

Abstract
Bacillibactin and enterobactin are hexadentate catecholate siderophores produced by bacteria upon iron limitation to scavenge ferric ion and seem to be the ultimate siderophores of their two respective domains: Gram-positive and Gram-negative. Iron acquisition mediated by these trilactone-based ligands necessitates enzymatic hydrolysis of the scaffold for successful intracellular iron delivery. The esterases BesA and Fes hydrolyze bacillibactin and enterobactin, respectively, as well as the corresponding iron complexes. Bacillibactin binds iron through three 2,3-catecholamide moieties linked to a tri-threonine scaffold via glycine spacers, whereas in enterobactin the iron-binding moieties are directly attached to a tri-l-serine backbone; although apparently minor, these structural differences result in markedly different iron coordination properties and iron transport behavior. Comparison of the solution thermodynamic and circular dichroism properties of bacillibactin, enterobactin and the synthetic analogs d-enterobactin, SERGlyCAM and d-SERGlyCAM has determined the role of each different feature in the siderophores' molecular structures in ferric complex stability and metal chirality. While opposite metal chiralities in the different complexes did not affect transport and incorporation in Bacillus subtilis, ferric complexes formed with the various siderophores did not systematically promote growth of the bacteria. The bacillibactin esterase BesA is less specific than the enterobactin esterase Fes; BesA can hydrolyze the trilactones of both siderophores, while only the tri-l-serine trilactone is a substrate of Fes. Both enzymes are stereospecific and cannot cleave tri-d-serine lactones. These data provide a complete picture of the microbial iron transport mediated by these two siderophores, from initial recognition and transport to intracellular iron release.
Keywords: Siderophore, Enterobactin, Bacillibactin, Esterase, Ferric Complex, Chirality, Trilactone, Iron transport
Introduction
The chemical activity of available ferric ion is limited to 10-18 M by the low solubility of ferric hydroxide in aerobic conditions at physiological pH,2 and yet a 10-6 M internal iron concentration is required by bacteria to achieve optimal growth.3 To overcome this discrepancy, a central microbial iron acquisition mechanism is the production of low molecular weight iron-chelating agents, called siderophores.4 Remarkable in their high specificity and affinity towards ferric ion, siderophores are exported from the cell in their apo form and can remove iron from minerals and host iron-binding proteins to form stable ferric complexes.5 Gram-negative bacteria incorporate ferric-siderophore complexes via specific outer membrane receptors in a process driven by the cytoplasmic membrane potential and mediated by the energy-transducing TonB-ExbB-ExbD system.3,6 Periplasmic binding proteins shuttle the complexes from the outer membrane receptors to cytoplasmic membrane ATP-binding cassette (ABC) transporters that, in turn, deliver the ferric-siderophores to the cytoplasm.7 Gram-positive bacteria, which lack the lipopolysaccharide layer encasing the cell, require only a lipoprotein tethered to the external surface of the cytoplasmic membrane and binding-protein-dependent ABC permeases to transport their siderophores into the cell.8 Bacteria often possess multiple membrane receptors, each providing the organism with specificity for different siderophores; it is also typical for bacterial strains to utilize xenobiotic siderophores, suggesting that the recognition process is common throughout the microbial world.9
Enterobactin (Ent) and bacillibactin (BB) are two remarkable hexadentate iron chelators:10 both ligands coordinate iron through three 2,3-catecholate units that are amide linked to a trilactone macrocycle (Figure 1); they have the highest known ferric complex formation constants (1049 and 1048, respectively), yet determined for siderophores.11,12 First isolated in 1970 from Escherichia coli13 and Salmonella typhimurium14, the tri-serine-based Ent is produced by a large number of enteric bacteria and was, for almost 30 years, the only example of a siderophore incorporating a trilactone. In contrast, the tri-threonine-based BB was recently isolated from Bacillus subtilis,15 Bacillus anthracis, Bacillus cereus and Bacillus thuringiensis,16-18 its hydrolyzed version was found in Bacillus licheniformis19 and genes encoding its production were reported in Bacillus amyloliquefaciens,20 indicating that this siderophore may be the counterpart of Ent in Gram-positive bacteria.
Figure 1.
