated cells, no change in the levels of p35 were detected in 2-ME2-treated Jurkat Bcl-2 cells (Fig. 2E). In contrast, no change in the expression levels of caspase-8 was detected using two different antibodies (data not shown). Immunoblotting also revealed that procaspase-3 was in its 32 kDa unprocessed form in untreated Jurkat and Jurkat Puro cells, but 2-ME2-treated cells presented procaspase-3 as 20 kDa and 17 kDa cleaved forms in addition to the inactive 32 kDa proform (Fig. 2E). In contrast, caspase-3 in 2-ME2-treated Bcl-2-expressing Jurkat cells remained in its inactive 32 kDa proform (Fig. 2E). Cleavage of poly-(ADP ribose)-polymerase (PARP-1), a caspase-3 substrate, is also a hallmark of cells undergoing apoptosis by a variety of apoptotic stimuli. Immunoblot analysis showed that whereas untreated and ethanol-treated Jurkat and Jurkat Puro cells expressed the intact 115 kDa enzyme, cells treated with 2-ME2 contained the intact 115 kDa band and an 89 kDa cleavage product (Fig. 2E). In contrast, in untreated, ethanol-treated or 2-ME2-treated Jurkat-Bcl-2 cells only the intact 115 kDa form of PARP-1 was detected (Fig. 2E). Taken together these data showed that 2-ME2-induced apoptosis by the mitochondrial pathway was effectively blocked by ectopic expression of Bcl-2 in Jurkat cells.
3.3. Bcl-2-mediated apoptotic block correlated with G1/S cell cycle arrest following 2-ME2 treatment
To investigate whether overexpression of Bcl-2 affected the proliferation of Jurkat cells in the presence or absence of 2-ME2, growth curves were constructed (Fig. 3A). Overexpression of Bcl-2 in Jurkat cells altered their growth rate compared to their vector-infected (Jurkat Puro) or uninfected (Jurkat) counterparts. Jurkat Bcl-2 cells grew slower than either Jurkat or Jurkat Puro cells (Fig. 3A) with a doubling time of 27.5 hr compared to 20.8 hr and 20.9 hr for their control counterparts, respectively. In contrast, cells treated with 2-ME2 presented a very different growth profile. While 2-ME2 treated Jurkat and Jurkat Puro cells decreased dramatically in cell numbers due to apoptosis, the cell numbers of Bcl-2-expressing Jurkat cells remained constant over a period of 9 days (Fig. 3A).
Fig. 3.

Bcl-2-mediated apoptotic block is linked to G1/S cell cycle arrest following treatment with 2-ME2. (A) Growth curves of uninfected and retrovirus-infected Jurkat cells in the presence or absence of 2-ME2, over a period of nine days. Each point represents the average of two wells in three independent experiments. (B) Flow cytometric analysis of asynchronous cultures of Puro- and Bcl-2-expressing Jurkat cells treated with 2-ME2. Jurkat Puro and Jurkat Bcl-2 cells treated with 2-ME2 or ethanol (Et) for 12 and 24 h were subjected to flow cytometric analysis. (C) Evaluation of apoptosis by annexin V/PI staining. Jurkat Puro and Jurkat Bcl-2 cells were treated with ethanol or 1.0 μM 2-ME2 for 24 h and subjected to annexin V/PI staining. Black bars represent viable cells and grey bars represent apoptotic cells.
Next we performed annexin V/PI staining to simultaneously quantify apoptotic and live cells (Fig. 3B) by flow cytometric analysis (Fig. 3C). Annexin V/PI staining showed that Jurkat Puro cells exhibited a dose-dependent decrease in live cells and an increase in the percentage of apoptotic cells from 5.8% to 34.35% and 37.25% after treatment with ethanol, 0.5 μM and 1.0 μM 2-ME2 for 24 h, respectively (Fig. 3B). In contrast, Jurkat Bcl-2 were more resistant to 2-ME2 treatment maintaing a higher percentage of live cells and exhibiting an increase in the percentage of apoptotic cells from 5.4% to 14.56% and 15,24% after treatment with ethanol or 0.5 μM and 1.0 μM 2-ME2 for 24 h, respectively (Fig. 3B).
Flow cytometric analysis of control and 2-ME2 treated Jurkat Puro and Bcl2 cells were in agreement with the above annexin V/PI results. For Jurkat Puro cells the sub-G1 peak specifiying apopotic cells increased from 5% in the ethanol control to 27.8% and 33.4% after 12 h in 0.5 and 1.0 μM 2-ME2 respectively, and from 3.6% in to 38.5% and 40.4% in response to the same 2-ME2 doses for 24 h (Fig. 3C). In sharp contrast, Bcl-2 over-expression protected Jurkat cells from 2-ME2 inducded apoptosis with their sub-G1 population increasing from 4% to only 5.7% and 13% after 12 h in to 0.5 and 1.0 μM 2-ME2 and up to 14.8% and 16.1% in response to 24 h of 0.5 and 1.0 μM 2-ME2 (Fig. 3C). Moreover, Bcl-2 also induced G1/S cell cycle phase arrest following treatment with 2-ME2 for 24 h compared to their control Jurkat Puro cells (Fig. 3C), consistent with cell growth curves (Fig. 3A).
Next, we screened for effects on effectors of cell cycle progression and apoptosis (including cyclins D3 and E, E2F1. pRb, p16INK4A, p21Cip1/Waf1 and p27Kip1) by immuoblotting (Fig. 4). Cyclin D3 levels were downregulated by 2-ME2 in Jurkat and Jurkat Puro cells, but were maintained in Bcl-2-expressing cells. In contrast, the levels of cyclin E and E2F1, but not pRb, were downregulated in all cell types in response to 2-ME2 (Fig. 4). 2-ME2 treatment of Jurkat cells also had differential effects on cyclin-dependent kinase inhibitors. p16INK4A expression was markedly induced by 2-ME2 in control cells, which was prevented by Bcl-2 over-expression. Both CKIs p21Cip1/Waf1 and p27Kip1 were detected in all cell types with p27Kip1 expressed at higher levels. 2-ME2 downregulated and induced the phosphorylation of p21Cip1/Waf1 in each Jurkat cell type. However, p27Kip1 levels remained unaffected by 2-ME2, although Jurkat Bcl-2 cells expressed ~2-3 fold more p27Kip1 compared to Jurkat or Jurkat Puro cells (Fig. 4).
Fig. 4.
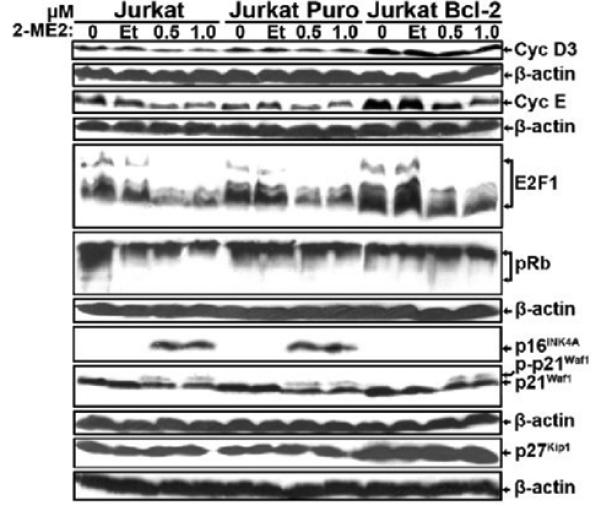
Expression of cell cycle regulatory proteins in control and Puro- or Bcl-2-expressing Jurkat cells following treatment with 2-ME2. Cells were left untreated or treated with ethanol, or 0.5 μM and 1.0 μM 2-ME2 for 24 h. Total proteins isolated from untreated and ethanol- or 2-ME2-treated cells were analyzed by immunoblotting for the expression of cyclin D3, cyclin E, E2F1, pRb p16INK4A, p21Cip1/Waf1 and p27Kip1 or β-actin. Experiments were repeated 2-3 times with comparable results.
