

Abstract
A series of position-6 substituted 2-amino-4-methylpyridine analogues was synthesized and compounds 9, 18, and 20 were identified as the inhibitors with the greatest potential to serve as PET tracers for imaging inducible nitric oxide synthase (iNOS). [18F]9 was synthesized and evaluated in a mouse model of lipopolysaccharide (LPS)-induced iNOS activation. In vivo biodistribution studies of [18F]9 indicate higher tracer uptake in the lungs of the LPS-treated mice when compared to control mice. Tracer uptake at 60 min post-injection was reduced in a blocking study using a known inhibitor of iNOS. The expression of iNOS was confirmed by Western blot analysis of lung samples from the LPS-treated mice. MicroPET studies also demonstrated accumulation of radiotracer in the lungs of the LPS-treated mice. Taken collectively, these data suggest that [18F]9 shows favorable properties as a PET tracer to image iNOS activation with PET.
Introduction
Nitric oxide (NO) is an important and unique mediator of a variety of physiological and pathological processes.1 NO is generated from the oxidation of L-arginine to L-citrulline in a two-step process by nitric oxide synthase (NOS) enzymes.2 In the NOS family, there are two constitutive isozymes of NOS, neuronal NOS (nNOS) and endothelial NOS (eNOS), and one inducible isozyme (iNOS). The three isozymes of NOS are expressed in different tissues to generate NO for specific physiological roles. nNOS generates NO as a neurotransmitter and neuromodulator, mainly in brain and peripheral nerve cells; eNOS regulates blood pressure, primarily in vascular endothelial cells;3 iNOS is induced by various inflammatory stimuli (e.g. endotoxin) in activated macrophages and other types of cells and plays an crucial role in the host defense and the inflammatory processes.
Normally, the basal level of NO in all parts of the body is very low, mainly due to the constitutive nNOS and eNOS. In contrast, once expressed, iNOS can continue to generate NO in large amounts (up to µM concentrations) for a prolonged period of time.4 Studies have shown that production of NO by iNOS is implicated in a variety of acute and chronic inflammatory diseases (e.g., sepsis, septic shock, vascular dysfunction in diabetes, asthma, arthritis, multiple sclerosis and inflammatory diseases of the gut)5; iNOS activity has also been found in many tumors.6 Because of the central role of iNOS in NO-related diseases, numerous efforts have been made to develop iNOS inhibitors as pharmaceuticals ranging from the non-selective L-arginine analogues7 to the selective inhibitors reported recently.8 Some inhibitors of iNOS have shown promising results in animal models of sepsis, lung inflammation, arthritis, and autoimmune diabetes.8c Therefore, the development of a radiolabeled iNOS inhibitor for probing iNOS expression in vivo using non-invasive positron emission tomography (PET) imaging will be of tremendous value to the study and treatment of NO-related diseases.
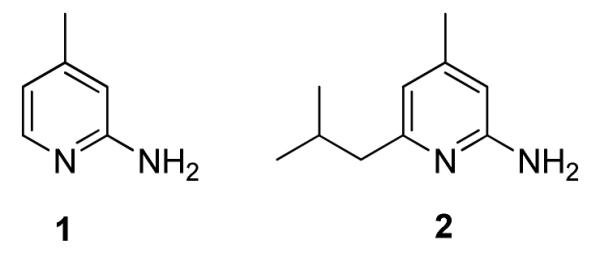
PET is being used more frequently in clinical and research studies because of its high sensitivity, good spatial resolution and ease in accurate quantification. Additionally, the absence of a physiologic effect from the radiotracers makes it a safe in vivo imaging tool. When short-lived positron-emitting radionuclides (18F t1/2 = 109.8 min and 11C t1/2 = 20.4 min) are incorporated into biologically active molecules (e.g. iNOS inhibitors), they can be used as tracers that target those physiological pathways. 2-amino-4-methylpyridine (1) has been reported as a non-selective NOS inhibitor with good potency;9 while the 6-substituted alkyl analogs of 1 have slightly improved potency and selectivity over the parent compound; analog 2 has the best potency (IC50 against iNOS = 28 nM).10 Computational calculations suggest that the position-6 is the most tolerant position to introduce a substitutent11 that would be suitable for radiolabeling with PET radionuclides 18F and 11C.
In the past decade, the development of radiolabeled PET tracers for iNOS has been limited12 compared with the relatively rapid development of novel iNOS inhibitors as pharmaceuticals. In this paper, we describe the synthesis and screening of a series of position-6 substituted 2-amino-4-methylpyridine analogues as potential PET tracers for imaging iNOS, the radiosynthesis of [18F]9, and the in vivo evaluation of [18F]9 in a mouse model of lipopolysaccharide (LPS)-induced iNOS activation.
Results and Discussion
Chemistry
The previously reported method was applied to synthesize the key intermediate 6 (Scheme 1).10 Compound 6 reacted with acetaldehyde to afford 7 in high yield (Scheme 2). Compound 7 was converted to 8 using diethylaminosulfur trifluoride (DAST) or perfluorobutane sulfonyl fluoride (PBSF) as the fluorinating agents. Compound 10 was obtained as a by-product in both cases and was formed as the major product when PBSF was used as the fluorinating agent. These results indicate the facile elimination to form a conjugated double bond adjacent to the pyridine ring. The conversion of the OH in 7 to Br using PPh3 and CBr4 failed to give the expected product (data not shown). Compounds 12 and 14 were synthesized from 7 via O-alkylation using CH3I and BrCH2CH2F, respectively in the presence of CaH2 (Scheme 2). The pyrrole protecting group in all the 2-amino pyridine analogues was removed by refluxing in an aqueous ethanol solution of hydroxylamine hydrochloride as previous reported.11 Although no details were given in the reference, we found that a 2:1 mixture of ethanol and water (containing 4 M NH2OH·HCl) at 110 °C afforded good results. The nucleophilic substitutions of BrCH2CH2F, BrCH2CH2CH2F, and (CH3)3SiCl by 6 afforded 17, 19, and 21, respectively in high yields (Scheme 3). Among the compounds synthesized, 9, 13, 15, 16, 27, 30, and 33 are racemic mixtures which were used without chiral resolution in the following studies.
Scheme 1.

Reagents and conditions: (a) HOAc, Toluene, reflux; (b) n-BuLi, Et2O, -20 to -10 °C.
Scheme 2.

Reagents and conditions: (a) CH3CHO, Et2O, -78 °C to RT; (b) DAST, CH2Cl2 or PBSF, (NEt3)(HF)3, Et3N, CH3CN; (c) NH2OH·HCl, EtOH, H2O; (d) CH3I or BrCH2CH2F, NaH, THF.
Scheme 3.

Reagents and conditions: (a) 17: BrCH2CH2F or 19: BrCH2CH2CH2F or 21: (CH3)3SiCl, Et2O; (b) NH2OH·HCl, EtOH, H2O.
The Peterson olefination reaction13 using α-trimethylsilyl carbanion 22 and the corresponding ketones was used to synthesize 23, 25, 28, and 31, respectively (Scheme 4). Compound 22 was synthesized by reacting 21 with one equivalent of n-butyl lithium in good yield, which was evidenced by the good yield of 23 following the reaction with acetone. E/Z isomers were observed in these reactions and the isomers were reduced either by ammonium formate/ethanol in the presence of palladium on carbon (25 and 28) or by magnesium in ethanol (31). The former method was unsuccessful for the synthesis of 32, most likely due to the poisoning of the palladium catalyst by the sulfur atom in 31.
Scheme 4.

Reagents and conditions: (a) n-BuLi, Et2O, -20 °C; (b) acetone, Et2O, - 78 °C to RT; (c) 1N HCl; (d) NH2OH·HCl, EtOH, H2O; (e) 25: CH3COCH2CH2OCH3; 28: CH3COCH2CH2F; or 31: CH3COCH2CH2SCH3, Et2O, -78 °C to RT; (f) HCOONH4, Pd/C, H2, EtOH, 80 °C (25, 28); or Mg, EtOH (31).
The synthesis of precursor 38 for the nucleophilic labeling of [18F]9 is shown in Scheme 5. The hydroxyl group in the 2-hydroxypropyl group had to be protected as the corresponding acetate ester during the reaction with di-tert-butyldicarbonate (Boc2O). Without protection of the -OH, 37 was only a minor product with the major product as the corresponding t-butylcarbonate and other by-products (data not shown). The acetylation to make 34 from 7 using acetyl chloride was slow and the yield was only 39%, probably due to steric hindrance from the secondary alcohol and the pyridine ring. A more efficient method of making 34 involved treating 6 with acetaldehyde followed by treatment with ethyl acetate to give 34 in an overall yield of 43%.
Scheme 5.

Reagents and conditions: (a) 6: CH3CHO, Et2O, -78 °C to RT; then AcOEt, Et2O; (b) 7: AcCl, Et3N, CH2Cl2; (c) NH2OH·HCl, EtOH, H2O; (d) Boc2O, t-BuOH; (e) K2CO3, MeOH, H2O; (f) MsCl, Et3N, CH2Cl2
The synthesis of the mesylate and tosylate precursors required for the radiosynthesis of [18F]18 failed to afford the desired products. It was found that the mesylate precursor formed initially but converted quickly even at room temperature to a cyclic 1-substituted pyridinium via an internal nucleophilic attack of the mesylate group by the pyridine nitrogen atom, and this cyclized product was confirmed by mass spectrometry, 1H NMR and elemental analysis (data not shown). Therefore, attempts to radiolabel 18 were not successful and our efforts focused on radiolabeling 9 with 18F.
Radiosynthesis of [18F]9
[18F]9 was synthesized via a nucleophilic substitution of the mesylate precursor 38 with [18F]fluoride, followed by deprotection with 1N HCl (Scheme 6). The incorporation of [18F]fluoride was only 10-20% at optimized conditions; however, after reversed phase HPLC purification, [18F]9 was obtained in high chemical and radiochemical purity. The specific activity was >1,000 mCi/μmol at the end of synthesis. The total synthesis and purification time was 120 min and the isolated yield was up to 10% (decay corrected). The low incorporation of [18F]fluoride should be due to the slow rate of nucleophilic substitution on the secondary carbon, and the base-catalyzed elimination to form the corresponding nonreactive olefin. The radiolabeling using acetonitrile as the solvent gave a higher yield than that in DMF; use of the corresponding tosylate and triflate precursors gave a much lower yield or no incorporation of 18F at all.
Scheme 6.

Reagents and conditions: (a) [18F]fluoride, K2CO3, K222, CH3CN, 110 °C; (b) 1N HCl, microwave.