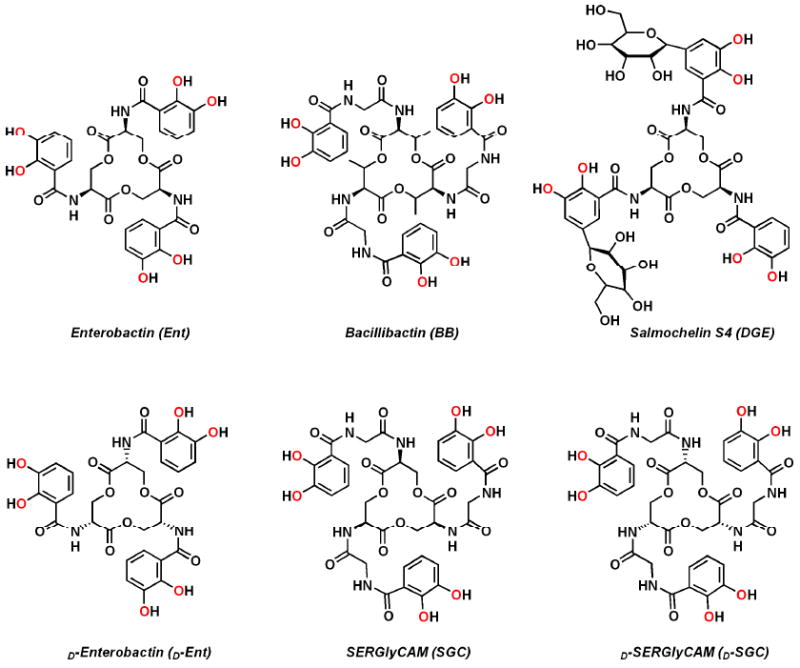
Molecular structures and abbreviations of the natural (top) and synthetic (bottom) siderophores discussed in this study. The iron-coordinating oxygen atoms are indicated in red.
The extremely high affinity for iron displayed by Ent and BB originates from the presence of the 2,3-dihydroxybenzamide motifs. However, incorporation of these powerful 2,3-catecholate iron-binding units in Ent and BB also makes the siderophores and their respective ferric complexes specific targets of the mammalian protein Siderocalin, a component of the antibacterial iron-depletion defense of the innate immune system.21,22 Siderocalin acts as a growth inhibitor of pathogens that rely solely on Ent-mediated iron acquisition and is speculated to have the same bacteriostatic effect on BB-dependent strains.22,23 The siderophore recognition patterns by Siderocalin or bacterial cognate proteins are likely different and are a determining factor of bacterial survival and infection. Understanding the processes involved in Ent- and BB-mediated bacterial iron acquisition is therefore important to further explore the relationship between siderophore production and pathogenicity. One strategy used by pathogenic Ent-producing strains, such as E. coli, Salmonella spp., and Klebsiella pneumoniae, is to evade siderophore sequestration by siderocalin via the production of C-glucosylated Ent analogs, called salmochelins.24-26 To date, Ent, BB and the triserine-based salmochelin S4 (diglucosyl enterobactin, DGE, Figure 1) are the only trilactone siderophores isolated from bacterial cultures and, due to their structural differences, all three siderophores require multiple, but partially overlapping, pathways for iron incorporation, as shown in uptake studies performed on S. typhimurium and B. subtilis.12,27 Nevertheless, a particular common pathway is taken by Ent, BB and DGE, once their ferric complexes are incorporated in the cytoplasm: in contrast to most ferric-siderophores that undergo reduction followed by spontaneous release or competitive sequestration of the reduced species,28 intracellular iron release from [FeIII(Ent)]3-, [FeIII(BB)]3- and [FeIII(DGE)]3- requires hydrolysis of the trilactone scaffolds by the specific esterases Fes, BesA and IroD, respectively.29,30 Cleavage of the ester functionalities of the siderophore backbone results in a dramatic loss of complex stability and corresponding increase in reduction potential,31 which facilitates subsequent removal of iron by other cellular iron-binding components.
Despite the close similarity of Ent- and BB-mediated iron uptake pathways, there are significant differences in the complexes' structures. What is the effect of the different molecular structures of these siderophores on the specific recognition of the ligands and their ferric complexes by biological receptors and enzymes? Especially, how does the incorporation of particular chiral amino acids in the trilactone scaffolds affect the iron-coordination properties of the chelators and the bacterial uptake and use of their respective ferric complexes? It has long been known that Ent effectively delivers iron for growth whereas its synthetic mirror image does not,32 yet both are recognized by the outer membrane receptor FepA.33 So where does chiral recognition occur?
To address these questions, we report solution thermodynamic, circular dichroism and receptor-binding studies on synthetic analogs of Ent and BB as well as bacterial uptake experiments and growth assays performed with B. subtilis strains. We also describe the specificity of the esterases BesA and Fes in the enzymatic hydrolysis of different trilactone backbones. The results presented herein demonstrate how molecular recognition is achieved by bacterial species through subtle changes in the structures of the two most powerful siderophores.
Results and Discussion
Enterobactin and bacillibactin: two siderophore archetypes
The architectures of Ent and BB make these siderophores predisposed to ferric ion binding and formation of highly stable octahedral complexes.10 Both iron chelators are three-fold symmetrical, hexadentate catecholate ligands; however, while the iron-binding motifs are directly attached to a tri-serine scaffold through amide linkages in Ent, BB is formed from a tri-threonine skeleton connected to the catecholamide subunits via glycine spacers. Though the wide range of Ent- or BB-producing microorganisms synthesize one or the other siderophore, some, such as Salmonella enterica serovar typhimurium34 and B. subtilis,12,35 can incorporate and use both in parallel for iron acquisition. By affecting the iron coordination chemistry properties of the chelators, the different structures of Ent and BB are responsible for the discrimination between their ferric complexes by bacterial strains. The coordination chemistry of Ent, BB and synthetic analogs, as well as their iron transport and release properties in selected organisms, are explored here to correlate the chemical structures of the siderophores to their biological recognition.