3.4. Nuclear-associated Bcl-2 enhanced the nuclear levels of p27Kip1
Because p27Kip1 was not altered by 2-ME2 and enhanced Bcl-2 expression has been associated with increased levels of p27Kip1 [53-55] (Fig. 4), we investigated the subcellular localization of Bcl-2 and p27Kip1 by indirect immunofluorescence and confocal laser scanning microscopy (supplementary information and Fig. 1S). Ethanol-treated Jurkat Puro cells expressed low levels of predominantly nuclear Bcl-2. However, after 2-ME2 treatment, Bcl-2 was detected in the nuclear compartment and outside the cell nucleus due to cell damage and nuclear disruption. In contrast, untreated Jurkat Bcl-2 cells expressed much higher levels of cytoplasmic and nuclear Bcl-2 protein (Fig. S1A). Following 2-ME2 treatment, Bcl-2 protein was still cytoplasmic but higher levels became associated with the nuclear membrane and nuclear matrix of intact nuclei (Fig. S1).
p27Kip1 was predominantly localized in the cytoplasm, around and loosely-associated with intact nuclei of control Jurkat Puro cells (Fig. S1B) and its cytoplasmic localizaton was disrupted by 2-ME2 treatment due to their compromised cell and nuclear integrity (Fig. S1B). In ethanol-control Jurkat Bcl-2 cells p27Kip1was strongly upregulated in the cytoplasm and was also more tightly-associated with intact nuclei (Fig. S1B). After 2-ME2 exposure Jurkat Bcl-2, p27Kip1 was still present in the cytoplasm but now at markedly higher levels in Jurkat Bcl-2 intact nuclei (Fig. S1B).
Proteins from sucrose gradient purified cell fractions of untreated, ethanol- or 2-ME2-treated Jurkat Puro and Jurkat Bcl-2 cells were also subjected to Bcl-2 immunobloting. Bcl-2 was predominantly nuclear in Jurkat Puro and Jurkat Bcl-2 cells (Fig. 5A). In response to 2-ME2, Phosphorylated Bcl-2 form was detected in cytoplasmic and nuclear extracts of both cell types and cytoplasmic Bcl-2 levels were reduced in control Jurkat Puro cells in response to 2-ME2 (Fig. 5A), suggesting differential regulation of these two different intracellular pools of Bcl-2 in Jurkat cell apoptosis [50]. Jurkat Bcl-2 cells presented higher levels of cytoplasmic and especially nuclear Bcl-2 protein in comparison to Jurkat Puro cells (Fig. 5A), which was also observed in nuclear extracts of Jurkat Puro and Jurkat Bcl-2 cells (Fig. 5B). Moreover the expression of nuclear phosphorylated and unphosphorylated forms of Bcl-2 was much higher in Bcl-2 over-expressing cells.
Fig. 5.
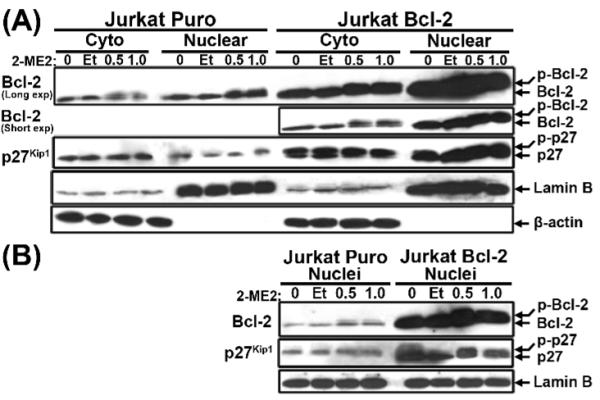
Subcellular expression of Bcl-2 and p27Kip1 in Puro- or Bcl-2-expressing Jurkat cells following 2-ME2 treatment. Cytoplasmic and nuclear fractions (A) or Nuclei (B) were isolated from Jurkat Puro and Jurkat Bcl-2 cells cultured in the absence or presence of ethanol or 2-ME2. The fractions were analyzed by immunoblotting for the expression of Bcl-2 or p27Kip1. Equal loading and verification of extracts was detected using an anti-Lamin B or anti-β-actin. No actin was detected in nuclear extracts or from isolated nuclei. All experiments were repeated multple times with similar results.
p27Kip1 immunobloting of proteins from specific cell fractions and isolated nuclei, confirmed that 2-ME2 did not affect p27Kip1 levels in either Jurkat Puro or Jurkat Bcl-2 and also that p27Kip1 expression was much higher in Jurkat Bcl-2 (Figs 5A and B). p27Kip1 was predominantly cytoplasmic in Jurkat Puro cells but in contrast was more prevalent in the nuclear extract of Jurkat Bcl-2 cells (Fig. 5A). The latter was in keeping with subcellular localization results (Fig. S1B) and was also confirmed with isolated nuclei of Jurkat Puro and Jurkat Bcl-2 cells (Fig. 5B). In addition, p27Kip1 migrated as a doublet band (particularly in Jurkat Bcl-2 cells), which may represent different sites of phosphorylation associated with either protein degradation (Thr187) or stabilization (Ser10) [56] (Figs 5A and B).
3.5. Involvement of NF-κB in the resistance of Jurkat Bcl-2 cells to 2-ME2
Although Bcl-2 overexpression has been associated with enhanced NF-κB activity [57-61], NF-κB’s contribution to 2-ME2-induced apoptosis has remained elusive [18,27]. To investigate the effects of 2-ME2 on NF-κB signaling, total cell lysates, cytoplasmic and nuclear extracts and proteins from isolated nuclei were analyzed by immunoblotting for expression of IκBα and canonical p50 and p65 NF-κB subunits (Fig. 6). No changes in the expression of the total levels of either p50 or p65 were detected in Jurkat, Jurkat Puro or Jurkat Bcl-2 cells in the absence or presence of 2-ME2 (Fig. 6A). However, after exposure to 2-ME2, phosphorylated p65 (Thr536) and phospho-IκBα levels increased in conjunction with IκBα being reduced in all Jurkat cell types. Total and phospho-IκBα levels were much lower in untreated and 2-ME2-treated Jurkat Bcl-2 than in Jurkat Puro cells most likely due to IκBα’s rapid phosphorylation and degradation in the context of Bcl-2 overexpression (Fig. 6A). Thus NF-κB was activated by 2-ME2 treatment and NF-κB was also more active in Jurkat Bcl-2 cells than their control counterparts. While both Jurkat Puro and Jurkat Bcl-2 cells contained much higher levels of p50 in nuclear than in cytoplasmic extracts, the opposite was observed for p65 (Fig. 6B) and the nuclear levels of p50 were higher in Jurkat Bcl-2 than in Jurkat Puro cells even in the presence of 2-ME2 (Figs 6B and S2). To verify these results, total proteins from isolated nuclei were probed for the expression of NF-κB p50 and p65 (Fig. 6C). Indeed, Jurkat Bcl-2 cells expressed higher nuclear levels of NF-κB p50 but the basal and 2-ME2-induced levels of nuclear p65 were similar to those detected in Jurkat Puro cells (Fig. 6C). The higher nuclear-associated expression of Bcl-2 in Jurkat Bcl-2 cells could have facilitated p50 nuclear transport. However, no specific binding of Bcl-2 to either p50 or p65 subunits was detected (data not shown), suggesting that higher levels of nuclear p50 in Jurkat Bcl-2 cells were most likely inidrecty due to their maintenance of nuclear membrane integrity. Confocal laser scanning microscopy showed that Jurkat Bcl-2 cells specifically contained higher levels of p50-p65 heterodimers in their nuclei and also that 2-ME2 induced the accumulation p50 and p65 in the nuclei of the few undamaged 2-ME2-treated Jurkat Puro cells and in the intact nuclei of most Jurkat Bcl-2 cells (supplementary information and Fig. S2).