In vitro enzyme assays
The inhibition potency for recombinant iNOS, eNOS and nNOS was determined using commercial Nitric Oxide Synthase Screening Kit (GE Healthcare Biosciences Corp., Piscataway, NJ) following the manufacturer’s protocol with minor modifications. The assay was validated using standard NOS inhibitors. Only the most potent iNOS inhibitors were further evaluated for eNOS and nNOS potency to determine the selectivity between iNOS and eNOS or nNOS.
As shown in Table 1, the most potent compound for iNOS was 18, with approximately 30-fold selectivity against eNOS and 10-fold against nNOS; 9 and 20 showed less potency for iNOS and less selectivity against eNOS and nNOS, but were comparable to the previously reported compound 2, which had a potency of IC50 = 193 nM for iNOS in our assay (The reported IC50 of 2 is 28 nM10). We cannot determine the reasons for the difference between our value and the reported data, but slow time-dependent inhibition has been frequently reported. 8b, 11, 14,15 The IC50 value may be influenced by the incubation time, longer incubation time may deliver more potent IC50 values. Additionally, the measured inhibition potency may depend on the concentrations of NADPH and L-arginine.15 Nevertheless, the assay was validated by standard iNOS inhibitors (Table 1), and 9, 18 and 20 were identified as potential PET tracers for imaging iNOS according to our assay results.
Table 1.
IC50 values of the 2-Amino-4-Methylpyridine analogsa
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NOS | IC50 (nM ± Standard Deviation, n ≥ 3)b |
|||||||||||
| 2 | 9 | 24 | 11 | 16 | 13 | 15 | 18 | 20 | 30 | 27 | 33 | |
| iNOS | 193±38 (28)c |
220±25 | 685±127 | 282±49 | 1776±395 | >5000 | >5000 | 57.6±5.3 | 170±26 | 731±87 | >5000 | >5000 |
| eNOS | (150)c | 1500±300 | -d | - | - | - | - | 1428±158 | - | - | - | - |
| nNOS | (100)c | 490±80 | - | - | - | - | - | 514±83 | - | - | - | - |
Note: Using recombinant human iNOS, eNOS and nNOS
Standard iNOS IC50 [measured (reported)]: SEITU = 30.5±4.6 (32), L-NIL = 1465±148 (1400), 2-AP = 108±35 (170)
ref. 10
“-” not determined.
It has been reported previously that the 6-position of the pyridine ring has the greatest amount of bulk tolerance with respect to potency for inhibiting iNOS. Therefore, several 6-substituted analogs were synthesized in order to further investigate this structure-activity relationship. When the 2′-methyl group in lead compound 2 was replaced with a fluorine atom (i.e., 9) the potency for iNOS remained the same. Introduction of a double bond to 2 to give an alkene analog, 24, resulted in a diminished potency for iNOS. Removal of the cis methyl from 24 (i.e., 11) regained the potency for iNOS (IC50 = 282 nM); this change in potency for iNOS from 24 and 11 implies that steric demand at the 6-position is high. The change from the methyl in 2 or fluorine in 9 to the hydroxy group in 16 resulted in a large reduction in potency for iNOS, suggesting an adverse electronic substituent effect in this position. The corresponding methoxy (i.e., 13) and 2-fluoroethoxy (i.e., 15) analogs were inactive. Compound 18, an isomer of 9 with the fluorine at the terminal position, had enhanced potency and selectivity for iNOS; however, extension of the alkyl chain in 18 by one methylene group (i.e., 20) resulted in a reduction of potency for iNOS. Addition of a methyl group in the 2′-position of 20 to give 30 resulted in a further decrease in potency for iNOS. The corresponding MeO or MeS analogs, 27 and 33, were inactive in the iNOS assay. In summary, there appear to be significant steric and electronic constraints for substituents at the 6-position of the pyridine ring with respect to potency for inhibiting iNOS.
In vitro stability and in vivo metabolism studies
An in vitro stability study was carried out using heparinized rat blood taken from an adult male Sprague-Dawley rat. [18F]9 was relatively stable in the whole blood for the duration of the experiment (2 h) at 37 °C. At 1 h, ~80% of the activity recovered from lysed whole blood was observed as [18F]9, and at 2 h, ~75% of the recovered activity was still the parent compound. These data suggest that the blood is not a major site for compound degradation, which is in contrast to the previously reported 18F and 11C labeled isothiourea analogues.12a
The in vivo metabolic stability of [18F]9 was evaluated in plasma samples obtained from an adult male Sprague-Dawley rat at 5 and 30 min post-injection. The supernatant extracts were analyzed by silica gel radio-TLC and reversed phase HPLC. After 30 min, the percent parent compound was only 20.2%. Additionally, within 5 min post-injection only 40.3% of the activity in the blood was [18F]9, which was confirmed by HPLC co-elution with non-radioactive 9. According to the HPLC analysis, the major metabolite, constituting 50% of the activity in blood at 5 min post-injection, was very polar but was not free [18F]fluoride. This observation is also consistent with the low bone uptake reported below in the biodistribution studies (Table 2). Since [18F]9 demonstrated reasonable in vitro stability in whole blood, the metabolite observed in vivo at 5 min in blood must be due to metabolism in peripheral organs.
Table 2.
Biodistribution of [18F]9 in control vs. LPS treated micea (data reported as mean %ID/g ± SD; n = 4)b
| %ID/g | 5 min | 30 min | 1 h | 2 h | ||||
|---|---|---|---|---|---|---|---|---|
| LPS | Control | LPS | Control | LPS | Control | LPS | Control | |
| Blood | 3.69±0.38 | 2.37±0.18 | 2.09±0.35 | 1.22±0.16 | 0.86±0.09 | 0.31±0.04 | 0.23±0.05 | 0.27±0.22 |
| Lung | 4.32±0.32 | 3.33±0.28 | 1.84±0.27 | 1.14±0.13 | 0.73±0.17 | 0.31±0.06 | 0.18±0.09 | 0.15±0.03 |
| Liver | 24.6±3.1 | 33.2±3.3 | 7.06±0.28 | 10.7±0.3 | 2.63±0.52 | 2.59±0.49 | 0.42±0.15 | 0.74±0.23 |
| Kidney | 26.7±4.1 | 20.6±2.4 | 10.9±2.8 | 7.30±0.66 | 3.95±0.53 | 2.20±0.25 | 0.91±0.63 | 0.80±0.27 |
| Muscle | 1.64±0.09 | 1.25±0.06 | 1.06±0.40 | 0.71±0.31 | 0.78±0.47 | 0.27±0.18 | 0.23±0.13 | 0.34±0.27 |
| Heart | 2.03±0.06 | 1.50±0.12 | 1.02±0.16 | 0.60±0.05 | 0.41±0.10 | 0.19±0.02 | 0.12±0.04 | 0.08±0.02 |
| Brain | 1.81±0.09 | 1.60±0.17 | 0.52±0.15 | 0.28±0.02 | 0.19±0.03 | 0.11±0.02 | 0.07±0.03 | 0.07±0.03 |
| Bone | 1.59±0.15 | 1.52±0.36 | 3.70±1.20 | 2.26±0.55 | 4.30±0.90 | 2.57±0.64 | 3.77±1.41 | 5.59±4.20 |
Note: LPS mice were treated with lipopolysaccharide (LPS) 10 mg/kg i.v. 6 h prior to tracer injection; all mice were injected with 50 μCi/110 μL [ Review.18F]9
SD: standard deviation.
In vivo biodistribution study
The biodistribution studies were performed in mature male C57BL/6 mice: one group was treated with bacterial lipopolysaccharide (LPS) (10 mg/kg, i.v.) to induce iNOS expression and one group without LPS-treatment as control. LPS has been well documented to induce iNOS mRNA and protein expression in both rats and mice,16 and administration of LPS resulted in an elevated iNOS expression at 6-7 h post-LPS-treatment. The iNOS distribution in organs was reported recently in male BALB/c mice.16c It has been demonstrated in that report that iNOS mRNA and protein expression 6 h after LPS stimulation is observed in many organs, including lungs, heart, liver, spleen, gut, and kidneys. Among them, the highest iNOS expression was in the lungs, with moderate expression in the spleen and kidneys, and the lowest in the heart, gut, and liver.16c Therefore, this endotoxin (LPS) lung injury mouse model was used to assess the efficiency of [18F]9 as an iNOS radiotracer.
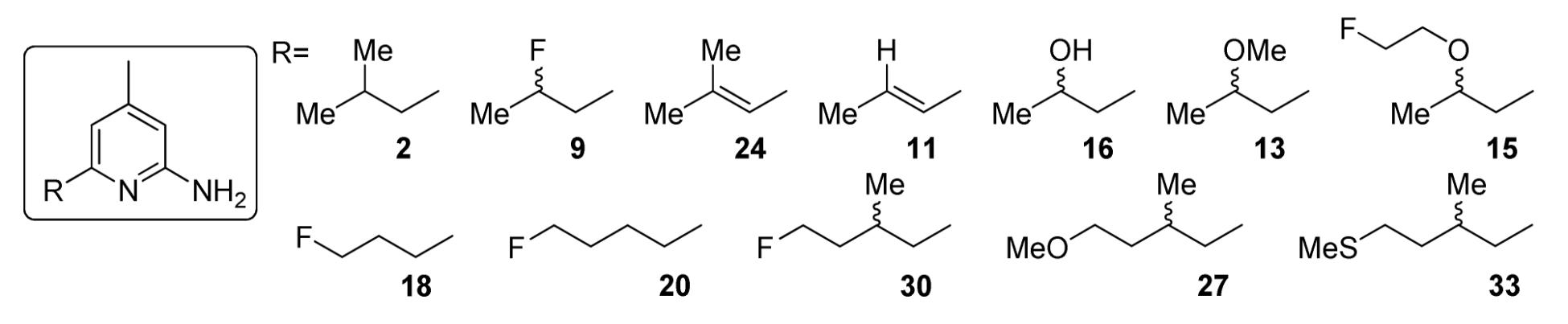
The results of in vivo biodistribution studies in the normal and LPS-treated mice are shown in Table 2 and Figure 1. The highest uptake in both groups was observed in the liver and kidneys, which are likely to be the major metabolic and/or excretory sites for the tracer. Excluding the potential metabolic sites, the highest uptake was observed in the lungs and blood, followed by muscle, heart, and brain in both groups. The low bone uptake suggested that [18F]9 had a high degree of stability towards defluorination, considering the facile elimination to form a conjugated double bond at this position. Compared to the control group, the LPS-treated mice had a ~30% increase in tracer uptake in most of the organs at 1 h. In the organ expected to have the highest iNOS expression, the lungs, the increase in injected dose/gram (ID/g) was even more pronounced, 30% at 5 min, 60% at 30 min and 130% at 1 h. Because systemic LPS-induced acute lung injury results in pulmonary edema, the difference in total lung uptake between control and treated animals (i.e., %I.D.) is more dramatic than the %ID/g in the lung (Figure 1). The overall higher uptake in tissues of the LPS-treated mice compared to the controls is consistent with the previously reported systemic iNOS response with LPS treatment.16c From 30 min to 1 h, the uptake in the LPS-treated mice washed out at a slower rate than that of the control mice, which again can be attributed to the specific binding of [18F]9 to iNOS. Based on the in vivo metabolic stability study in the blood of normal rat (Table 2), there should be little circulating [18F]9 left at 2 h post-injection. At 2 h post-injection, all the activity was washed out to the same low level and the uptake at 2 h post-injection should be due to the non-specific uptake of the metabolites. In previously reported biodistribution studies in rats injected with less potent and less selective 18F and 11C labeled isothiourea analogues, about 30% higher uptake of 18F labeled analog was observed in lungs, blood, liver, kidneys, and heart of LPS-treated rats than that of normal rats at 10 min post-injection. However, the difference in uptake between the two groups the differences became insignificant at 30 min post-injection, possibly due to the rapid metabolism of these radiotracers. The more stable 11C labeled analog showed 40% higher uptake at 30 min post-injection in the lungs of the LPS-treated rats than that of the control.12a Compared to the previously described 18F labeled isothiourea analogue, [18F]9, with prolonged retention in the LPS-treated mice, is a suitable PET tracer for iNOS.