Thermodynamic stability and induced chirality of trilactone-based ferric siderophore complexes
The structural changes between Ent and BB (Figure 1) result in these two siderophores having different affinities for iron.12,36 The trilactone backbone of Ent appears perfect for the size of the ferric ion, and addition of the glycine spacer seems detrimental to the overall stability, as suggested by the lowered thermodynamic stability of ferric BB compared to ferric Ent.12 However, another important change is the addition of methyl groups on the BB trilactone. The conformation of the 12 membered rings in Ent and BB can be viewed as three fused cyclohexane-like rings in which the central atom is missing. There are two solutions to the requirement that each ring must be in a chair conformation: one in which the amide functionalities are in axial positions ([FeIII(Ent]3-, Figure S1) and one where they are in equatorial positions ([FeIII(BB)]3-, Figure S1).37 In order to confirm that the thermodynamic behavior of BB is dominated by the presence of the glycine spacers and not by the incorporation of threonine units, thermodynamic measurements were performed on the intermediate synthetic analog: SERglyCAM (SGC, also known as serine-corynebactin and serine-bacillibactin, Figure 1).36,37 This ligand incorporates three 2,3-catecholamide moieties linked to a tri-serine lactone through glycine spacers; its synthesis has been described earlier and it was prepared similarly.37 Formation of a ferric siderophore complex requires deprotonation of the catecholate phenol groups, so determination of ligand pKa values is crucial to evaluating the stability of ferric complexes. Ligand protonation constants were determined for SGC and the ferric formation and protonation constants were evaluated. Previous studies of Ent and its analogs established the protonation constants (pKa1 − pKa3) of the meta-hydroxyl oxygen atoms are well separated from the ortho-hydroxyl oxygen atoms (pKa4 − pKa6).11 Thus, three of the six stepwise protonation constants (K0ln, for n = 1-6), as defined by the equations (1) and (2), were determined potentiometrically.
| (1) |
| (2) |
The stability constant (eq. 1) of the SGC ferric complex was measured via a competition experiment with ethylene-diamine tetraacetic acid (EDTA), by monitoring the changes in UV-Vis absorbance (Table 1). This spectrophotometric titration exploits the broad ligand-to-metal-charge-transfer (LMCT) band which increases in intensity as iron exchanges from the colorless EDTA complex to the red [FeIII(SGC)]3- complex (Figure 2). The protonation constants obtained for SGC (Table 1) confirm that the insertion of a glycine spacer between the trilactone backbone and the catecholamide arms slightly alters the acidity of the ligand when compared to Ent. The addition of the amino acid spacer is also detrimental to the overall thermodynamic stability of the iron complex. However, methylation of the trilactone in BB provides a more stable iron complex than the ferric SGC. The calculated pM values1 of all three chelators Ent,11 BB12 and SGC (Table 1) corroborate that insertion of a threonine trilactone optimizes the iron-coordination in a structure containing glycinyl arms but does not compensate for the loss of iron-complex stability in comparison to the more compact Ent molecule. Similar studies on an additional analog containing three catecholamide units directly linked to a tri-threonine backbone are necessary to complete this analysis and assess the importance of glycine insertion in the BB structure. Unfortunately and despite several individual efforts (Ramirez et al.38 and unpublished results), the synthesis of the threonine trilactone precursor has not been performed successfully yet.
Table 1.
Solution Thermodynamic Parameters for the Trilactone-Based Ent, BB and SGC.
| SERglyCAM | Enterobactina | Bacillibactinb | ||
|---|---|---|---|---|
| [L]6- | log K1 | 12.1 | 12.1 | 12.1 |
| [LH]5- | log K2 | 12.1 | 12.1 | 12.1 |
| [LH2]4- | log K3 | 12.1 | 12.1 | 12.1 |
| [LH3]3- | log K4 | 8.37(3) | 8.6 | 8.43 |
| [LH4]2- | log K5 | 7.54(2) | 7.5 | 7.43 |
| [LH5]- | log K6 | 6.68(3) | 6.0 | 6.77 |
| Σlog K1-6 | 58.89 | 58.4 | 58.93 | |
| [FeL]3- | log β110 | 44.1(2) | 49 | 47.6 |
| pM(FeIII) | 29.6 | 34.3 | 33.1 |
Figure 2.