Fig. 6.

Expression of NF-κB and Pim-2 in Puro- and Bcl-2-expressing Jurkat cells following 2-ME2 treatment. Total proteins (A), Cytoplasmic and nuclear fractions (B) or Nuclei (C) isolated from untreated, ethanol- or 2-ME2-treated Jurkat Puro and Jurkat Bcl-2 cells were subjected to immunoblotting for the expression of the indicated proteins. (D) Expression of Pim-2, a NF-κB target protein. Total proteins isolated from untreated and ethanol- or 2-ME2-treated Jurkat Puro and Jurkat Bcl-2 cells were analyzed by immunoblotting for the expression of Pim-2. Equal loading and verification of extracts was detected using a rabbit polyclonal anti-Lamin B or a mouse monoclonal anti-β-actin. Experiments were repeated twice producing similar results.
To investigate whether the higher levels of nuclear NF-κB p50-p65 heterodimers in Jurkat Bcl-2 cells correlated with increased NF-κB transcriptional activity, total cells lysates from untreated and 2-ME2-treated Jurkat Puro and Jurkat Bcl-2 cells were probed for the expression of Pim-2, a serine/threonine kinase and a direct NF-κB target gene with putative anti-apoptotic properties [43,62] (Fig. 6D). Whereas, Pim-2 expression was downregulated in Jurkat Puro in response to 2-ME2, its expression was sustained in Jurkat Bcl-2 cells (Fig. 6D), in agreement with the activated state of NF-κB in these cells as also shown by confocal microscopy (Fig. S2).
3.6. Suppression of canonical NF-κB activity sensitized Jurkat cells to apoptosis
In order to further confirm that NF-κB activity contributed to increased resistance of Jurkat Bcl-2 to 2-ME2-induced apoptosis, an IκBα super repressor (IκBαSR) [43] was used to suppress NF-κB signaling pathway in both Jurkat and Jurkat Bcl-2 cells. As expected cells retrovirally transduced with IκBαSR expressed higher levels of IκBα compared to their control counterparts (Fig. 7A). As the human p27Kip1 gene promoter contains an NF-κB-responsive element [63], we investigated whether there was a link between NF-κB and p27Kip1 expression, which both contribute to increased resistance of cells to apoptosis. The levels of p27Kip1 and Pim-2 were reduced, albeit modestly, in proliferating Jurkat Bcl-2/IκBαSR compared to Jurkat Bcl-2 cells but such reductions were not as obvious in confluent cultures (Fig. 7B). Furthermore, DNA analysis showed that IκBαSR expressing Jurkat Bcl-2 cells were more sensitive to 2-ME2-induced apoptosis (Fig. 7C).
Fig. 7.
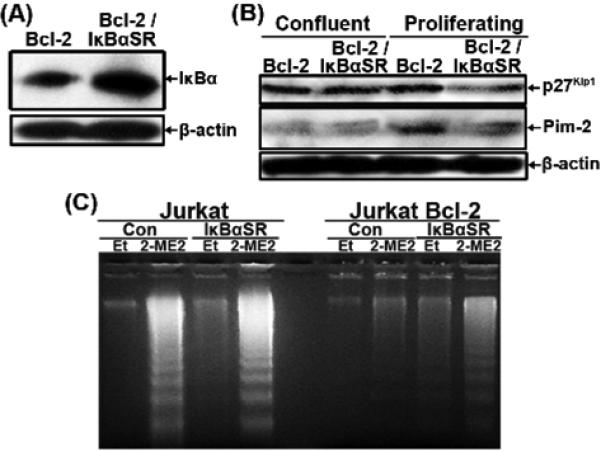
Suppression of NF-κB signaling pathway in Jurkat cells. Jurkat and Jurkat Bcl-2 cells were infected with recombinant retrovirus carrying α IκBαSR to generate Jurkat IκBαSR and Jurkat Bcl-2/IκBαSR cells. (A) Total lysates from all the different cell types were analyzed by immunoblotting for the expression of IκBα or β-actin. (B) Total lysates from all the different cell types were analyzed by western immunoblot for the expression of p27Kip1 and Pim-2 or β-actin. (C) DNA was isolated from untreated, ethanol- or 2-ME2-treated cells as indicated and analyzed by agarose gel electrophoresis. Similar results were obtained in another independent experiment (data not shown.
3.7. p27Kip1 knockdown sensitized Jurkat cells to 2-ME2-induced apoptosis
To determine if higher p27Kip1 levels were a critical factor contributing to the resistance of Jurkat Bcl-2 cells to 2-ME2-induced apoptosis [49,56], we knocked-down p27Kip1 expression by RNA interference [42]. Immunoblotting showed that the stable introduction of a p27Kip1 shRNA into Jurkat and Jurkat Bcl-2 cells markedly reduced p27Kip1 expression compared to their control counterparts (Fig. 8A). Analysis of low MW DNA on agarose gels revealed that p27Kip1 KD Jurkat cells were sensitized to spontaneous apoptosis (Fig. 8B) and importantly p27Kip1 KD Jurkat Bcl-2 cells were also more prone to 2-ME2-induced apoptosis (Fig. 8C). Thus, on the basis of the above results and our other findings with IκBαSR expressing cells , p27Kip1 is functionally linked to the resistance of Jurkat Bcl-2 cells to 2-ME2-induced apoptosis.
Fig. 8.
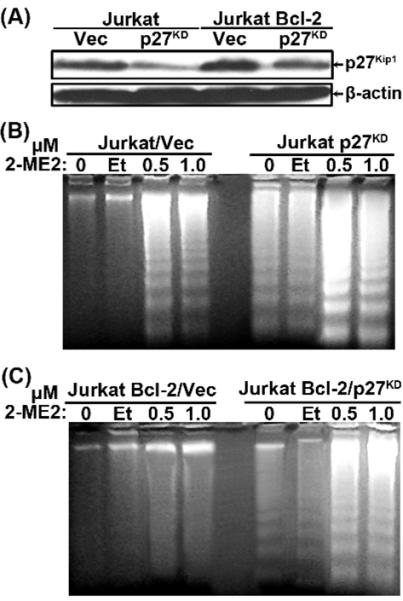
Reduction of p27Kip1 levels sensitized Jurkat and Jurkat Bcl-2 cells to apoptosis. Jurkat and Jurkat Bcl-2 cells were infected with recombinant retrovirus bearing shRNA to p27Kip1 or a control vector to generate Jurkat Vec Jurkat p27KD, Jurkat Bcl-2/Vec and Jurkat Bcl-2/p27KD cells. (A) Total lysates from all the different cell types were analyzed by immunoblotting for the expression of p27Kip1 or β-actin. (B) and (C) Low molecular weight DNA was isolated from untreated, ethanol- or 2-ME2-treated Jurkat/Vec and Jurkat p27KD cells (B) or Jurkat Bcl-2/Vec and Jurkat Bcl-2/p27KD cells (C), as indicated and analyzed by agarose gel electrophoresis. Vec, vector pSR-Puro. All experiments were repeated twice with comparable results.