Figure 1.
Comparison of total lung activity post-injection of [18F]9 in LPS-pretreated mice vs. control mice.
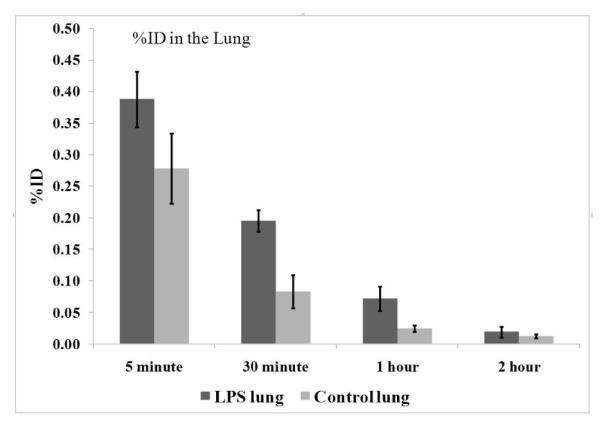
A standard Western blot was performed to compare the iNOS induction in lungs upon LPS stimulation versus untreated controls and the results are shown in Figure 2. Protein for the Western blot was obtained from the lungs harvested from treated and untreated animals in the biodistribution study. The first lane is the purified iNOS protein as a positive control. Protein from two control lungs and six LPS-treated lungs were evaluated. As indicated in Figure 2, neither of the control samples demonstrated iNOS expression whereas the LPS-treated samples all showed some degree of iNOS induction due to the response toward LPS stimulation. This result is consistent with the higher tracer uptake in the lungs of the LPS-treated mice in the biodistribution studies, suggesting iNOS-specific uptake of [18F]9.
Figure 2.
Representative Western blots showing the levels of iNOS expression in lungs from the control and LPS treated mice. The first lane is purified iNOS as a positive control (iNOS); no iNOS expression was observed in the lungs of normal mice (C); different degrees of iNOS expression were found in the lungs of the LPS-treated mice (T).
Blocking study
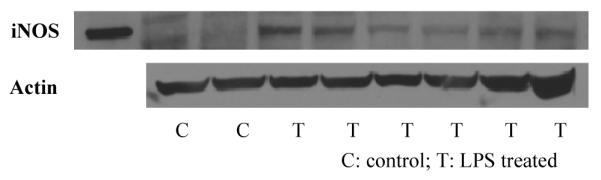
In order to further demonstrate that the increased uptake in the LPS-treated mice was due to specific binding of [18F]9 to iNOS, blocking studies were carried out. First, the non-selective NOS inhibitor 2-amino-4-methylpyridine (1) (10 mg/kg, i.v.) failed to block the increased uptake in the LPS-treated mice (data not shown). A failure to block tracer uptake using non-selective NOS inhibitors S-methyl or S-ethyl isothiourea has been reported previously;12a the failure was contributed to the blood pressure change caused by the blocking agents in the animal, which may alter the tracer uptake function. 2-amino-4-methylpyridine (1) elevates blood pressure9a,10,17 and blocking of eNOS expression may also reduce the blood flow.18 Therefore, a highly selectively iNOS inhibitor is preferred in order to avoid the side-effects from non-selective inhibitors. N-(3-(aminomethyl)benzyl)acetamidine (1400W) is a slow, tight binding, and highly selective inhibitor of iNOS in vitro and in vivo,8b and has been used commonly as an iNOS inhibitor in many reported studies. However, due to its high toxicity,19 only 5 mg/kg of 1400W was injected intravenously immediately before the injection of [18F]9. The results are shown in Figure 3. At 1 h post injection of [18F]9, 32% of the tracer uptake in the lungs and blood of the LPS-treated mice was blocked by 1400W (Figure 3). This reduced uptake in the lungs of the LPS-treated mice should be due to the specific blocking of iNOS by 1400W, and is consistent with that the uptake of [18F]9 in the LPS-treated mice is iNOS specific.
Figure 3.
Comparison of activity in the lung 1 h post-injection of [18F]9 in the control, LPS treated, and LPS-treated-1400W-blocked mice (mean %ID/g ± SD, n = 4, p < 0.05)
MicroPET study
A microPET imaging study using [18F]9 was carried out on an intratracheally LPS-treated mouse and a normal mouse as control. The microPET images (0-60 min dynamic scan) are shown in Figure 4. As shown in the microPET images (Figure 4), accumulation of [18F]9 was observed in the target organ, the lungs, of the LPS-treated mouse, whereas no such accumulation was observed in the lungs of normal mouse. The difference in the tracer uptake in the lungs of the LPS-treated and control mice is consistent with iNOS expression induced by LPS treatment and is similar to the results of biodistribution study.
Figure 4.

Whole-body MicroPET images of [18F]9 in the LPS-treated mice (left) and the control mice (right). (a) transaxal, (b)coronal. Imagines were summed from 0 to 60 min after injection of [18F]9. Accumulation of the activity was observed in the lungs of the LPS-treated mouse, whereas no such accumulation in the normal control.
Conclusion
We have identified 6-(2-fluoropropyl)-4-methylpyridin-2-amine (9), 6-(3-fluoropropyl)-4-methylpyridin-2-amine (18), and 6-(4-fluorobutyl)-4-methylpyridin-2-amine (20) as iNOS inhibitors with the greatest potential to serve as PET radiotracers. [18F]9 was synthesized in modest yield (~10%), but with high chemical and radiochemical purities. In the biodistribution study of a mouse model of LPS-induced iNOS activation, higher uptake of [18F]9 was observed in the lungs of the LPS-treated mice than those in the control mice. The higher uptake in the lungs of the LPS-treated animals correlated well with iNOS expression, which was confirmed by Western blot analysis of the lung samples from control and LPS-treated mice. The increased uptake in the lungs of the LPS-treated mice at 60 min post injection of [18F]9 was reduced by injection of the highly selective iNOS inhibitor, 1400W. The blocking study suggests that the higher uptake of 18F]9 in the lungs of the LPS-treated mice is due to specific binding of [18F]9 to [iNOS. MicroPET study of the LPS-treated mouse using [18F]9 demonstrated an accumulation of [18F]9 in the lungs of the LPS-treated mice, in sharp contrast to those of the control mouse. In conclusion, [18F]6-(2-Fluoropropyl)-4-methylpyridin-2-amine ([18F]9) is a potential radiotracer for PET imaging of iNOS expression.
Experimental Section
General methods and materials
All chemicals were obtained from standard commercial sources and used without further purification. All reactions were carried out by standard air-free and moisture-free techniques under an inert argon atmosphere with dry solvents unless otherwise stated. Flash column chromatography was conducted using Scientific Adsorbents, Inc. silica gel, 60A, “40 Micron Flash” (32-63 μm). Melting points were uncorrected. Routine 1H NMR spectra were recorded at 300 MHz. All chemical shifts were reported as a part per million (ppm) downfield from tetramethylsilane (TMS) or when chloroform-d was used as solvent and the solvent peak at δ 7.25 ppm was used as an internal standard. All coupling constants (J) are given in Hz. Splitting patterns are typically described as follows: s, singlet; d, doublet; t, triplet; m, multiplet. 19F NMR spectra were recorded at 282.2 MHz, and chemical shifts are reported as Hz upfield from an external CFCl3 standard. ESI/MS was performed on a Waters ZQ 4000 single quadrupole mass spectrometer equipped with an electrospray ionization (ESI) LC-MS interface. High performance liquid chromatography (HPLC) was performed with an ultraviolet detector operating at 251 nm and a well-scintillation NaI (Tl) detector and associated electronics for radioactivity detection. Alltech Platinum EPS C18 250 × 10 mm semi-preparative column and Alltech Platinum EPS C18 250 × 4.6 mm analytical column were used for preparation and analysis respectively. 4-Methoxybutan-2-one was synthesized according to literature.20 The purities of final compounds are ≥95%, which were confirmed by examination of elemental analysis results or by reversed phase HPLC (for compound 13, 15, 18, 24, and 27).
H2 18O was purchased from Rotem Industries (Israel). [18F]Fluoride was produced in Washington University by the 18O(p,n)18F reaction through proton irradiation of enriched (95%) 18O water using RDS111 cyclotron. Materials were heated using a custom-designed microwave cavity, model 420BX (Micro-Now Instruments, Skokie, IL). Screw-cap test tubes used for microwave heating were purchased from Fisher Scientific (Pyrex No. 9825). HLB Sep-Pak cartridges were purchased from Waters Corporation. For the TLC analyses, EM Science Silica Gel 60 F254 TLC plates were purchased from Fisher Scientific (Pittsburgh, PA). Radio-TLC was accomplished using a Bioscan 200 imaging scanner (Bioscan, Inc., Washington, DC). Radioactivity was counted with a Beckman Gamma 6000 counter containing a NaI crystal (Beckman Instruments, Inc., Irvine, CA).
Typical procedure for the synthesis of 6
Into a solution of 5 (2.0 g, 10 mmol) in dry Et2O (20 mL) at -20 °C was added dropwise n-BuLi (1.6 M in Hexane, 6.9 mL, 10 mmol) within 5 min, then the reaction mixture was stirred at -20 to -10 °C for 1 h to complete the reaction. Orange solids precipitated gradually during the experiment, and the reaction mixture of 6 was used directly for further reactions.