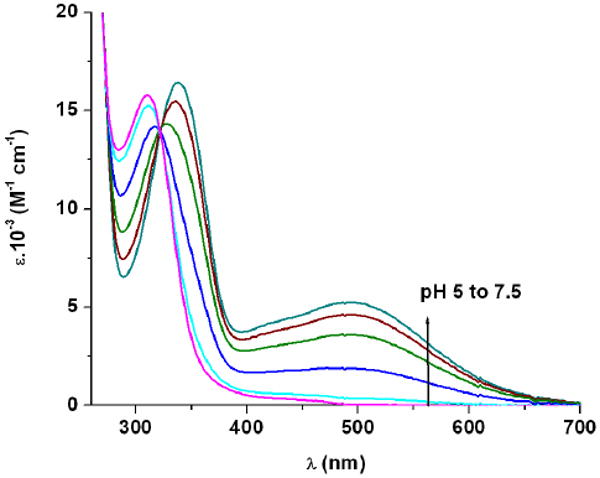
Spectrophotometric competition titration of SGC against EDTA ([Fe3+] = 0.09 mM, [SGC] = 0.10 mM, [EDTA] = 1.0 mM, pH from 5 to 7.5, 48 h, 0.1 M KCl, 25 °C, 1 cm cell).
A notable feature of metal complexes formed with these trilactone-based siderophores is their distinct chirality at the metal center, as evidenced by circular dichroism in previous studies.39 All three complexes [FeIII(Ent)]3-, [FeIII(BB)]3- and [FeIII(SGC)]3- exhibit intense high energy CD bands (< 390 nm) corresponding to ligand centered π-π* transitions, as well as characteristic ferric catechol transitions in the visible region. The latter arise from LMCT transitions and are sensitive to the chirality at the metal center. While the absolute configuration of [FeIII(Ent)]3- was established as Δ,40 both ferric complexes of SGC and BB adopt the opposite Λ chirality,39 similar to d-enterobactin (d-Ent, enantio-enterobactin), the synthetic enantiomer of Ent composed of a d-serine trilactone, which perforce forms a Λ ferric complex. To provide a complete set of compounds to the following study, d-SERglyCAM (d-SGC), the enantiomer of SGC incorporating a tri-d-serine lactone, has been prepared following the same synthetic procedure as for SGC: coupling of the tri-hydrochloride salt of the d-serine trilactone to three equivalents of 2,3-bis(benzyloxy)-glycinylfluoride benzoylamide, and subsequent hydrogenation with a Pd/C catalyst afforded the enantiomerically pure hexadentate ligand d-SGC (Scheme S1).
The ferric complexes [FeIII(SGC)]3- and [FeIII(d-SGC)]3- display opposite chiralities, as shown by circular dichroism (Figure 3, Table 2). Incorporation of chiral amino acids in the trilactone scaffold of siderophores such as Ent and BB induces the chirality of the corresponding ferric complexes. However, insertion of glycine spacers on each catecholate arms of BB or SGC inverts the ferric complex configuration as compared to Ent. In contrast, methylation of the trilactone backbone alone does not affect the metal center chirality. Comparison of the π-π* transitions in [FeIII(BB)]3- and [FeIII(SGC)]3- may also provide some information about the chirality of the tri-threonine lactone. The identical splitting of the high energy CD bands (in two bands with peaks at 311 and 354 nm) for both complexes is indicative of similar S chiral configuration adjacent to the amide functionalities linking the trilactone to the arms.41 This conformation is consistent with scaffolds formed with l-serine or l-threonine. Nevertheless, the dimodular non-ribosomal peptide synthetase DhbF adenylates threonine during the biosynthesis of BB in B. subtilis and has selectivity not only for the predicted substrate l-threonine (2S, 3R) but also for the miscognate substrate allo-l-threonine (2S, 3S).15 Thus, BB could be based on either l-threonine or allo-l-threonine units and the absolute configuration of the second stereocenter in each threonine unit remains to be established.
Figure 3.
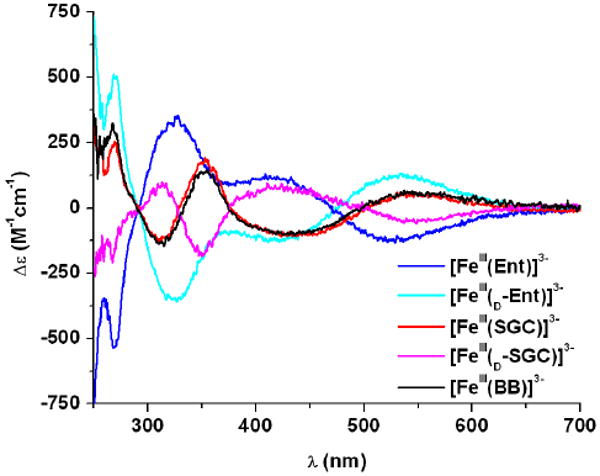
Circular dichroism spectra of the studied ferric complexes ([FeL] = 0.1 mM, pH 7.4, 1 cm path, 25 °C).
Table 2.