4. Discussion
2-ME2 has been shown to inhibit the growth and induce apoptosis of tumor cells but not of normal cells, through several mechanisms studied in different cellular systems including phosphorylation and inactivation of Bcl-2 [7,15,27-29]. However, the mechanism(s) involved in 2-ME2-induced apoptosis in a particular setting and more specifically the role(s) of Bcl-2 in the effects of 2-ME2 remain unclear. In this report, the effects of 2-ME2 on human Jurkat T-leukemic cells and the mechanistic roles of Bcl-2 in 2-ME2-induced apoptosis were investigated. Our findings reveal an unexpected degree of complexity in the protective roles of Bcl-2 against 2-ME2 induced cell death including: (1) 2-ME2 induced dose- and time-dependent apoptosis of human T-leukemic Jurkat cells; (2) The induction of apoptosis by 2-ME2 through the mitochondrial pathway correlated with downregulation and phosphorylation, hence inactivation, of Bcl-2, correlated with JNK/SAPK activation, and up-regulation of Bak, leading to activation of caspase-9, caspase-3 and PARP-1 cleavage; (3) enforced expression of Bcl-2 blocked 2-ME2-induced apoptosis of Jurkat cells by inducing G1/S cell cycle phase arrest that correlated with changes in the expression of proteins involved in cell cycle progression and apoptosis; (4) Bcl-2 protein was found to be nuclear-associated leading to higher NF-κB activity, as documented by the sustained expression of Pim-2, and to higher nuclear levels of p27Kip1 in Jurkat Bcl-2 cells following treatment with 2-ME2, which occurred at least in part due to their maintenance of nuclear integrity; (5) suppression of NF-κB signaling sensitized Jurkat Bcl-2 cells to 2-ME2-induced apoptosis, through downregulation of p27Kip1; and (6) knocking-down the expression levels of p27Kip1 resulted in spontaneous Jurkat cell apoptosis and to the loss of Bcl-2 anti-apoptotic activity in Jurkat Bcl-2 cells following treatment with 2-ME2. Collectively, our data have dissected the molecular basis by which 2-ME2 induced apoptosis of Jurkat leukemia cells and also by which Bcl-2 suppresses these 2-ME2-induced effects on cell physiology. 2-ME2 induced apoptosis of Jurkat cells in a dose- and time-dependent manner as demonstrated by DNA fragmentation and flow cytometric analysis. The induction of apoptosis by 2-ME2 was p53-independent as Jurkat cells bear a mutant p53 allele [64] and correlated with downregulation and phosphorylation of Bcl-2. Treatment of several cell lines with chemotherapeutic agents that perturb tubulin microtubules resulted in phosphorylation of Bcl-2 during apoptosis [34,38,39,65]. However, Bcl-2 phosphorylation was not detected in cells treated with pro-apoptotic drugs that do not affect microtubule dynamics suggesting that microtubule damage may trigger Bcl-2 phosphorylation and that one of the functions of Bcl-2 may be to monitor microtubule integrity [38,39].
2-ME2-induced apoptosis in leukemia cells involved effects on multiple parameters: JNK activation and Bcl-2:Bak ratio
2-ME2-induced apoptosis of leukemic [26,29] and prostate cancer [15,27] cells has been associated with JNK-dependent phosphorylation, Akt inactivation [26] along with Bcl-2 phosphorylation; but the precise roles of these events in the ensuing apoptotic responses have remained elusive [34]. The role of Bcl-2 phosphorylation in the regulation of apoptosis is unclear, since some studies showed inactivation of its anti-apoptotic function [36-39,65], while others showed potentiation of its anti-apoptotic function [35,55,66]. The data presented here were in support of the latter, in that 2-ME2 induced Bcl-2 phosphorylation was well correlated with the accumulation of the phosphorylated, activated forms of JNK/SAPK in Jurkat cells [15,26-29]. Members of the Bcl-2 family are major regulators of mitochondrial apoptotic events. Anti- (Bcl-2 and Bcl-XL) and pro- (Bax and Bak) apoptotic Bcl-2 proteins regulate apoptosis in part by controlling cytochrome c release from mitochondria. The ratio between pro- and anti-apoptotic proteins determines in part the susceptibility of cells to a death signal [31-33]. Previous studies showed that Bak played a key role in the mitochondrial apoptotic response in of Jurkat cells induced by UV or anticancer drugs [47,48]. 2-ME2 induced the expression levels of Bak, albeit modestly, in Jurkat and Jurkat Puro cells but not in Bcl-2-expressing cells. In addition, no changes in the protein levels of Bax and PUMAα were detected in any cell type following treatment with 2-ME2 (data not shown). Thus, whether 2-ME2 either interfered with the dimerization of Bcl-2 to Bak [37,38] or resulted in the inactivation of Bcl-2, through the induction of Bcl-2 phosphorylation (and downregulation), allowing Bak to initiate apoptosis [31-33], it changed the ratio of anti- to pro-apoptotic (Bcl-2:Bak) members of the Bcl-2 family towards the latter thus driving the cell towards cell death. The latter most likely occurred viathe mitochondrial pathway as shown by the activation of caspases-9 and -3 but not -8 (data not shown) resulting in the cleavage of the caspase-3 substrate PARP-1, a hallmark of apoptotic cells. In contrast, in Bcl-2-overexpressing cells the ratio of anti- to pro-apoptotic (Bcl-2:Bak) Bcl-2 family members was shifted towards the former, hence these cells were more resistance to 2-ME2-induced apoptosis [31-33], Thus we suggest that the severe apoptotic reaction of Jurkat cells to 2-ME2 involved JNK activation mediating Bcl-2 phosphorylation, hence inactivation, leding to an altered Bcl-2:Bak ratio that simultaneously yield to mitochondrial-dependent programmed cell death.
The molecular basis for Bcl-2 mediated protection of leukemia cells to 2-ME2-induced apoptosis involved a G1/S cell cycle arrest
Interestingly, Bcl-2-mediated apoptotic block to 2-ME2 was linked to G1/S cell cycle arrest following 2-ME2-treatment of the cells, through changes in the expression of cell cycle regulators. Whereas in Jurkat and Jurkat Puro cells treatment with 2-ME2 downregulated the expression of the G1 cyclin, cyclin D3, in Jurkat Bcl-2 cells the levels of cyclin D3 were higher than their control counterparts and were sustained following 2-ME2-treatment. Downregulation of cyclin D3 in Jurkat and Jurkat Puro cells by 2-ME2 was due to apoptosis leading to fewer cells progressing through the cell cycle as documented by flow cytometry. In contrast, higher levels of cyclin D3 detected in Bcl-2-expressing cells were most likely due to accumulation of live cells in the G1 phase following treatment with 2-ME2, and to higher NF-κB activity in these cells. The Cyclin D3 gene is rearranged and the protein is overexpressed in several human lymphoid malignancies [67]. Cyclin D3-/- animals fail to undergo normal expansion of immature T lymphocytes and show greatly reduced susceptibility to T cell malignancies triggered by specific oncogenic pathways. Further, knock-down of cyclin D3 inhibited proliferation of acute lymphoblastic leukemias deriving from immature T lymphocytes, suggesting a requirement of cyclin D3 in the development of T cell leukemias [67]. The finding that 2-ME2 downregulated cyclin D3 in human Jurkat T leukemic cells and its sustained expression in Bcl-2-expressing Jurkat cells would be in keeping with these studies. A reduction in the levels of cyclin E by 2-ME2 in all the different cell types suggested that 2-ME2 affected G1 to S cell cycle progression [68].
Cell cycle progression relies on the activation of cyclins and CDKs which successively act together in G1 to initiate S phase and in G2 to initiate mitosis. To prevent abnormal proliferation, cyclin-CDK complexes are precisely regulated by two families of cell cycle inhibitors that block their catalytic activity [69]. The first class of inhibitors includes the INK4a proteins that bind to Cdk4/6 kinases and not to cyclins and are therefore specific for early G1 phase. The second family of inhibitors is composed of Cip/Kip proteins, such as p21Cip1/Waf1 and p27Kip1, that inhibit all cyclin-CDK complexes and are not specific for a particular phase. Unlike INK4a, Cip/Kip proteins do not dissociate cyclin-CDK complexes [69]. However, other studies suggested that p21Cip1/Waf1 and p27Kip1 might have new activities, that are unrelated to their function as CDK inhibitors, such as the regulation of apoptosis and in transcriptional activation [70].