1-(6-(2,5-Dimethyl-1H-pyrrol-1-yl)-4-methylpyridin-2-yl)propan-2-ol (7)
Into an Et2O solution of 6 at -78 °C, which was made from 5 (5.6 g, 28.0 mmol), n-BuLi (1.6 M in Hexane, 19.5 mL, 31.2 mmol) in Et2O (30 mL), was added CH3CHO (2 mL, 35.6 mmol) via a syringe. The reaction mixture was allowed to warm up to room temperature and stirring was continued for 10 min. Then, the reaction mixture was treated with H2O (100 mL), and extracted with ethyl acetate (3 × 50 mL). The organic solution was washed with brine (2 × 30 mL), dried over NaSO4 and the solvent was evaporated under reduced pressure. The crude product was purified with silica gel chromatograph using 1:4 ethyl acetate/hexanes to afford 7 (5.6 g) in 80 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 6.97 (s, 1H), 6.89 (s, 1H), 5.87 (s, 2H), 4.51 (s, br, 1H), 4.23 (m, 1H), 2.94-2.80 (m, 2H), 2.39 (s, 3H), 2.12 (s, 6H), 1.25 (d, 3H, J = 6.3 Hz).
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(2-fluoropropyl)-4-methylpyridine (8)
Using diethylaminosulfur trifluoride (DAST) as fluorinating agent
Into a solution of 7 (0.5 g, 2.05 mmol) in CH2Cl2 (5 mL) at 0 °C was added DAST (0.4 mL, 3.05 mmol) dropwise. The reaction mixture became brown in color, and the starting material was consumed within 30 min. Saturated NaHCO3 solution (10 mL) was added to quench the reaction, and the aqueous solution was extracted with ethyl acetate (2 × 30 mL). The organic layers was washed with brine (2 × 30 mL) and dried over Na2SO4. Solvents were evaporated under reduced pressure and the crude product was purified with silica gel chromatograph using 1:10 ethyl acetate/hexanes to afford 8 (0.13 g) in 26 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 7.08 (s, 1H), 6.93 (s, 1H), 5.91 (s, 2H), 5.05-5.60 (m, 1H), 3.20 (m, 2H), 2.43 (s, 3H), 2.15 (s, 6H), 1.43 (dd, 3H, J = 23.4, 6.3 Hz); ESI/MS m/z 247.06 [M+H+].
Using perfluorobutane sulfonyl fluoride (PBSF) as fluorinating agent
Into a solution of 7 (0.5 g, 2.05 mmol) in CH3CN (5 mL) were added PBSF (0.74 mL, 4.1 mmol), Et3N (1.73 mL, 12.4 mmol) and (NEt3)(HF)3 (0.67 mL, 4.11 mmol). The reaction mixture was stirred quickly and became homogenous after 40 min. The reaction was complete in 2.5 h according to TLC analysis, and the solvent was evaporated under reduced pressure. Silica gel chromatograph purification of the residue using 1:10 ethyl acetate/hexanes afforded 8 (0.13 g) in 26 % yield as colorless liquid.
6-(2-Fluoropropyl)-4-methylpyridin-2-amine (9): Typical procedure for the deprotection
Into a 50 mL round-bottom flask equipped with a magnetic stirring bar and a condenser were added 8 (1.10 g, 4.1 mmol) and NH2OH·HCl (10 g, 144 mmol), followed by ethanol (25 mL) and water (12.5 mL). The reaction mixture was stirred at refluxed and the reaction was monitored by TLC. Upon the completion of the reaction in about 3 h, the reaction mixture was treated with saturated Na2CO3 solution (10 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers was washed with brine (2 × 20 mL) and dried over Na2SO4. After removal of volatiles under reduced pressure, the crude product was purified with silica gel chromatograph using ethyl acetate to afford 9 (0.61 g) in 81 % yield as light yellow solid (mp. 51.0-52.5 °C; oxalate 154.0-155.9 °C). The oxalate was precipitated using 1:1 of 9 and oxalic acid in ethyl acetate to afford the salt as a white solid. 1H NMR (CD3Cl, 300 MHz) δ 6.40 (s, 1H), 6.18 (s, 1H), 5.20-4.90 (m, 1H), 4.37 (s, br, 2H), 3.00-2.70 (m, 2H), 2.19 (s, 3H), 1.37 (dd, 3H, J = 24.0, 6.3 Hz).
(E)-2-(2,5-Dimethyl-1H-pyrrol-1-yl)-4-methyl-6-(prop-1-enyl)pyridine (10)
10 was obtained as a by-product during the synthesis of 9 using PBSF as fluorinating agent in 49 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 7.00 (s, 1H), 6.83 (s, 1H), 6.85-6.78 (m, 1H), 6.51-6.44 (s, 1H), 5.88 (s, 2H), 2.38 (s, 3H), 2.15 (s, 6H), 1.92 (d, 3H, J = 6.9 Hz).
(E)-4-Methyl-6-(prop-1-enyl)pyridin-2-amine (11)
11 was synthesized from 10 using the standard deprotection procedure in 53 % yield as a solid (mp. 66.4-67.0 °C; oxalate 137.5-138.7 °C)). 1H NMR (CD3Cl, 300 MHz) δ 6.67-6.56 (m, 1H), 6.40 (s, 1H), 6.28 (d, 1H, J = 15.3 Hz), 6.14 (s, 1H), 4.42 (s, br, 2H), 2.17 (s, 3H), 1.86 (d, 3H, J = Hz).
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(2-methoxypropyl)-4-methylpyridine (12)
Into a solution of 7 (0.9 g, 3.68 mmol) in freshly distilled THF (20 mL) was added 60% NaH in mineral oil (0.3 g, 12.6 mmol). After 5 min, MeI (0.5 mL, 8.03 mmol) was added, and the reaction mixture was stirred at room temperature for 5 h. The mixture was then quenched with an aqueous 1 N HCl solution, followed by neutralization with saturated solution of aqueous Na2CO3. The product was extracted with ethyl acetate (3 × 20 mL) and the combined organic layers was washed with brine (30 mL) and dried over MgSO4. After evaporation of solvents under reduced pressure, silica gel chromatograph purification of the residue using 1:4 ethyl acetate/hexanes afforded 12 (0.89 g) in 94% yield as a light yellow liquid. 1H NMR (CD3Cl, 300 MHz) δ 7.01 (s, 1H), 6.86 (s, 1H), 5.87 (s, 2H), 3.84 (m, 1H), 3.30 (s, 3H), 3.05-2.70 (m, 2H), 2.39 (s, 3H), 2.11 (s, 6H), 1.17 (d, 3H, J = 6.0 Hz).
6-(2-Methoxypropyl)-4-methylpyridin-2-amine (13)
13 was synthesized from 12 using the standard deprotection method. The corresponding oxalate was precipitated using 1:1 of 13 and oxalic acid in ethyl acetate to afford the salt as a as white solid (mp. oxalate 123-125 °C).1H NMR (CD3Cl, 300 MHz) δ 6.35 (s, 1H), 6.13 (s, 1H), 4.49 (s, br, 2H), 3.71 (m, 1H), 3.30 (s, 3H), 2.89-2.49 (m, 2H), 2.16 (s, 3H), 1.12 (d, 3H, J = 6 Hz).
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(2-(2-fluoroethoxy)propyl)-4-methylpyridine (14)
Into a solution of 7 (1.0 g, 4.1 mmol) in freshly distilled THF (20 mL) were added 60 % NaH in mineral oil (0.35 g, 8.8 mmol) and 1-bromo-2-fluoroethane (1.0 mL, 8 mmol). The reaction mixture was stirred at room temperature for 6 h, then at 60 °C overnight. The reaction mixture was filtered and the organic solution was washed with 1 N HCl solution, saturated Na2CO3, and brine and then dried over Na2SO4. Solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatograph using 1:8 ethyl acetate/hexanes to afford 14 (0.37 g) in 31 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 7.04 (s, 1H), 6.86 (s, 1H), 5.87 (s, 2H), 4.54- 4.35 (m, 2H), 3.97 (m, 1H), 3.79-3.53 (m, 2H), 3.09-2.81 (m, 2H), 2.38 (s, 3H), 2.10 (s, 6H), 1.20 (d, 3H, J = 6.3 Hz).
6-(2-(2-Fluoroethoxy)propyl)-4-methylpyridin-2-amine (15)
15 was synthesized from 14 using the standard deprotection procedure in 81% yield as solid (mp. 60.2-63.0 °C; oxalate 163- 164 °C).1H NMR (CD3Cl, 300 MHz) δ 6.40 (s, 1H), 6.16 (s, 1H), 4.56-4.37 (m, 2H), 4.35 (s, br, 2H), 3.88 (m, 1H), 3.67-3.53 (m, 2H), 2.92-2.55 (m, 2H), 2.18 (s, 3H), 1.18 (d, 3H, J = 6.3 Hz).
1-(6-Amino-4-methylpyridin-2-yl)propan-2-ol (16)
16 was synthesized from 7 using the standard deprotection procedure in 66 % yield as a light yellow solid (mp. 95-96 °C).1H NMR (CD3Cl, 300 MHz) δ 6.31 (s, 1H), 6.18 (s, 1H), 4.30 (s, br, 1H), 4.12 (m, 1H), 2.62 (m, 2H), 2.19 (s, 3H), 1.23 (d, 3H, J = 6.0 Hz).
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(3-fluoropropyl)-4-methylpyridine (17)
Into an Et2O solution of 5 at -78 °C, which was made from 3 (2.0 g, 10 mmol), n-BuLi (1.6 M in Hexane, 8.0 ml, 12.8 mmol)) in Et2O (20 ml), was added BrCH2CH2F (2 .0 g, 15.7 mmol) via a syringe. The reaction mixture was allowed to warm up to room temperature and continued to stir for 30 min. Then, the reaction mixture was treated with saturated Na2CO3 solution and then brine. The organic solution was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude product was purified with silica gel chromatograph using 1:8 ethyl acetate/hexanes to afford 29 (1.6 g) in 61 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 7.04 (s, 1H), 6.91 (s, 1H), 5.92 (s, 2H), 4.53 (dt, 2H, J = 6.0, 47.4 Hz), 2.95 (t, 2H, J = 7.5 Hz), 2.44 (s, 3H), 2.3-2.1 (m, 2H), 2.16 (s, 6H); 19F NMR (CD3Cl, 282.2 MHz) δ: -42.9.
6-(3-Fluoropropyl)-4-methylpyridin-2-amine (18).