Chiral Features of the Ligand and Ferric Complexes Studied.
| Chirality | π-π* transitions | LMCT transitions | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Ligand | Trilactone Amino Acid | Metal Center ([FeIII(L)]3-) | λmax (nm) | Δε (M-1 cm-1) | λmax (nm) | Δε (M-1 cm-1) |
| Ent | L | Δ | 326 | + 348 | 528 | − 133 |
| d-Ent | D | Λ | 325 | − 345 | 530 | + 132 |
| SGC | L | Λ | 310, 353 | − 126, + 192 | 548 | + 58 |
| d-SGC | D | Δ | 313, 352 | + 101, − 180 | 548 | − 58 |
| BB | L or L-alloa | Λ | 311, 354 | −137, + 142 | 545 | + 65 |
The configuration of the threonine second carbon stereocenter (numbered 3) has not been established.
Transport and incorporation of trilactone-based catecholate siderophores by B. subtilis
Both BB and its precursor Itoic acid (DHBG, the glycine conjugate of 2,3-dihydroxybenzoic acid) have previously been characterized as the two endogenous siderophores of B. subtilis.15 However, due to its relatively weak affinity for iron, DHBG is unlikely to have physiological relevance as a siderophore under natural conditions and BB has been established as the main extracellular ferric ion scavenger produced by B. subtilis under iron limitation.35,42 Furthermore, B. subtilis, like many bacteria, can utilize a range of exogenous siderophores, such as the hydroxamates ferrichrome, ferrioxamine, coprogen, schizokinen and arthrobactin, or the catecholate Ent.35,42 Recent mutational analyses and transport studies have revealed at least two catecholate transport mechanisms are operative in B. subtilis. The major catecholate feuABC-encoded ABC transporter coupled to the yusV-encoded ATPase promotes the uptake of both [FeIII(BB)]3- and [FeIII(Ent)]3-, as shown by growth stimulation assays35 and fluorescence spectroscopy,30 whereas another unidentified secondary receptor does not recognize [FeIII(BB)]3- and transports only [FeIII(Ent)]3-.12,43 It has been proposed that the opposite chirality of the ferric complexes formed with Ent and BB accounts for the discrimination between both siderophores by receptors.36,37 However, not only does d-Ent have the same transport properties as Ent,12 both [FeIII(SGC)]3- and [FeIII(d-SGC)]3- are transported into B. subtilis at levels similar to that of [FeIII(BB)]3-, as shown by 55Fe-siderophore uptake experiments performed on B. subtilis ATCC 6051 cells cultured in iron-limited medium (Figure 4). Since chirality at the metal center does not affect ferric siderophore incorporation, the larger size and elongated shape of [FeIII(BB)]3-, [FeIII(SGC)]3- and [FeIII(d-SGC)]3- (resulting from the insertion of glycine spacers) are therefore more likely to be the discriminating features of the second Ent receptor.
Figure 4.
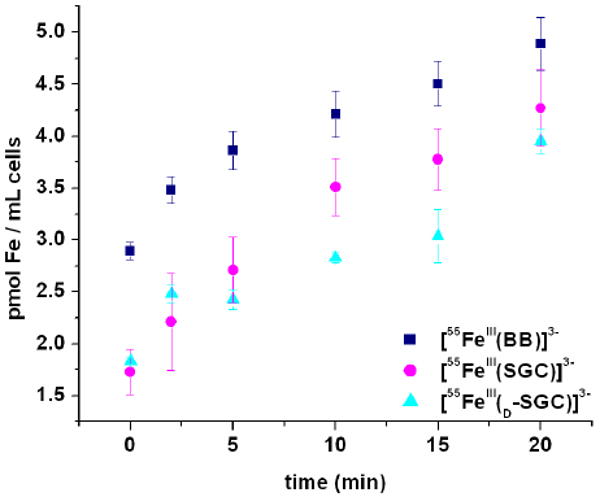
Iron transport mediated by [55FeIII(BB)]3-, [55FeIII(SGC)]3-, and [55FeIII(d-SGC)]3- in B. subtilis at 37 °C in iron-limited medium. Data presented are the average of three independent experiments for BB and SGC, and two independent experiments for d-SGC.
To confirm that all five ferric-siderophore complexes can be incorporated through the major catecholate transporter FeuABC, the interactions of each complex with the substrate-binding protein FeuA were characterized using fluorescence spectroscopy. FeuA was recombinantly produced as a C-terminal His6-tag fusion and purified by Ni affinity chromatography, following a modified described procedure.30 To determine the equilibrium dissociation constant of FeuA for each ferric-siderophore complex, aliquots of freshly prepared solutions of the complex (6 μM) were added successively to a freshly isolated FeuA-His6 solution (100 nM) and the fluorescence intensity of the mixture (λexc = 281 nm, λem = 340 nm) was measured after 5 min of equilibration. The Kd values calculated by non-linear least-squares analysis of the fluorescence titration curves (Figure 5) are reported in Table 3. As previously discussed by Miethke et al.,30 both [FeIII(BB)]3- and [FeIII(Ent)]3- specifically bind to FeuA, with similar affinities (Kd = 15 ± 4 nM and 19 ± 5 nM, respectively). Correspondingly, all three synthetic analogs [FeIII(d-Ent)]3-, [FeIII(SGC)]3- and [FeIII(d-SGC)]3- exhibit specific quenching of the receptor protein, with equilibrium dissociation constants between 10 nM and 52 nM, in the same order of magnitude as those observed for the two complexes derived from natural siderophores. Significantly, the ferric complexes formed with the d-serine trilactone-based ligands, [FeIII(d-Ent)]3- and [FeIII(d-SGC)]3-, showed slightly higher affinities for FeuA than their respective l-serine counterparts. The same trend was observed in the binding of [FeIII(Ent)]3- and [FeIII(d-Ent)]3- to the E. coli periplasmic protein FepB,44 which transports ferric-Ent from the periplasm into the cytoplasm. These spectroscopic measurements confirm that there is no significant discrimination in the recognition and binding of the different ferric complexes of trilactone-based catecholate siderophores by the FeuA substrate-binding protein, the entrance point of the FeuABC transporter.