The induction of p16INK4A by 2-ME2 in Jurkat and Jurkat Puro cells probably contributed to two outcomes: inhibition of cell proliferation and also cell apoptosis induced by 2-ME2. However, because p16INK4A was not induced by 2-ME2 in Bcl-2-expressing cells, up-regulation of p16INK4A would seem to be more likely involved in 2-ME2-induced apoptosis rather than in to growth inhibition, and several other studies have pointed towards a role of p16INK4A in drug-induced apoptosis of tumor cells [71,72]. 2-ME2 treatment resulted in the downregulation and phosphorylation of p21Cip1/Waf1. p21Cip1/Waf1 functions both as a positive and negative regulator of the cell cycle by regulating both CDK activity and DNA synthesis, but it also appears to play a role in enhancing cell survival [69,70,73,74]. Further, it appears that the switch between cell cycle promotion and inhibition by p21Cip1/Waf1 may occur by virtue of the subcellular localization of this protein through phosphorylation, [73,74]. Treatment of Jurkat cells with 2-ME2 resulted in the downregulation and phosphorylation of p21Cip1/Waf1 in all the different Jurkat cell types, which did not correlate with apoptosis. To clarify the role of p21Cip1/Waf1 in 2-ME2-induced apoptosis, p21Cip1/Waf1 was ectopically overexpressed in Jurkat cells and it neither induced growth arrest nor protected the cells from 2-ME2-induced apoptosis (data not shown). Hence, phosphorylation of p21Cip1/Waf1 following treatment with 2-ME2 probably affected its function as an assembly factor of cyclin D-CDK4/6 complexes, which function as sensors of growth factors at G1/S phase [69,70,73,74].
In contrast to p16INK4A and p21Cip1/Waf1, 2-ME2 did not affect the expression of p27Kip1 in all the different Jurkat cell types. However, Jurkat Bcl-2 cells expressed elevated levels of p27Kip1 compared to their control counterparts, accounting for their reduced growth rates. This negative effect of Bcl-2 on cell proliferation was in agreement with previous studies [49,75-79], through elevation of p27Kip1 [49,53-55] and to its role in T cell survival [80,81].
In summary, our results showed that in control cells 2-ME2 downregulated the expression of cyclin D3, cyclin E, and E2F1, downregulated and phosphorylated p21Cip1/Waf1, and also induced the expression of p16INK4A without affecting the levels of pRb and p27Kip1. In contrast, in Jurkat Bcl-2 cells the levels of cyclin D3 and p27Kip1 were elevated and sustained, and no induction of p16INK4A was detected following treatment with 2-ME2. These data taken together with the growth curves and flow cytometry analyses suggested that Jurkat Bcl-2 cells were arrested in G1/S phase of the cell cycle, most likely at the restriction point.
Bcl-2-induced cell cycle arrest is caused by the NF-κB-dependent enhancement of p27Kip1 expression
To get some insight into the mechanism by which Bcl-2 arrested growth following treatment with 2-ME2, we analyzed the expression and subcellular localization of Bcl-2 and p27Kip1. We demonstrated that Bcl-2 and p27Kip1 were predominantly nuclear-associated and that Bcl-2 expression most likely enhanced nuclear levels and perhaps the stability of p27Kip1. Given the role of p27Kip1 in the regulation of cell cycle progression and apoptosis [56,81] the elevated levels of p27Kip1 in Bcl-2-expressing cells could account for both the reduced growth rate or G1/S arrest [49] following treatment with 2-ME2 but also for the increased resistance [56] to 2-ME2-induced apoptosis.
p27Kip1 expression is regulated at the posttranscriptional level, both at the level of protein translation and stability. The cyclin E/CDK2 complexes are the major target of p27Kip1 inhibitory activity, but on the other hand p27Kip1 can act as a substrate for cyclin E/CDK2 complexes. Once activated, cyclin E/CDK2 complexes phosphorylate p27Kip1 at Thr187, thereby triggering its Skp2-dependent ubiquitination and degradation by the proteasome complex [56]. Initially, p27Kip1 binds with low affinity acting as a substrate, and then slowly the binding shifts to high affinity and p27Kip1 becomes an inhibitor. At equilibrium, p27Kip1 inhibits cyclin E/CDK2 activity and this provides a negative regulatory feedback loop that makes G1/S transition irreversible [56]. In addition, p27Kip1 is phosphorylated at Ser10 accounting for 70% of the total phosphorylation of the protein. In contrast to Thr187, Ser10 phosphorylation increases the stability of the protein [56]. Whereas, the S-phase degradation of p27Kip1 is Thr187 and Skp2-dependent, the degradation of p27Kip1 at the G1 restriction point is independent of these but dependent on mitogen stimulation.
Previous studies showed that stable overexpression of Bcl-2 enhanced [57,58], preserved [59] or had no effect [60,61] on NF-κB activity. Further, nuclear compartment-associated Bcl-2 was previously reported in lymphoid cells [50], during aging and oxidative stress [51] and nuclear localization of transfected Bcl-2 interfered with the nuclear import of NF-κB subunits in non-lymphoid cells, thus inhibiting their activity [52], but nuclear Bcl-2 was not detected in untransfected cells [51,52]. Biochemical and immunostaining techniques showed that Bcl-2-expressing Jurkat cells contained higher levels and activity of NF-κB, as documented by the maintenance of Pim-2 expression following treatment with 2-ME2, compared to their control counterparts. However because immunoprecipitations coupled to immunoblot analysis did not reveal interactions between Bcl-2 and NF-κBs (data not shown), Bcl-2’s enhancing effect on nuclear NF-κB levels would appear to be indirect. One plausible explanation for this intriguing (albeit indirect) connection between Bcl-2 and NF-κB could perhaps be that higher levels of nuclear-associated Bcl-2 protein help to maintain nuclear stability, which then somehow contribute to higher nuclear levels and activity of NF-κB and also to higher nuclear levels and stability of p27Kip1. However the indirect effect of nuclear Bcl-2 on NF-κB (and also p27Kip1) appear to be at least somewhat specific, because other transcription and cell cycle regulators are not similarly affected [52] (see our observations on E2F1 along with several other effectors of cell cycle progression in Fig. 4).
To investigate a link between the roles of NF-κB and p27Kip1 in 2-ME2-induced apoptosis but also their contribution to resistance to 2-ME2 exhibited by Jurkat Bcl-2 cells, we interfered with NF-κB signaling pathway and p27Kip1 expression. Suppression of NF-κB signaling using a super repressor, IκBαSR, sensitized Jurkat Bcl-2 cells to 2-ME2-induced apoptosis, through downregulation of p27Kip1. Knocking-down p27Kip1 expression led to spontaneous Jurkat cell apoptosis and sensitized Jurkat Bcl-2 to 2-ME2-induced apoptosis.
Collectively these data, show that Bcl-2 protected Jurkat cells from 2-ME2-induced apoptosis by multiple mechanisms including: inhibition of the mitochondrial apoptotic pathway, maintenance of nuclear integrity via its nuclear-association, maintaining active nuclear NF-κB resulting in higher levels of Pim-2, (a direct NF-κB target with potent anti-apoptotic properties) and finally to enhanced levels and stability of p27Kip1, which was recently reported to be a direct NF-κB-target gene [82]. On the basis of these results we have achieved a much better understanding of the penetrance and mechanistic complexity of Bcl-2 dependent anti-apoptotic pathways in cancer cells and why Bcl-2 inactivation is so critical for the efficacy of apoptosis and anti-proliferative inducing drugs like 2-ME2.