18 was synthesized from 17 using the standard deprotection procedure in 55 % yield as light yellow solid (mp. oxalate 112 °C decomposition). 1HMR (CD3Cl, 300 MHz) δ 6.35 (s, 1H), 6.14 (s, 1H), 4.45 (dt, 2H, J = 6.0, 47.1 Hz), 4.37 (s, br, 2H), 2.66 (t, 2H, J = 7.6 Hz), 2.18 (s, 3H), 2.20-2.00 (m, 2H); 19F NMR (CD3Cl, 282.2 MHz) δ -42.6.
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(4-fluorobutyl)-4-methylpyridine (19)
Into an Et2O solution of 6 at -78 °C, which was made from 5 (3.0 g, 15 mmol), n-BuLi (1.6 M in Hexane, 11.0 ml, 17.6 mmol)) in Et2O (30 ml), was added BrCH2CH2CH2F (2.1 g, 15.0 mmol) via a syringe. The reaction mixture was allowed to warm up to room temperature and continued to stir for 30 min. The reaction mixture was then treated with saturated Na2CO3 solution and brine. The organic was dried over NaSO4 and the solvent was evaporated under reduced pressure. The crude product was purified with silica gel chromatograph using 1: 10 ethyl acetate/hexanes to afford 19 (2.5 g) in 63 % yield as colorless liquid. 1HMR (CD3Cl, 300 MHz) δ 6.98 (s, 1H), 6.85 (s, 1H), 5.87 (s, 2H), 4.47 (dt, 2H, J = 6.0, 47.1 Hz), 2.82 (t, 2H, J = 7.5 Hz), 2.39 (s, 3H), 2.11 (s, 6H), 1.9-1.7 (m, 4H); 19F NMR (CD3Cl, 282.2 MHz) δ - 41.7.
6-(4-Fluorobutyl)-4-methylpyridin-2-amine (20)
20 was synthesized from 19 using the standard deprotection procedure in 57 % yield as light yellow solid (mp. Oxalate 133.0-135.0 °C).1HMR (CD3Cl, 300 MHz) δ 6.33 (s, 1H), 6.14 (s, 1H), 4.44 (dt, 2H, J = 6.0, 47.4 Hz), 4.38 (s, br, 2H), 2.58 (t, 2H, J = 7.4 Hz), 2.18 (s, 3H), 1.8-1.65 (m, 4H). 19F NMR (CD3Cl, 282.2 MHz) δ: - 41.5.
2-(2,5-Dimethyl-1 H-pyrrol-1-yl)-4-methyl-6-((trimethylsilyl)methyl)pyridine (21)
Into an Et2O solution of 6 at -78 °C, which was made from 5 (5.78 g, 28.9 mmol), n-BuLi (1.6 M in Hexane, 21.6 mL, 34.6 mmol) in Et2O (50 mL), was added Me3SiCl (4.4 mL, 34.6 mmol) via a syringe. The reaction mixture was allowed to warm to room temperature and continued to stir for 30 min. Then, the reaction mixture was treated with saturated Na2CO3 solution and washed with brine. The organic solution was dried over NaSO4 and the solvent was evaporated under reduced pressure. The crude product was purified with silica gel chromatograph using 1:4 ethyl acetate/hexanes to afford 21 (7.4 g) in 94 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 6.79 (s, 1H), 6.72 (s, 1H), 5.84 (s, 2H), 2.35 (s, 2H), 2.07 (s, 6H), 0.03 (s, 9H); ESI/Ms: m/z 273.2 (M+H+).
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-4-methyl-6-(2-methylprop-1-enyl)pyridine (23)
Into a solution of 21 (1.0 g, 3.67 mmol) in Et2O (10 mL) at -20 °C was added dropwise n-butyl lithium (1.6 M in hexane, 2.75 mL, 4.4 mmol). The reaction solution turned brown in color and was stirred at -20 to -10 °C for 2 h to afford a solution of ((6-(2,5-dimethyl-1H-pyrrol-1-yl)-4-methylpyridin-2-yl)(trimethylsilyl)methyl)lithium (22). The solution was then cooled to -78 °C, and 2 ml acetone was added. The solution became clear in slightly yellow color, and it was allowed to warm up to room temperature during 20 min. The organic solution was acidified with 1 N HCl (6 mL), followed by washing with saturated Na2CO3 solution, brine, and dried over Na2SO4. Solvent was evaporated under reduced pressure and the crude product was purified with silica gel chromatograph using 1:15 ethyl acetate/hexanes to afford 23 (0.56 g) in 64 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 6.97 (s, 1H), 6.79 (s, 1H), 6.28 (s, 1H), 5.86 (s, 2H), 2.38 (s, 3H), 2.13 (s, 6H), 2.12 (s, 3H), 1.94 (s, 3H); ESI/MS: m/z 241.2 (M+H+).
4-Methyl-6-(2-methylprop-1-enyl)pyridin-2-amine (24)
24 was synthesized from 23 using the standard deprotection procedure in 64 % yield as slightly yellow liquid (mp. oxalate 154-156 °C). 1H NMR (CD3Cl, 300 MHz) δ 6.38 (s, 1H), 6.13 (s, 1H), 6.12 (s, 1H), 4.37 (s, br, 2H), 2.17 (s, 3H), 1.99 (s, 3H), 1.87 (s, 3H).
(E/Z)2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(4-methoxy-2-methylbut-1-enyl)-4-methylpyridine (25)
Into an Et2O solution of 22 at -78 °C, which was made from 21 (1.0 g, 3.67 mmol), n-BuLi (1.6 M in Hexane, 2.75 mL, 4.4 mmol) in Et2O (10 mL), was added 4-methoxybutan-2-one (0.45 g, 4.4 mmol). After the addition, the reaction mixture was allowed to warm up to room temperature during 30 min. The mixture was then acidified with 1 N HCl (6 mL), neutralized by saturated Na2CO3 solution and diluted with ethyl acetate (50 mL). The organic solution was washed with brine and dried over Na2SO4. Solvent was evaporated under reduced pressure and the crude product was purified with silica gel chromatograph using 1:15 and 1:8 ethyl acetate/hexanes to afford E and Z isomers of 23 (0.25 g and 0.22 g) in 45 % total yield as colorless liquid.1H NMR (CD3Cl, 300 MHz) δ 7.01 (s, 1H), 6.81 (s, 1H), 6.35 (s, 1H), 5.85 (s, 2H), 3.55 (t, 2H, J = 7.2 Hz), 3.25 (s, 3H), 2.90 (t, 2H, J = 7.2 Hz), 2.38 (s, 3H), 2.11 (s, 6H), 1.97 (s, 3H) and δ 7.00 (s, 1H), 6.80 (s, 1H), 6.33 (s, 1H), 5.87 (s, 2H), 3.58 (t, 2H, J = 6.6 Hz), 3.37 (s, 3H), 2.48 (t, 2H, J = 6.90 Hz), 2.38 (s, 3H), 2.16 (s, 3H), 2.13 (s, 6H).
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(4-methoxy-2-methylbutyl)-4-methylpyridine (26)
Into a 100 mL round-bottom flask equipped with a magnetic stirring bar were loaded the isomeric mixture of 25 (0.47 g, 1.7 mmol) and absolute EtOH (20 mL), followed by 10% Pd/C (0.25 g). The reaction mixture was stirred in an 80° C oil bath, and the reaction was completed in 1 h according to TLC analysis. Solids were removed by filtration and solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate and the organic solution was treated with saturated Na2CO3 solution (10 mL) and washed with brine, dried over MgSO4. Solvent was evaporated under reduced pressure and the crude product was purified with silica gel chromatograph using 1:10 and 1:4 ethyl acetate/hexanes to afford 26 (0.38 g) in 81 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 6.94 (s, 1H), 6.83 (s, 1H), 5.86 (s, 2H), 3.41 (m, 2H), 3.30 (s, 3H), 2.82- 2.55 (m, 2H), 2.37 (s, 3H), 2.15 (m, 1H), 2.10 (s, 6H), 1.69-1.44 (m, 2H), 0.90 (d, 3H, J = 6.60 Hz).
Synthesis of 6-(4-methoxy-2-methylbutyl)-4-methylpyridin-2-amine (27)
27 was synthesized from 26 using the standard deprotection procedure in 71 % yield as slightly yellow solid (mp. 48.0-50.0 °C). 1H NMR (CD3Cl, 300 MHz) δ 6.32 (s, 1H), 6.14 (s, 1H), 4.35 (s, br, 2H), 3.42 (m, 2H), 3.30 (s, 3H), 2.59-2.33 (m, 2H), 2.18 (s, 3H), 2.05 (m, 1H), 1.70-1.40 (m, 2H), 0.89 (d, 3H, J = 6.90 Hz).
(E/Z>)-2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(4-fluoro-2-methylbut-1-enyl)-4-methylpyridine (28)
28 was synthesized from 22 using the standard deprotection method in 50% yield as a colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 6.97 (s, 1H), 6.79 (s, 1H), 6.38 (s, 1H), 4.61 (dt, 2H, J = 6.0, 47.1 Hz), 2.60 (dt, 2H, J = 6.1, 24.0 Hz), 2.38 (s, 3H), 2.10 (s, 6H), 2.00 (s, 3H) and δ 7.01 (s, 1H), 6.82 (s, 1H), 6.36 (s, 1H), 4.63 (dt, 2H, J = 6.3, 47.1 Hz), 3.09 (dt, 2H, J = 6.1, 25.2 Hz), 2.39 (s, 3H), 2.18 (s, 3H), 2.13 (s, 6H).
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-6-(4-fluoro-2-methylbutyl)-4-methylpyridine (29)
29 was synthesized from a mixture of 28 using ammonium formate in the presence of 10% Pd/C in 30% yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 6.95 (s, 1H), 6.85 (s, 1H), 5.86 (s, 2H), 4.50 (dt, 2H, J = 6.0, 47.4 Hz), 2.83-2.59 (m, 2H), 2.38 (s, 3H), 2.28-2.18 (m, 1H), 2.06 (s, 6H), 1.87-1.48 (m, 2H), 0.93 (d, 3H, J = 6.9 Hz).
6-(4-Fluoro-2-methylbutyl)-4-methylpyridin-2-amine (30)
30 was synthesized from 29 using the standard deprotection procedure in 47% yield as light yellow solid (oxalate: mp. 136.6-137.1 °C). 1H NMR (CD3Cl, 300 MHz) δ 6.31 (s, 1H), 6.15 (s, 1H), 4.67 (s, br, 2H), 4.50 (dt, 2H, J = 6.3, 47.4 Hz), 2.61-2.37 (m, 2H), 2.19 (s, 3H), 2.15-2.05 (m, 1H), 1.90-1.45 (m, 2H), 0.93 (d, 3H, J = 6.3 Hz); 19F NMR (CD3Cl, 282.2 MHz) δ: -41.1.