Figure 5.
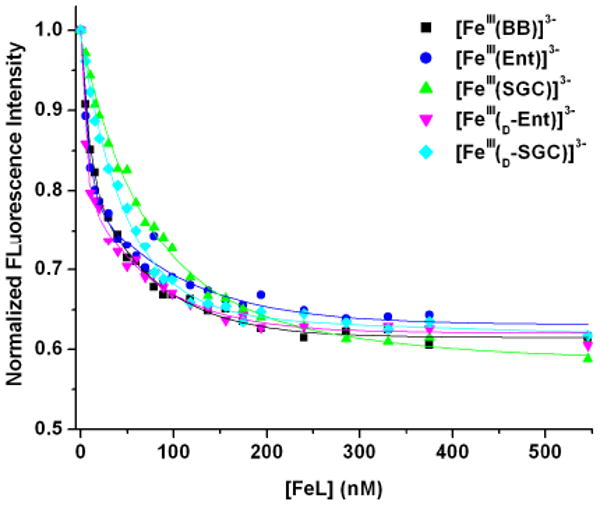
Fluorescence quenching analyses of the substrate-binding receptor protein FeuA-His6 with the five studied ferric complexes at pH 7.4. Symbols give the fluorescence data at 340 nm, and lines give the non-linear least squares calculated fits.
Table 3.
Kd Values Determined by Fluorescence Quenching for the Binding of Ferric Complexes by the Receptor Protein FeuA at pH 7.4.a
| FeuA substrate | Kd nM (esd's) |
|---|---|
| [FeIII(Ent)]3- | 19(5)b |
| [FeIII(d-Ent)]3- | 10(2) |
| [FeIII(SGC)]3- | 52(8) |
| [FeIII(d-SGC)]3- | 32(4) |
| [FeIII(BB)]3- | 15(4)b |
Uncertainties were determined from the standard deviation of three independent titrations.
The discrepancy in the absolute Kd values determined for these complexes in the present study, as compared to those reported earlier,30 are attributed to the different analytical methods used (least-squares analysis vs. midpoint estimate). A significant decrease in ferric-siderophore complex affinity was observed when stock solutions of the protein were not prepared on the day of the fluorescence experiment, which could also impair comparison between these and the previously reported values.
Utilization of trilactone-based catecholate siderophores by B. subtilis
Active transport of a ferric-siderophore complex through the bacterial membrane does not necessarily imply that the iron is easily available to the bacteria. To probe whether all five trilactone-based siderophores and synthetic analogs provide iron usable by B. subtilis, growth promotion assays were performed with B. subtilis HB5600 (dhbA∷spc), a strain lacking the biosynthetic enzymes for its own endogenous DHBG and BB.35 Growth of this strain in the presence of the strong iron chelator 2,2′-dipyridyl requires transport and utilization of an exogenously provided siderophore. While the natural ligands Ent and BB and the synthetic SGC all promoted growth of B. subtilis HB5600, no significant zone of growth stimulation was observed when discs infused with d-Ent and d-SGC were overlaid on the bacterial cultures (Table 4). Furthermore, deletion of the receptor feuA in B. subtilis HB5625 (feuA∷spc dhbA∷mls) prevented growth stimulation by not only BB and Ent, as reported by Helmann and coworkers,35 but also by SGC. Thus, FeuABC is the major transporter used to incorporate the ferric complexes of all five trilactone-based ligands into the bacterial intracellular space, where discrimination between iron transporters occurs. These growth stimulation experiments did not however provide any evidence of the secondary Ent receptor detected through direct uptake assays by Dertz et al.12 Similar results have been reported for Ent-producing Gram-negative bacteria: in S. typhimurium, both outer membrane receptors FepA and IroN participate in Ent-mediated iron transport but only IroN recognizes [FeIII(BB)]3-.34 Correspondingly, [FeIII(d-Ent)]3- is also incorporated in E. coli through FepA but does not promote growth of the microorganism.32,33 In the ferric complexes tested, the chiralities of the trilactone stereocenters and of the metal centers must be distinguished. Both [FeIII(BB)]3- and [FeIII(SGC)]3- exhibit Λ chirality at the ferric center, the opposite configuration to that of [FeIII(Ent)]3- (Δ), and still deliver iron to the bacteria as efficiently as [FeIII(Ent)]3-. In contrast, d-SGC forms a Δ ferric complex but does not stimulate growth of the bacterium. Thus there is a clear picture: the chirality of the ligand trilactone, not that of the metal center, is the selective feature of these complexes for successful bacterial iron uptake.