Supplementary Material
Acknowledgements
We are grateful to Dr. D. Tang, University of Montreal, Canada for providing pBabe-Puro/hBcl-2, Dr. J. Chen, H. Lee Moffitt Cancer Center & Research Institute, The University of South Florida, USA, for pSR-Puro and pSR-Puro#p27Kip1, and Drs C. J. Sherr and M. Roussel, Department of Genetics, St. Jude Children’s Research Hospital, Memphis, Tennessee, USA, for providing p21Cip1/Waf1 and p27Kip1 retroviral vectors. We also thank Drs S. Georgatos, C. Murphy and P. Pappas, University of Ioannina Medical School, Greece, for providing rabbit polyclonal anti-lamin B and reagents. This work was supported by a bilateral agreement grant from the General Secretariat of Research and Technology, Greece (E. Kolettas, 61/1722) and the Cyprus Research Promotion Foundation (A. Constantinou; ENTAKS/0505/52). And USA NIH grant GM066882 awarded to KBM.
Footnotes
The authors have no conflicting financial interests.
REFERENCES
- [1].Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, Nawroth PP, et al. The endogenous estrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature. 1994;368:237–39. doi: 10.1038/368237a0. [DOI] [PubMed] [Google Scholar]
- [2].D’Amato RJ, Lin CM, Flynn E, Folkman J, Hamel E. 2-methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicine site. Proc Natl Acad Sci USA. 1994;91:3964–68. doi: 10.1073/pnas.91.9.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Klauber N, Parangi S, Flynn E, Hamel E, D’Amato RJ. Inhibition of angiogenesis and breast cancer in mice by microtubule inhibitors 2-methoxyestradiol and taxol. Cancer Res. 1997;57:81–86. [PubMed] [Google Scholar]
- [4].Pribluda VS, Gubish ER, Jr, LaVallee TM, Treston A, Swartz GM, Green SJ. 2-methoxyestradiol: an endogenous antiangiogenic and antiproliferative drug candidate. Cancer Metastasis Rev. 2000;19:173–79. doi: 10.1023/a:1026543018478. [DOI] [PubMed] [Google Scholar]
- [5].Ryoo JJ, Cole CE, Anderson KC. Novel therapies for multiple myeloma. Blood Rev. 2002;16:167–74. doi: 10.1016/s0268-960x(02)00009-7. [DOI] [PubMed] [Google Scholar]
- [6].Lakhani NJ, Sarkar MA, Venitz J, Figg WD. 2-methoxyestradiol, a promising anticancer agent. Pharmacotherapy. 2003;23:165–72. doi: 10.1592/phco.23.2.165.32088. [DOI] [PubMed] [Google Scholar]
- [7].Mooberry SL. New insights into 2-methoxyestradiol, a promising antiangiogenic and antitumor agent. Curr Opin Oncol. 2003;15:425–30. doi: 10.1097/00001622-200311000-00004. [DOI] [PubMed] [Google Scholar]
- [8].Mooberry SL. Mechanism of action of 2-methoxyestradiol: new developments. Drug Resist Updat. 2003;6:355–61. doi: 10.1016/j.drup.2003.10.001. [DOI] [PubMed] [Google Scholar]
- [9].Attalla H, Mäkelä TP, Adlercreutz H, Anderson LC. 2-methoxyestradiol arrests cells in mitosis without depolymerising tubulin. Biochem Biophys Res Commun. 1996;228:467–73. doi: 10.1006/bbrc.1996.1683. [DOI] [PubMed] [Google Scholar]
- [10].Dingli D, Timm M, Russell SJ, Witzig TE, Rajkumar SV. Promising preclinical activity of 2-methoxyestradiol in multiple myeloma. Clin Cancer Res. 2002;8:3948–54. [PubMed] [Google Scholar]
- [11].Zoubine MN, Weston AP, Johnson DC, Campbell DR, Banerjee SK. 2-methoxyestradiol-induced growth suppression and lethality in estrogen responsive MCF-7 cells may be mediated by downregulation of p34cdc2 and cyclin B1 expression. Int J Oncol. 1999;15:639–46. doi: 10.3892/ijo.15.4.639. [DOI] [PubMed] [Google Scholar]
- [12].La Vallee TM, Zhan XH, Herbstritt CJ, Kough FC, Green SJ, Pribluda VS. 2-methoxyestradiol inhibits proliferation and induces apoptosis independently of estrogen receptors α and β. Cancer Res. 2002;62:3691–97. [PubMed] [Google Scholar]
- [13].Kumar AP, Garcia GE, Slaga TJ. 2-methoxyestradiol blocks cell cycle progression at G2/M phase and inhibits growth of human prostate cancer cells. Mol Carcinog. 2001;31:111–24. doi: 10.1002/mc.1046. [DOI] [PubMed] [Google Scholar]
- [14].Qadan LR, Perez-Stable CM, Abderson C, D’Ippolito G, Herron A, Howard GA, et al. 2-methoxystradiol induces G2/M arrest and apoptosis in prostate cancer. Biochem Biophys Res Commun. 2001;285:1259–66. doi: 10.1006/bbrc.2001.5320. [DOI] [PubMed] [Google Scholar]
- [15].Bu S, Blaukat A, Fu X, Heldin N-E, Landström M. Mechanisms for 2-methoxyestradiol-induced apoptosis of prostate cancer cells. FEBS Lett. 2002;531:141–51. doi: 10.1016/s0014-5793(02)03478-6. [DOI] [PubMed] [Google Scholar]
- [16].Lin HL, Liu TY, Wu VW, Chi CW. 2-methoxyestradiol-induced caspase-3 activation and apoptosis occurs through G2/M arrest dependent and independent pathways in gastric carcinoma cells. Cancer. 2001;92:500–9. doi: 10.1002/1097-0142(20010801)92:3<500::aid-cncr1348>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- [17].Ghosh R, Ott AM, Seetharam D, Slaga TJ, Kumar AP. Cell cycle block and apoptosis induction in a human melanoma cell line following treatment with 2-methoxyestradiol: therapeutic implications. Melanoma Res. 2004;13:119–27. doi: 10.1097/00008390-200304000-00003. [DOI] [PubMed] [Google Scholar]
- [18].Kumar AP, Garcia GE, Orsborn J, Levin VA, Slaga TJ. 2-methoxyestradiol interferes with NF-κB transcriptional activity in primitive neuroectodermal brain tumors: implications for management. Mol Carcinog. 2003;24:209–16. doi: 10.1093/carcin/24.2.209. [DOI] [PubMed] [Google Scholar]
- [19].Seegers JC, Lottering ML, Grobler CJ, van Papendorp DH, Habbersett RC, Shou Y, et al. The mammalian metabolite, 2-methoxyestradiol, affects p53 levels and apoptosis induction in transformed cells but not in normal cells. J Steroid Biochem Mol Biol. 1997;62:253–67. doi: 10.1016/s0960-0760(97)00043-5. [DOI] [PubMed] [Google Scholar]
- [20].Schumacher G, Kataoka M, Roth JA, Mukhopadhyay T. Potent antitumor activity of 2-methoxyestradiol in human pancreatic cancer cell lines. Clin Cancer Res. 1999;5:493–99. [PubMed] [Google Scholar]
- [21].Golebiewska J, Rozwadowski P, Spodnik JH, Knap N, Wakabayashi Y, Wozniak M. Dual effect of 2-methoxyestradiol on cell cycle events in human osteosarcoma 143b cells. Acta Biochim Pol. 2002;49:59–65. [PubMed] [Google Scholar]
- [22].Qanungo S, Basu A, Das M, Haldar S. 2-methoxyestradiol induces mitochondrial dependent apoptotic signaling in pancreatic cancer cells. Oncogene. 2002;21:4149–57. doi: 10.1038/sj.onc.1205508. [DOI] [PubMed] [Google Scholar]
- [23].Mukhopadhay T, Roth JA. Induction of apoptosis in human cancer cells after wild-type p53 activation by methoxyestradiol. Oncogene. 1997;14:379–84. doi: 10.1038/sj.onc.1200835. [DOI] [PubMed] [Google Scholar]