(E/Z)-2-(2,5-Dimethyl-1H-pyrrol-1-yl)-4-methyl-6-(2-methyl-4-(methylthio)but-1-enyl)pyridine (31)
Into an Et2O solution of 22 at -78 °C, which was made from 21 (1.6 g, 5.87 mmol), n-BuLi (1.6 M in Hexane, 4.8 mL, 7.68 mmol) in Et2O (25 mL), was added 4-(methylthio)butan-2-one (0.90 g, 7.61 mmol). The reaction mixture was allowed to warm to room temperature over 30 min. The reaction solution was then acidified with 1 N HCl (10 mL), followed by neutralization by saturated Na2CO3 solution. The mixture was diluted with ethyl acetate (50 mL), and the organic solution was washed with brine, and dried over Na2SO4. Solvent was evaporated under reduced pressure and the crude product was purified with silica gel chromatograph using 1:10 ethyl acetate/hexanes to afford E/Z isomer mixture of 31 (1.38 g) in 78 % yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 6.94 (s, 1H), 6.80 (s, 1H), 6.28 (d, 1H, J = 1.5 Hz), 5.83 (s, 2H), 2.90 (m, 2H), 2.62 (m, 2H), 2.37 (s, 3H), 2.09 (s, 6H), 1.95 (d, 3H, J = 1.5 Hz), 1.87 (s, 3H) and δ 7.00 (s, 1H), 6.80 (s, 1H), 6.32 (d, 1H, J = 1.2 Hz), 5.86 (s, 2H), 2.70 (m, 2H), 2.48 (m, 2H), 2.38 (s, 3H), 2.14 (s, 3H), 2.14 (d, 3H, J = 1.2 Hz), 2.12 (s, 6H).
2-(2,5-Dimethyl-1H-pyrrol-1-yl)-4-methyl-6-(2-methyl-4-(methylthio)butyl)pyridine (32)
Into a solution of 31 (1.38 g, 4.6 mmol) in EtOH (30 mL) at 0 °C was added Mg turnings (1.25 g, 51 mmol). The reaction mixture was stirred at 0 °C for 1 h, and then at room temperature overnight. All Mg turnings were consumed. The reaction mixture was acidified with 1 N HCl, and then treated with saturated Na2CO3 solution, extracted with ethyl acetate (3 × 150 mL). The organic layers were combined and the solvent was evaporated under reduced pressure. The residue was purified with silica gel chromatograph using 1:10 ethyl acetate/hexanes to afford 32 (0.42 g) in 30% yield as colorless liquid. 1H NMR (CD3Cl, 300 MHz) δ 6.97 (s, 1H), 6.86 (s, 1H), 5.88 (s, 2H), 2.80-2.50 (m, 4H), 2.40 (s, 3H), 2.15 (m, 1H), 2.12 (s, 6H), 2.07 (s, 3H), 1.7-1.5 (m, 2H), 0.92 (d, 3H, J = 6.90 Hz).
4-Methyl-6-(2-methyl-4-(methylthio)butyl)pyridin-2-amine (33)
33 was synthesized from 32 using the standard deprotection procedure in 84% yield as a light yellow solid (oxalate, mp. 103.0-103.5 °C). 1H NMR (CD3Cl, 300 MHz) δ 6.32 (s, 1H), 6.15 (s, 1H), 4.62 (s, br, 2H), 2.62-2.34 (m, 4H), 2.20 (s, 3H), 2.08 (s, 3H), 2.04 (m, 1H), 1.75-1.42 (m, 2H), 0.90 (d, 3H, J = 6.6 Hz).
1-(6-(2,5-Dimethyl-1H-pyrrol-1-yl)-4-methylpyridin-2-yl)propan-2-yl acetate (34)
Method 1
Into a solution of 7 (1.1 g, 4.50 mmol) in CH2Cl2 (20 mL) were added triethyl amine (0.94 mL, 6.75 mmol) and acetyl chloride (0.48 ml, 6.79 mmol). A white solid was formed instantly and the reaction was stopped at 10 min by addition of methanol (1 mL). The reaction mixture was treated with saturated NaHCO3 solution, and the product was extracted with CH2Cl2 (2 × 50 mL) and dried over Na2SO4. Solvent was removed under reduced pressure and the residue was purified by silica gel chromatograph using 1:4 ethyl acetate/hexanes to afford 34 (0.5 g) in 39% yield as colorless liquid. 1HMR (CD3Cl, 300 MHz) δ 7.00 (s, 1H), 6.89 (s, 1H), 5.87 (s, 2H), 5.31 (m, 1H), 4.48 (s, br, 2H), 3.11-2.93 (m, 2H), 2.40 (s, 3H), 2.10 (s, 6H), 1.97 (s, 3H), 1.28 (d, 3H, J = 6.0 Hz).
Method 2
Into an Et2O solution of 6 at -78 °C, which was made from 5 (8.1 g, 40.5 mmol), n-BuLi (1.6 M in Hexane, 27.8 mL, 44.5 mmol) in Et2O (120 mL), was added CH3CHO (2.5 mL, 44.5 mmol) via a syringe. The reaction mixture was allowed to warm to room temperature over 30 min. The reaction mixture was the cooled down to -78 °C and ethyl acetate (10 mL) was added. The mixture was allowed to warm to room temperature and stirring continued for an additional 30 min. The reaction mixture was treated with H2O (100 mL), and the products were extracted with ethyl acetate (100 mL). The organic solution was washed with brine and water, dried over NaSO4 and the solvent was evaporated under reduced pressure. The crude product was purified with silica gel chromatograph using 1:6, 1:4, and 1:1 ethyl acetate/petroleum ether to afford 7 (3.9 g) and 34 (4.85 g) as colorless liquids.
1-(6-Amino-4-methylpyridin-2-yl)propan-2-yl acetate (35)
35 was synthesized from 34 using the standard deprotection procedure in 55% yield as a light yellow solid (mp. 83-85 °C). 1HMR (CD3Cl, 300 MHz) δ 6.37 (s, 1H), 6.19 (s, 1H), 5.25 (m, 1H), 4.48 (s, br, 2H), 2.92-2.68 (m, 2H), 2.20 (s, 3H), 1.99 (s, 3H), 1.25 (d, 3H, J = 6.0 Hz).
1-(6-(tert-Butoxycarbonylamino)-4-methylpyridin-2-yl)propan-2-yl acetate (36)
A solution of 35 (0.5 g, 2.4 mmol) and di-tert-butyl dicarbonate (1.2 g, 5.5 mmol) in t-BuOH (15 mL) was stirred in a 65 °C oil bath for 66 h. Solvent was removed under reduced pressure and the residue was purified by silica gel chromatograph using 1:4 ethyl acetate/hexanes to afford 36 (0.5 g) in 68% yield as colorless liquid. 1HMR (CD3Cl, 300 MHz) δ 7.61 (s, 1H), 6.65 (s, 1H), 5.26 (m, 1H), 2.95-2.71 (m, 2H), 2.30 (s, 3H), 1.97 (s, 3H), 1.50 (s, 9H), 1.23 (d, 3H, J = 6.0 Hz).
tert-Butyl 6-(2-hydroxypropyl)-4-methylpyridin-2-ylcarbamate (37)
Into a solution of 36 (0.9 g, 2.9 mmol) in methanol (15 mL) was added a solution of K2CO3 (0.8 g, 5.7 mmol) in water (6 mL). The reaction was stirred at room temperature for 2 h, then diluted in water (100 mL) and extracted with ethyl acetate (2 × 50 mL). The organic solution was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by silica gel chromatograph using 1:1 ethyl acetate/hexanes for afford 37 (0.6 g) in 78% yield as white solid (mp. 118.0-120.0 °C). 1HMR (CD3Cl, 300 MHz) δ 7.66 (s, 1H), 6.63 (s, 1H), 4.15 (m, 1H), 2.81-2.65 (m, 2H), 2.32 (s, 3H), 1.52 (s, 9H), 1.24 (d, 3H, J = 6.0 Hz).
1-(6-(tert-Butoxycarbonylamino)-4-methylpyridin-2-yl)propan-2-yl methanesulfonate (38)
Into a solution of 37 (0.25 g, 0.94 mmol) was added triethyl amine (200 μL, 1.44 mmol), followed by methanesulfonyl chloride (80 μL, 1.0 mmol). The reaction mixture was stirred at room temperature for 1 h, then diluted with water (20 mL). The product was extracted with CH2Cl2 (2 × 50 mL), and the combined organic layer was dried over Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by silica gel chromatograph using 1:2 ethyl acetate/hexanes to afford 38 (0.27 g) in 83% yield as white solid (mp. 61-63 °C). 1HMR (CD3Cl, 300 MHz) δ 7.66 (s, 1H), 6.69 (s, 1H), 5.16 (m, 1H), 3.03-2.82 (m, 2H), 2.62 (s, 3H), 2.31 (s, 3H), 1.52 (s, 9H), 1.48 (d, 3H, J = 6.3 Hz).
Radiosynthesis of [18F]9
~180 mCi [18F]fluoride was dried by azeotropic distillation using CH3CN (3 × 1 mL) in the presence of K2CO3 (0.75 mg) and K222 (5 mg) at 110 °C under a flow of N2, then a solution of 38 (2.5 mg) in CH3CN (400 μL) was added. After the reaction mixture was heated in an oil bath (110 °C) for 10 min [incorporation: 17.3 ± 4.4% (n = 10) according to radio-TLC analysis: silica, 1:1 ethyl acetate/hexanes], it was passed through a silica gel SepPak (Waters) and CH3CN (2 × 1 mL) was used to rinse the reaction vial and the SepPak. The elution was concentrated to less than 500 μL in the presence of 1N HCl (100 μL) at 110 °C under a flow of N2, then 1N HCl (500 μL) was added. The reaction mixture was irradiated under microwave for 30 and 25 sec with an interval of 30 sec between each irradiation, and then was diluted in water (3 mL) for HPLC injection. [18F]9 was purified by reversed phased HPLC using an Alltech Platinum EPS C18 column (250 × 10 mm, 10 μ) eluted with 15% CH3CN, 85% water with 0.1% trifluoroacetic acid (TFA) at a flow rate of 4 mL/min and UV at 272 nm. The radioactivity (~8 mCi) corresponding to [18F]9 was collected at 17 min, and the collection fraction was concentrated under reduced pressure to less than 0.5 mL and then diluted in water (40 mL). [18F]9 was separated from the dilution by passing the dilution through an Oasis HLC cartridge (Waters) and eluted from the cartridge with ethanol (1~2 mL). If necessary, the ethanol solution was concentrated under a flow of N2 in order to make a final dose for animal study with <10% ethanol in saline. The total synthesis and purification time was 120 min; decay-corrected radiochemical yield was 6.2 ± 2.1% (n = 10); Radiochemical purity was >99.9% and specific activity was 2,160 ± 1,660 mCi/μmol (n = 10) at the end of synthesis, analyzed by an analytical HPLC column (Alltech Platinum EPS C18 250 × 4.6 mm, 10μ, 20% CH3CH, 80% water, 0.1% TFA, 2 mL/min, 272 nm) and determined by comparison of the integrated UV absorbance with a calibrated mass/UV absorbance curve of 9. The identity of [18F]9 was confirmed by the coelution of [18F]9 with nonradioactive standard 9 on the analytical HPLC system.