Table 4.
Growth Promotion of B. subtilis HB5600 (dhbA∷spc) and B. subtilis HB5625 (feuA∷spc dhbA∷mls) by Natural and Synthetic Trilactone-Based Siderophores.a
| Growth of B. subtilis straina (diameter of growth zone, mm) | |||
|---|---|---|---|
|
|
|||
| Siderophore | HB 5600 | HB 5625 | |
| dhbA | − | − | |
| feuA | + | − | |
| Ent | + (21) | − | |
| d-Ent | − | − | |
| SGC | + (21) | − | |
| d-SGC | − | − | |
| BB | + (20) | − | |
Growth promotion of B. subtilis strains was assessed on LB agar plates supplemented with 0.5 mM 2,2′-dipyridyl. The genotypes of the strains are shown as follows: +, wild type; -, mutant. Symbols: +, a halo of growth around filter paper discs is observed; -, no growth.
The chiral recognition of trilactone-based siderophores by esterases
The use of BB for iron uptake by Bacilli species requires enzymatic hydrolysis of the tri-threonine backbone.30 The B. subtilis gene locus yuiI (recently named besA)30 encodes the production of the esterase BesA, which was found to hydrolyze both BB and [FeIII(BB)]3-, with a higher catalytic efficiency observed for the ferric complex.30 The linear derivatives of BB formed after cleavage of the ester bonds in the lactone are based on 2,3-dihydroxybenzoyl-glycine-threonine units (2,3-DHBGT). Miethke and coworkers have shown that five BB-producing B. subtilis related species, including the human pathogens B. anthracis and B. cereus, possess a yuiI ortholog downstream of their feuABC(feuD) transport genes.30 The yuiI orthologs were found highly conserved among these species and may correspondingly be essential for hydrolysis of the BB scaffold and subsequent intracellular iron release in all respective species. The esterase BesA was recombinantly produced as a C-terminal His6-tag fusion and purified by Ni affinity chromatography. Hydrolysis reactions by the protein were carried out at pH 7.4, with 72 nM enzyme and 64 μM siderophore, and followed by HPLC (Figure 6). The single substrate concentrations used in this enzymatic assay did not permit quantitative determination of the kinetic parameters but were sufficient to describe the scope of substrates for Bes-catalyzed hydrolysis. The three ligands BB, Ent and SGC were hydrolyzed sequentially into the linear trimeric, dimeric and monomeric derivatives of 2,3-DHBGT, 2,3-DHBS (2,3-dihydroxybenzoyl-serine) and 2,3-DHBGS (2,3-dihydroxybenzoyl-glycine-serine) respectively. However BesA had no effect on the tri-d-serine lactone of d-Ent and d-SGC. In addition, the reactions between the ferric complexes formed with all five siderophores and BesA were monitored by UV-Vis spectroscopy (Figures 7 and S2). When BesA was added to [FeIII(BB)]3-, [FeIII(Ent)]3- or [FeIII(SGC)]3-, the UV-Vis spectra changed, as expected, to the characteristic spectra of the corresponding tris-bidentate complexes [FeIII(2,3-DHBGT)]3-, [FeIII(2,3-DHBS)3]3- and [FeIII(2,3-DHBGS)3]3-, respectively.29 No change in the absorption spectra was observed when Bes was added to [FeIII(d-Ent)]3- or [FeIII(d-SGC)]3-.
Figure 6.