- [24].Chauhan D, Li G, Hideshima T, Podar K, Mitsiades C, Mitsiades N, et al. Superoxide-dependent and independent mitochondrial signalling during apoptosis in multiple myeloma. Oncogene. 2003;22:6296–300. doi: 10.1038/sj.onc.1206734. [DOI] [PubMed] [Google Scholar]
- [25].Mabjeesh NJ, Escuin D, La Valle TM, Pribluda VS, Stewart GM, Johnson MS, et al. 2-ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–75. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- [26].Gao N, Rahmani M, Dent P, Grant S. 2-methoxyestradiol-induced apoptosis in human leukaemia cells proceeds through a reactive oxygen species and Akt-dependent process. Oncogene. 2005;24:3797–809. doi: 10.1038/sj.onc.1208530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shimada K, Nakamura M, Ishida E, Kishi M, Konishi N. Roles of p38- and c-jun NH2-terminal kinase-mediated pathways in 2-methoxyestradiol-induced p53 induction and apoptosis. Carcinogenesis. 2003;24:1067–75. doi: 10.1093/carcin/bgg058. [DOI] [PubMed] [Google Scholar]
- [28].Bu SZ, Huang Q, Jiang YM, Min HB, Hou Y, Guo ZY, et al. p38 Mitogen-activated protein kinases is required for counteraction of 2-methoxyestradiol to estradiol-stimulated cell proliferation and induction of apoptosis in ovarian carcinoma cells via phosphorylation Bcl-2. Apoptosis. 2006;11:413–25. doi: 10.1007/s10495-006-4064-z. [DOI] [PubMed] [Google Scholar]
- [29].Attalla H, Westberg JA, Andersson LC, Adlercreutz H, Mäkelä TP. 2-methoxyestradiol-induced phosphorylation of Bcl-2: Uncoupling from JNK/SAPK activation. Biochem Biophys Res Commun. 1998;247:616–19. doi: 10.1006/bbrc.1998.8870. [DOI] [PubMed] [Google Scholar]
- [30].Basu A, Haldar S. Identification of a novel Bcl-XL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett. 2003;538:41–47. doi: 10.1016/s0014-5793(03)00131-5. [DOI] [PubMed] [Google Scholar]
- [31].Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–76. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- [32].Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Verma YK, Gangenahalli GU, Singh VK, Gupta P, Chandra R, Sharma RK, et al. Cell death regulation by B-cell lymphoma protein. Apoptosis. 2006;11:459–71. doi: 10.1007/s10495-006-5702-1. [DOI] [PubMed] [Google Scholar]
- [35].Deng X, Kornblau SM, Ruvolo PP, May WS., Jr. Regulation of Bcl-2 phosphorylation and potential significance for leukemic cell chemoresistance. J Nat Cancer Inst Monogr. 2000;28:30–37. doi: 10.1093/oxfordjournals.jncimonographs.a024254. [DOI] [PubMed] [Google Scholar]
- [36].Yamamoto K, Ichijo H, Korsmeyer SJ. Bcl-2 is phosphorylated and inactivated by ASK1/jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–78. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci USA. 1999;96:3775–80. doi: 10.1073/pnas.96.7.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Haldar S, Chintapalli J, Croce CM. Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res. 1996;56:1253–55. [PubMed] [Google Scholar]
- [39].Pathan N, Aime-Sempe C, Kitada S, Basu A, Haldar S, Reed JC. Microtubule-Targeting drugs induce Bcl-2 phosphorylation and association with Pin1. Neoplasia. 2001;3:550–59. doi: 10.1038/sj.neo.7900213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Huang P, Feng L, Oldham A, Keating MJ, Plunkett W. Superoxide dismutase as a target for killing of cancer cells. Nature. 2000;407:390–95. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- [41].Tang D, Okada H, Ruland J, Liu L, Stambolic V, Mak TW, et al. Akt is activated in response to an apoptotic signal. J Biol Chem. 2001;276:30461–66. doi: 10.1074/jbc.M102045200. [DOI] [PubMed] [Google Scholar]
- [42].Wang C, Hou X, Mohapatra S, Ma Y, Cress WD, Pledger WJ, et al. Activation of p27Kip1, expression by E2F1: a negative feedback mechanism. J Biol Chem. 2005;280:12339–43. doi: 10.1074/jbc.C400536200. [DOI] [PubMed] [Google Scholar]
- [43].Li J, Peet GW, Balzarano D, Li X, Massa P, Barton RW, et al. Novel NEMO/IκB kinase and NF-κB target genes at the pre-B to immature B cell transition. J Biol Chem. 2001;276:18579–90. doi: 10.1074/jbc.M100846200. [DOI] [PubMed] [Google Scholar]
- [44].Kolettas E, Skoufos I, Kontargiris E, Markopoulou S, Tzavaras Th, Gonos ES. Bcl-2 but not clusterin/apolipoprotein J protected human diploid fibroblasts and immortalised keratinocytes from ceramide-induced apoptosis: Role of p53 in the ceramide response. Arch Biochem Biophys. 2006;445:184–95. doi: 10.1016/j.abb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- [45].Dimri GP, Hara E, Campisi J. Regulation of two E2F-related genes in presenescent and senescent human fibroblasts. J Biol Chem. 1994;269:16180–86. [PubMed] [Google Scholar]
- [46].Reddy KB, Karode MC, Harmony JAK, Howe PH. Transforming growth factor β (TGFβ)-induced nuclear localization of apolipoprotein J/clusterin in epithelial cells. Biochemistry. 1996;35:6157–63. doi: 10.1021/bi952981b. [DOI] [PubMed] [Google Scholar]
- [47].Wang G-Q, Gastman BR, Wieckowski E, Goldstein LA, Gambotto A, Kim T-H, et al. A role for mitochondrial Bak in apoptotic response to anticancer Drugs. J Biol Chem. 2001;276:34307–17. doi: 10.1074/jbc.M103526200. [DOI] [PubMed] [Google Scholar]
- [48].Wang G-Q, Wieckowski E, Goldstein LA, Gastman BR, Rabinovitz A, Gambotto A, et al. Resistance to granzyme B-mediated cytochrome c release in Bak-deficient cells. J Exp Med. 2001;194:1325–37. doi: 10.1084/jem.194.9.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006;13:1351–59. doi: 10.1038/sj.cdd.4401987. [DOI] [PubMed] [Google Scholar]
- [50].Scheel-Toellner D, Raza K, Assi L, Pilling D, Ross EJ, Lee WY, et al. Differential regulation of nuclear and mitochondrial Bcl-2 in T cell apoptosis. Apoptosis. 2008;13:109–117. doi: 10.1007/s10495-007-0143-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kaufmann JA, Perez M, Zhang WR, Bickford PC, Holmes DB, Taglialatela G. Free radical-dependent nuclear localization of Bcl-2 in the central nervous system of aged rats is not associated with Bcl-2-mediated protection from apoptosis. J Neurochem. 2003;87:981–94. doi: 10.1046/j.1471-4159.2003.02092.x. [DOI] [PubMed] [Google Scholar]
- [52].Massaad CA, Portier BP, Taglialatela G. Inhibition of transcription factor activity by nuclear compartment-associated Bcl-2. J Biol Chem. 2004;279:54470–78. doi: 10.1074/jbc.M407659200. [DOI] [PubMed] [Google Scholar]