NOS Enzyme Assays
All assays were performed using the Nitric Oxide Synthase Screening Kit (GE Healthcare Biosciences Corp., Piscataway, NJ) following the manufacturer’s protocol with minor modifications. Recombinant inducible nitric oxide synthase (iNOS), endothelial nitric oxide synthase (eNOS), and neuronal nitric oxide synthase (nNOS) (Cayman Chemical Company, Ann Arbor, MI) were assayed to establish optimal enzyme concentrations and conditions. In short, all assays were carried out in reaction buffer containing 50 mM Tris pH 7.4, 1.0 mM NADPH, 3.8 μM FMN, 3.8 μM FAD, 3.8 μM tetrahydro-L-biopterin, 2.0 mM dithiothreitol, and 2.0 μM L-arginine, with the exception of the eNOS and nNOS assays which included 20 μg/mL calmodulin and 1 mM CaCl2 in the reaction buffer to stabilize the enzymes. All chemicals used in the reaction buffer were purchased from EMD Chemicals Inc. (Gibbstown, NJ). Assays were performed at 80 μL per well in 96-well format. All experimental inhibitors were diluted in methanol or water to ensure compatibility with the assay. 100,000 cpm or 0.1 μCi [3H]Arginine was added to the enzyme-inhibitor mixture to initiate the reaction. The reaction was allowed to incubate for 30 min at room temperature before it was terminated by the addition of 40 μL Yttrium-Silicate SPA Arginine binding beads in ‘stop solution’ (50 mg/mL in 50 mM NaOH solution). The arginine binding beads were allowed to settle for 2 h before the plates were counted on a Micro-Beta (PerkinElmer Life and Analytical Sciences, Waltham, MA). Inhibition curves were determined using Kaleidagraph (Synergy Software, Reading, PA). All IC50 values were recorded as mean ± S.D. (N ≥ 3).
Western Blot Analysis
Lungs from non-treated and LPS-treated C57BL/6N mice (Charles River Laboratories, Wilmington MA) were harvested and homogenized in T-Per® tissue protein extraction reagent (Pierce Biotechnology, Rockford, IL) containing Complete™ protease Inhibitor cocktail tablets (Roche Applied Science, Indianapolis, IN) on ice. Homogenates were sonicated 5-10 seconds 3 times on ice and centrifuged at 12,000 rpm at 4 °C for 15 min. Aliquots of protein (200 μg) from each sample were analyzed using standard immunoblotting procedures. The presence of iNOS was probed with monoclonal anti-iNOS primary antibody (Sigma-Aldrich, Inc., St. Louis, MO) at a 1:1,000 dilution and horseradish peroxidase-conjugated goat anti-rabbit IgG (Cell Signaling Technology, Danvers, MA) at 1:3,000 dilution. The SuperSignal WestDura Extended Duration Substrate assay kit (Pierce Biotechnology, Rockford, IL) was used to detect the secondary antibody. Loading control was performed with β-actin (Cell Signaling Technology, Danvers, MA).
In vitro stability study
An in vitro stability study was carried out in heparinized whole rat blood (mature male Sprague-Dawley rat). The whole blood (5 mL) was incubated with ~400 μCi [18F]9 in 150 μL saline for 5 min, 30 min, 1 h and 2 h at 37 °C. An aliquot of blood was treated with 3 volumes of ethanol at each time point, and the lysed sample was centrifuged to separate the supernatant from the pellet. The radioactivity in the supernatant and the pellet was counted separately on a Beckman Gamma 8000 well counter. The radioactive species in the supernatant was analyzed by Silica TLC using ethyl acetate as the developing solvent (Rf = 0.7 for [18Br]9) and co-elution with non-radioactive 9.
In vivo blood metabolism study
The metabolism of [18F]9 was evaluated in a male Sprague-Dawley rat by iv injection of [18F]9 (400 μCi) in 10% ethanol/saline via the tail vein of an anesthetized rat. Blood samples (1.2 mL) were obtained via cardiac puncture under anesthesia at 5 and 30 min post-injection. The plasma was separated from the red blood cells by centrifugation (14,000 rpm), and the radioactivity in both samples was measured. After the plasma (400 μL) was treated with 3 equivalents of acetonitrile, supernatant and pellet were separated by centrifugation (14,000 rpm) and the radioactivity in both samples was measured. The supernatant was filtered through a 0.45 μm nylon filter, and acetonitrile (1mL) was used to rinse the filter. This supernatant solution was concentrated under a flow of N2 at room temperature to less than 400 μL. The radioactive species in the supernatant was analyzed by silica gel radio-TLC (developed in ethyl acetate and co-spotted with non-radioactive 9) and reverse phase HPLC (co-eluted with 9). All the samples were kept on ice to prevent degradation.
In vivo biodistribution time-course study
All animal experiments were conducted in compliance with the Guidelines for the Care and Use of Research Animals established by Washington University’s Animal Studies Committee. The biodistribution study was performed in mature male C57BL/6N mice (age 6-8 weeks) in two experimental groups: one group was intravenously injected with lipopolysaccharide (LPS) (Escherichia coli O127:B8, Sigma-Aldrich Co., MO) dissolved in PBS (10mg/kg, μL per mouse, i.v.) 6 h prior to the tracer injection to induce iNOS expression; untreated mice were used as control. Reverse phase HPLC purified [18F]9 was injected (~50 μCi in 120 μL 10% ethanol/saline, i.v.) via the tail vein. At the specified time points post-injection (5 min, 30 min, 1 h, and 2 h), the mice were sacrificed, and blood, tissues, and organs were removed, weighed, and counted in a Beckman Gamma 8000 counter with standard diluted aliquots of the injectate. The percent injected dose per gram of tissue (%ID/g) was presented as mean ± standard deviation.
The blocking study was performed following the same procedure described above with the addition of a blocking group (injected with 1400W, 5 mg/kg in saline, i.v.). [18F]9 was injected (~5 μCi in 120 μL 10% ethanol/saline, i.v.) via the tail vein. Based on the tracer uptake profile exhibited in the initial biodistribution study and the kinetics of the blocking agent, uptake was evaluated 1 hour post-injection. A lower dose of [18F]9 with higher specificity (4,300 mCi/μmol) was used in this study than in the initial evaluation.
MicroPET study
Two groups of mature male C57BL/6N mice (age 6-8 weeks) were used for the MicroPET study: one set was intratracheally injected with LPS (Escherichia coli O127:B8, Sigma-Aldrich Co., MO, 10 mg/kg) 6 h prior to the tracer injection (n=2); the other set received no treatment and was used as control (n=2). The mice were anesthetized with isoflurane and injected with ~100 μCi/100 μL of [18F]9 via the tail vein. The imaging sessions were carried out as 1 h dynamic scan using the MicroPET® Focus (Siemens Medical Solutions USA, Inc.) scanner. The MicroPET data was then processed using filter back projection algorithm with attenuation and scatter corrections.
Supplementary Material
Chart.
Acknowledgement
This research was supported by the National Institute of Health grants HL13851 and CA 86307.
a Abbreviations:
- NO
nitric oxide
- NOS
nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- nNOS
neuronal nitric oxide synthase
- LPS
lipopolysaccharide
- PET
positron emission tomography
- DAST
diethylaminosulfur trifluoride
- PBSF
perfluorobutane sulfonyl fluoride
- SEITU
S-ethylisothiourea
- L-NIL
L-N6-(1- iminoethyl)lysine
- 2-AP
2-aminopyridine
Footnotes
Supporting Information Available: Combustion analysis data of compound 9, 11, 13, 15, 16, 18, 20, 24, 27, 30, 33, and 38. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1 (a).For the latest reviews, see: Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol. Rev. 2005;57:217–252. doi: 10.1124/pr.57.2.1.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593.
- 2 (a).Stuehr DJ. Mammalian nitric oxide synthases. Biochim. Biophys. Acta. 1999;1411:217–30. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]; (b) Marletta MA. Nitric oxide synthase structure and mechanism. J. Biol. Chem. 1993;268:12231–12234. [PubMed] [Google Scholar]
- 3 (a).Marletta MA, Hurshman AR, Rusche KM. Catalysis by nitric oxide synthase. Curr. Opin. Chem. Biol. 1998;2:656–663. doi: 10.1016/s1367-5931(98)80098-7. [DOI] [PubMed] [Google Scholar]; (b) Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 5 (a).Titheradge MA. Nitric oxide in septic shock. Biochim. Biophys. Acta. 1999;1411:437–455. doi: 10.1016/s0005-2728(99)00031-6. [DOI] [PubMed] [Google Scholar]; (b) Hobbs AJ, Higgs A, Moncada S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annu. Rev. Pharmacol. Toxicol. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]; (c) Vallance P, Leiper J. Blocking NO synthesis: how, where and why? Nat. Rev. Drug Discov. 2002;1:939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]; (d) Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]; (e) Li H, Förstermann U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000;190:244–254. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]; (f) Xu W, Liu LZ, Loizidou M, Ahmed M, Charles IG. The role of nitric oxide in cancer. Cell Res. 2002;12:311–320. doi: 10.1038/sj.cr.7290133. [DOI] [PubMed] [Google Scholar]
- 6.Lechner M, Lirk P, Rieder J. Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Semin. Cancer Biol. 2005;15:277–289. doi: 10.1016/j.semcancer.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 7 (a).Narayanan K, Spack L, McMillan K, Kilbourn RG, Hayward MA, Masters BS, Griffith OW. S-Alkyl-L-thiocitrullines. Potent stereoselective inhibitors of nitric oxide synthase with strong pressor activity in vivo. J. Biol. Chem. 1995;270:11103–11110. doi: 10.1074/jbc.270.19.11103. [DOI] [PubMed] [Google Scholar]; (b) Moore WM, Webber RK, Jerome GM, Tjoeng FS, Misko TP, Currie MG. L-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J. Med. Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]; (c) Moore WM, Webber RK, Fok KF, Jerome GM, Connor JR, Manning PT, Wyatt PS, Misko TP, Tjoeng FS, Currie MG. 2-Iminopiperidine and other 2-iminoazaheterocycles as potent inhibitors of human nitric oxide synthase isoforms. J. Med. Chem. 1996;39:669–672. doi: 10.1021/jm950766n. [DOI] [PubMed] [Google Scholar]; (d) McCall TB, Feelisch M, Palmer RM, Moncada S. Identification of N-iminoethyl-L- ornithine as an irreversible inhibitor of nitric oxide synthase in phagocytic cells. Br. J. Pharmacol. 1991;102:234–238. doi: 10.1111/j.1476-5381.1991.tb12159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Furfine ES, Harmon MF, Paith JE, Knowles RG, Salter M, Kiff RJ, Duffy C, Hazelwood R, Oplinger JA, Garvey EP. Potent and selective inhibition of human nitric oxide synthases. Selective inhibition of neuronal nitric oxide synthase by S-methyl-L-thiocitrulline and S-ethyl-L-thiocitrulline. J. Biol. Chem. 1994;269:26677–26683. [PubMed] [Google Scholar]; (f) Narayanan K, Spack L, McMillan K, Kilbourn RG, Hayward MA, Masters BS, Griffith OW. S-Alkyl-L- thiocitrullines. Potent stereoselective inhibitors of nitric oxide synthase with strong pressor activity in vivo. J. Biol. Chem. 1995;270:11103–11110. doi: 10.1074/jbc.270.19.11103. [DOI] [PubMed] [Google Scholar]; (g) Southan GJ, Szabo C, Thiemermann C. Isothioureas: potent inhibitors of nitric oxide synthases with variable isoform selectivity. Br. J. Pharmacol. 1995;14:510–516. doi: 10.1111/j.1476-5381.1995.tb13256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8 (a).Young RJ, Beams RM, Carter K, Clark HA, Coe DM, Chambers CL, Davies PI, Dawson J, Drysdale MJ, Franzman KW, French C, Hodgson ST, Hodson HF, Kleanthous S, Rider P, Sanders D, Sawyer DA, Scott KJ, Shearer BG, Stocker R, Smith S, Tackley MC, Knowles RG. Inhibition of inducible nitric oxide synthase by acetamidine derivatives of hetero-substituted lysine and homolysine. Bioorg. Med. Chem. Lett. 2000;10:597–600. doi: 10.1016/s0960-894x(00)00055-x. [DOI] [PubMed] [Google Scholar]; (b) Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]; (c) Strub A, Ulrich WR, Hesslinger C, Eltze M, Fuchss T, Strassner J, Strand S, Lehner MD, Boer R. The novel imidazopyridine 2-[2-(4-methoxy-pyridin-2-yl)-ethyl]-3H-imidazo[4,5-b]pyridine (BYK191023) is a highly selective inhibitor of the inducible nitric-oxide synthase. Mol. Pharmacol. 2006;69:328–337. doi: 10.1124/mol.105.017087. [DOI] [PubMed] [Google Scholar]; (d) Beaton H, Hamley P, Nicholls DJ, Tinker AC, Wallace AV. 3,4-Dihydro-1-isoquinolinamines: a novel class of nitric oxide synthase inhibitors with a range of isoform selectivity and potency. Bioorg. Med. Chem. Lett. 2001;11:1023–1026. doi: 10.1016/s0960-894x(01)00119-6. [DOI] [PubMed] [Google Scholar]; (e) Naka M, Nanbu T, Kobayashi K, Kamanaka Y, Komeno M, Yanase R, Fukutomi T, Fujimura S, Seo HG, Fujiwara N, Ohuchida S, Suzuki K, Kondo K, Taniguchi N. A potent inhibitor of inducible nitric oxide synthase, ONO-1714, a cyclic amidine derivative. Biochem. Biophys. Res. Commun. 2000;270:663–667. doi: 10.1006/bbrc.2000.2474. [DOI] [PubMed] [Google Scholar]
- 9 (a).Faraci WS, Nagel AA, Verdries KA, Vincent LA, Xu H, Nichols LE, Labasi JM, Salter ED, Pettipher ER. 2-Amino-4-methylpyridine as a potent inhibitor of inducible NO synthase activity in vitro and in vivo. Br. J. Pharmacol. 1996;119:1101–1108. doi: 10.1111/j.1476-5381.1996.tb16010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pettipher ER, Hibbs TA, Smith MA, Griffiths RJ. Analgesic activity of 2-amino-4-methylpyridine, a novel NO synthase inhibitor. Inflamm. Res. 1997;46:S135–S136. doi: 10.1007/s000110050142. [DOI] [PubMed] [Google Scholar]
- 10.Hagmann WK, Caldwell CG, Chen P, Durette PL, Esser CK, Lanza TJ, Kopka IE, Guthikonda R, Shah SK, MacCoss M, Chabin RM, Fletcher D, Grant SK, Green BG, Humes JL, Kelly TM, Luell S, Meurer R, Moore V, Pacholok SG, Pavia T, Williams HR, Wong KK. Substituted 2-aminopyridines as inhibitors of nitric oxide synthases. Bioorg. Med. Chem. Lett. 2000;10:1975–1978. doi: 10.1016/s0960-894x(00)00389-9. [DOI] [PubMed] [Google Scholar]
- 11.Connolly S, Aberg A, Arvai A, Beaton HG, Cheshire DR, Cook AR, Cooper S, Cox D, Hamley P, Mallinder P, Millichip I, Nicholls DJ, Rosenfeld RJ, StGallay SA, Tainer J, Tinker AC, Wallace AV. 2-Aminopyridines as highly selective inducible nitric oxide synthase inhibitors. Differential binding modes dependent on nitrogen substitution. J. Med. Chem. 2004;47:3320–3323. doi: 10.1021/jm031035n. [DOI] [PubMed] [Google Scholar]
- 12 (a).Zhang J, McCarthy TJ, Moore WM, Currie MG, Welch MJ. Synthesis and evaluation of two positron-labeled nitric oxide synthase inhibitors, S-[11C]methylisothiourea and S-(2-[18F]fluoroethyl)isothiourea, as potential positron emission tomography tracers. J. Med. Chem. 1996;39:5110–5118. doi: 10.1021/jm960481q. [DOI] [PubMed] [Google Scholar]; (b) Tian H, Lee Z. Radiosynthesis of 8-fluoro-3-(4-[18F]fluorophenyl)-3,4-dihydro-1-isoquinolinamine ([18F]FFDI), a potential PET radiotracer for the inducible nitric oxide synthase. Current Radiopharmaceuticals. 2008;1:49–53. [Google Scholar]; (c) de Vries EF, Vroegh J, Dijkstra G, Moshage H, Elsinga PH, Jansen PL, Vaalburg W. Synthesis and evaluation of a fluorine-18 labeled antisense oligonucleotide as a potential PET tracer for iNOS mRNA expression. Nucl. Med. Biol. 2004;31:605–612. doi: 10.1016/j.nucmedbio.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Peterson DJ. Carbonyl olefination reaction using silyl-substituted organometallic compounds. J. Org. Chem. 1968;33:780–784. [Google Scholar]
- 14.Bryk R, Wolff DJ. Pharmacological modulation of nitric oxide synthesis by mechanism-based inactivators and related inhibitors. Pharmacol. Ther. 1999;84:157–178. doi: 10.1016/s0163-7258(99)00030-3. [DOI] [PubMed] [Google Scholar]
- 15.Tiso M, Strub A, Hesslinger C, Kenney CT, Boer R, Stuehr DJ. BYK191023 (2-[2-(4-methoxy-pyridin-2-yl)-ethyl]-3h-imidazo[4,5-b]pyridine) is an NADPH- and time-dependent irreversible inhibitor of inducible nitric-oxide synthase. Mol. Pharmacol. 2008;73:1244–1253. doi: 10.1124/mol.107.041319. [DOI] [PubMed] [Google Scholar]
- 16.Speyer CL, Neff TA, Warner RL, Guo RF, Sarma JV, Riedemann NC, Murphy ME, Murphy HS, Ward PA. Regulatory effects of iNOS on acute lung inflammatory responses in mice. Am. J. Pathol. 2003;163:2319–2328. doi: 10.1016/S0002-9440(10)63588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (a) Liu SF, Adcock IM, Old RW, Barnes PJ, Evans TW. Differential regulation of the constitutive and inducible nitric oxide synthase mRNA by lipopolysaccharide treatment in vivo in the rat. Crit. Care Med. 1996;24:1219–1225. doi: 10.1097/00003246-199607000-00026. [DOI] [PubMed] [Google Scholar]; (b) Buttery LD, Evans TJ, Springall DR, Carpenter A, Cohen J, Polak JM. Immunochemical localization of inducible nitric oxide synthase in endotoxin-treated rats. Lab Invest. 1994;7:755–764. [PubMed] [Google Scholar]; (c) Kan W, Zhao KS, Jiang Y, Yan W, Huang Q, Wang J, Qin Q, Huang X, Wang S. Lung, spleen, and kidney are the major places for inducible nitric oxide synthase expression in endotoxic shock: role of p38 mitogen-activated protein kinase in signal transduction of inducible nitric oxide synthase expression. Shock. 2004;21:281–287. doi: 10.1097/01.shk.0000113314.37747.55. [DOI] [PubMed] [Google Scholar]
- 17.Moncada S, Higgs EA. Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J. 1995;9:1319–1330. [PubMed] [Google Scholar]
- 18 (a).Kovách AG, Szabó C, Benyó Z, Csáki C, Greenberg JH, Reivich M. Effects of NG-nitro-L-arginine and L-arginine on regional cerebral blood flow in the cat. J. Physiol. 1992;449:83–196. doi: 10.1113/jphysiol.1992.sp019081. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Benyó Z, Kiss G, Szabó C, Csáki C, Kovách AG. Importance of basal nitric oxide synthesis in regulation of myocardial blood flow. Cardiovasc. Res. 1991;25:700–703. doi: 10.1093/cvr/25.8.700. [DOI] [PubMed] [Google Scholar]; (c) Gardiner SM, Compton AM, Bennett T, Palmer RM, Moncada S. Control of regional blood flow by endothelium-derived nitric oxide. Hypertension. 1990;15:486–492. doi: 10.1161/01.hyp.15.5.486. [DOI] [PubMed] [Google Scholar]
- 19.Alderton WK, Angell AD, Craig C, Dawson J, Garvey E, Moncada S, Monkhouse J, Rees D, Russel RJ, Schwartz S, Waslidge N, Knowles RG. GW274150 and GW273629 are potent and highly selective inhibitors of inducible nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 2005;145:301–312. doi: 10.1038/sj.bjp.0706168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman Melvin S., Waltcher I, Ginsberg HF. The synthesis and reduction of some polyfunctional acetylene compounds. J. Org. Chem. 1952;17:962–970. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.