- [53].Vairo G, Soos TJ, Upton TM, Zalvide J, DeCaprio JA, Ewen ME, et al. Bcl-2 retards cell cycle entry through p27Kip1, pRB relative p130 and altered E2F regulation. Mol Cell Biol. 2002;20:4745–53. doi: 10.1128/mcb.20.13.4745-4753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Greider C, Chattopadhyay A, Parkhurst C, Yang E. Bcl-XL and Bcl-2 delay myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene. 2002;21:7765–75. doi: 10.1038/sj.onc.1205928. [DOI] [PubMed] [Google Scholar]
- [55].Deng X, Gao F, May WS., Jr. Bcl2 retards G1/S cell cycle transition by regulating intracellular ROS. Blood. 2003;102:3179–85. doi: 10.1182/blood-2003-04-1027. [DOI] [PubMed] [Google Scholar]
- [56].Borriello A, Cucciolla V, Oliva A, Zappia V, Ragione FD. p27Kip1 metabolism. Cell Cycle. 2007;6:1053–1061. doi: 10.4161/cc.6.9.4142. [DOI] [PubMed] [Google Scholar]
- [57].Ricca A, Biroccio A, Del Bufalo D, Mackay AR, Santoni A, Cippitelli M. Bcl-2 overexpression enhances NF-κB activity and induces mmp-9 transcription in human MCF7ADR breast cancer cells. Int J Cancer. 2000;86:188–96. doi: 10.1002/(sici)1097-0215(20000415)86:2<188::aid-ijc7>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- [58].Jang J-H, Surh Y-J. Bcl-2 attenuation of oxidative cell death is associated with up-regulation of γ-glutamylcysteine ligase via constitutive NF-κB activation. J Biol Chem. 2004;279:38779–86. doi: 10.1074/jbc.M406371200. [DOI] [PubMed] [Google Scholar]
- [59].Crawford MJ, Krishnamoorthy RR, Rudick VL, Collier RJ, Kapin M, Aggarwal BB, et al. Bcl-2 overexpression protects photooxidative stress-induced apoptosis of photoreceptor cells via NF-κB preservation. Biochem Biophys Res Comm. 2001;281:1304–12. doi: 10.1006/bbrc.2001.4501. [DOI] [PubMed] [Google Scholar]
- [60].Albrecht H, Tschopp J, Jongenel CV. Bcl-2 protects from oxidative damage and apoptotic cell death without interfering with activation of NF-κB by TNF. FEBS Lett. 1994;351:45–48. doi: 10.1016/0014-5793(94)00817-5. [DOI] [PubMed] [Google Scholar]
- [61].Herrmann JL, Beham AW, Sarkiss M, Chiao PJ, Rands MT, Bruckheimer EM, et al. Bcl-2 suppresses apoptosis resulting from disruption of the NF-κB survival pathway. Exp Cell Res. 1997;237:101–9. doi: 10.1006/excr.1997.3737. [DOI] [PubMed] [Google Scholar]
- [62].Hammerman PS, Fox CJ, Cinalli RM, Xu A, Wagner JD, Lindsten T, et al. Lymphocyte transformation by Pim-2 is dependent on nuclear factor-κB activation. Cancer Res. 2004;64:8341–48. doi: 10.1158/0008-5472.CAN-04-2284. [DOI] [PubMed] [Google Scholar]
- [63].Minami S, Ohtani-Fujita N, Igata E, Tamaki T, Sakai T. Molecular cloning of the human p27kip1 gene promoter. FEBS Lett. 1997;411:1–6. doi: 10.1016/s0014-5793(97)00660-1. [DOI] [PubMed] [Google Scholar]
- [64].Cheng J, Haas M. Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Mol Cell Biol. 1990;10:5502–9. doi: 10.1128/mcb.10.10.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Blagosklonny MV, Bishop PC, Robey R, Fojo T, Bates SE. Loss of cell cycle control allows selective microtubule-active drug-induced Bcl-2 phosphorylation and cytotoxicity in autonomous cancer cells. Cancer Res. 2000;60:3425–28. [PubMed] [Google Scholar]
- [66].Deng X, Gao F, Flagg T, May WS., Jr. Mono- and multisite phosphorylation enhances Bcl2’s antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci USA. 2004;101:153–58. doi: 10.1073/pnas.2533920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. 2003;4:451–61. doi: 10.1016/s1535-6108(03)00301-5. [DOI] [PubMed] [Google Scholar]
- [68].Gladden AB, Diehl JA. Cell cycle progression without cyclin E-CDK2: Breaking down the walls of dogma. Cancer Cell. 2003;4:160–62. doi: 10.1016/s1535-6108(03)00217-4. [DOI] [PubMed] [Google Scholar]
- [69].Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1 phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- [70].Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2002;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- [71].Lagresle C, Gardie B, Eyquem S, Fassen M, Vieville J-C, Pla M, et al. Transgenic expression of the p16INK4A cyclin-dependent kinase inhibitor leads to enhanced apoptosis and differentiation arrest of CD4-CD8- immature thymocytes. J Immunol. 2002;168:2325–31. doi: 10.4049/jimmunol.168.5.2325. [DOI] [PubMed] [Google Scholar]
- [72].Sachs Z, Sharpless NE, DePinho RA, Rosenberg N. p16INK4A interferes with Abelson virus transformation by enhancing apoptosis. J Virol. 2004;78:3304–11. doi: 10.1128/JVI.78.7.3304-3311.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gartel AL, Tyner AL. The role of cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–49. [PubMed] [Google Scholar]
- [74].Weiss RH. p21Waf1/Cip1 as a therapeutic target in breast and other cancers. Cancer Cell. 2003;4:425–29. doi: 10.1016/s1535-6108(03)00308-8. [DOI] [PubMed] [Google Scholar]
- [75].O’Reilly LA, Huang DCS, Strasser A. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 1996;15:6979–90. [PMC free article] [PubMed] [Google Scholar]
- [76].Borner C. Diminished cell proliferation associated with the death protective activity of bcl-2. J Biol Chem. 1996;271:12695–98. doi: 10.1074/jbc.271.22.12695. [DOI] [PubMed] [Google Scholar]
- [77].Huang DCS, O’Reilly LA, Strasser A, Cory S. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 1997;16:4628–38. doi: 10.1093/emboj/16.15.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Johnson VL, Cooper IR, Jenkins JR, Chow SC. Effects of differential overexpression of Bcl-2 on apoptosis, proliferation and telomerase activity in Jurkat cells. Exp Cell Res. 1999;251:175–84. doi: 10.1006/excr.1999.4557. [DOI] [PubMed] [Google Scholar]
- [79].Janumyan YM, Sansam CG, Chattopadhay A, Cheng N, Soucie EL, Penn LZ, et al. Bcl-XL/Bcl-2 coordinately regulate apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003;22:5459–70. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Cheng N, Janumyan YM, Didion L, van Holfwegen C, Yang E, Knudson CM. Bcl-2 inhibition of T-cell proliferation is related to prolonged T-cell survival. Oncogene. 2004;23:3770–80. doi: 10.1038/sj.onc.1207478. [DOI] [PubMed] [Google Scholar]
- [81].Hiromura K, Pippin JW, Fero ML, Roberts JM, Shankland SJ. Modulation of apoptosis by the cyclin-dependent kinase inhibitor p27Kip1. J Clin Invest. 1999;103:597–04. doi: 10.1172/JCI5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Prasad RC, Wang XL, Law BK, Davis B, Green G, Boone B, Sims L, Law M. Identification of genes, including the gene encoding p27Kip1, regulated by serine 276 phosphorylation of the p65 subunit of NF-κB. Cancer Lett. 2009;275:139–49. doi: 10.1016/j.canlet.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.