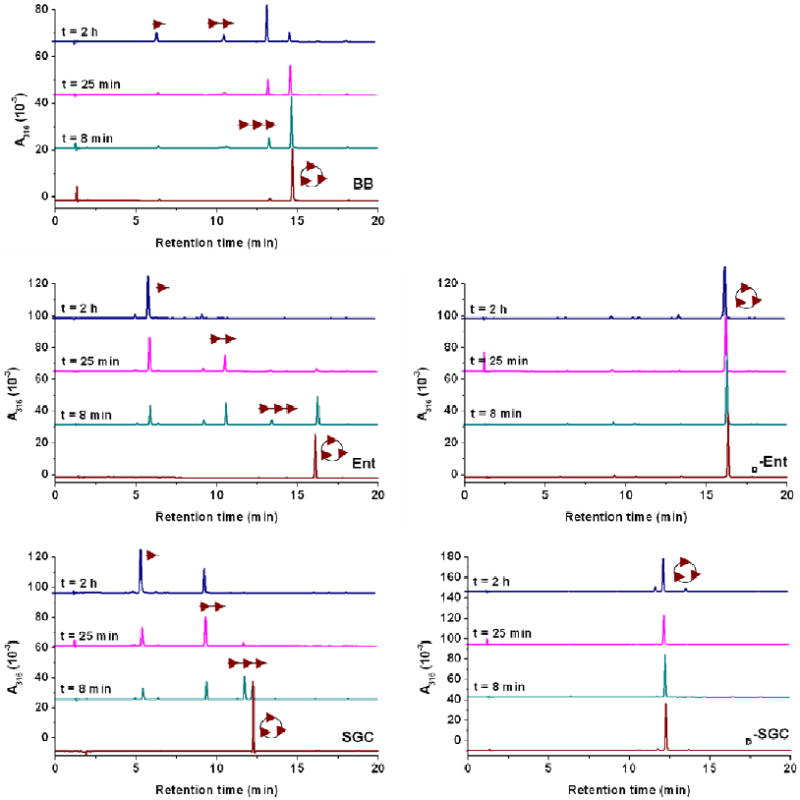
Reaction time courses of Bes-catalyzed hydrolysis of BB (top), Ent (middle left) and SGC (bottom left). The presence of Bes in solution did not affect d-Ent (middle right) and d-SGC (bottom right). Reaction aliquots were quenched at different time points and analyzed by HPLC. The assignment of the hydrolysis products is based on the corresponding mass spectrometry data (Figures S4-S6), and the schematic representations of the hydrolysis products (cyclic trimer, linear trimer, dimer and monomer) are shown consistently with similar previous studies.29
Figure 7.
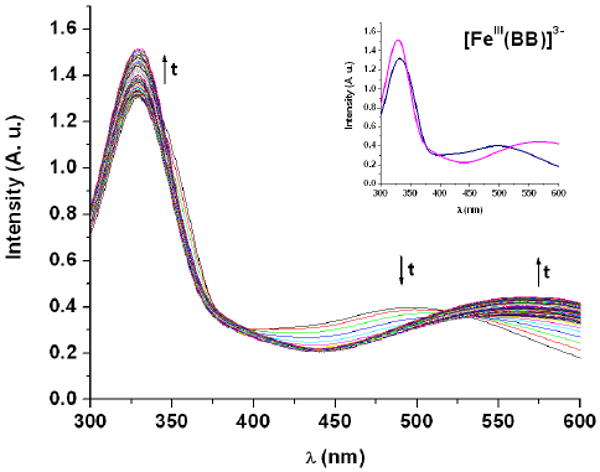
Bes-catalyzed hydrolysis of [FeIII(BB)]3- to [FeIII(2,3-DHBGT)3]3- followed by UV-vis spectroscopy ([Fe(BB)] = 100 μM, [Bes] = 1 μM, 75mM HEPES, pH 7.5, 25 °C, 1 cm cell). The inset shows the spectra at t = 0 h (blue) and t = 12 h (pink).
In Ent-producing species, the apo form of Ent and its ferric complex are subject to enzymatic sequential hydrolysis by the designated cytosolic esterase Fes, with the catalytic efficiency being much higher for [FeIII(Ent)]3- than for apo-Ent.29 On the other hand, neither apo-d-Ent nor [FeIII(d-Ent)]3- are hydrolyzed by Fes.45 Enzymatic reactions with Fes from Shigella flexneri were followed for the remaining siderophores: SGC was hydrolyzed whereas d-SGC and BB did not react (Figure S3). Thus, Fes appears more specific than BesA: while the l-serine scaffold is an essential feature for Fes hydrolysis and intracellular iron release in enterobacteric species, the esterase BesA used by Bacilli species can also hydrolyze the threonine scaffold of BB.
Ent- and BB-mediated iron transport
As known to date, microorganisms producing Ent (or BB, respectively) do not synthesize BB (or Ent, respectively). However, bacteria commonly develop several membrane receptors to appropriate xenobiotic siderophores for their own use. As mentioned above, some Ent-producing bacterial strains, such as S. typhimurium, possess two receptor proteins, FepA and IroN, for catecholate siderophores and can transport both [FeIII(Ent)]3- and [FeIII(BB)]3-.34 Likewise, B. subtilis can utilize Ent and acquires its ferric complex through FeuA and a specific receptor different from that designed for [FeIII(BB)]3-.12,35 Nevertheless, once taken up, the ferric complexes must be hydrolyzed to release iron. While BesA enzymatically cleaves both threonine and l-serine trilactones, only the tri-l-serine macrocycle is a substrate for Fes. Enteric strains utilizing BB must therefore encode an ortholog of yuiI. Pathogenic E. coli and Salmonella strains that encode the iroA gene cluster produce the esterases IroE and IroD,29 which show approximately 37% (Basic Local Alignment Search Tool,46,47 E = 5×10-26) and 30% (E = 2×10-05) identity to BesA, respectively, and contain the conserved G×S×G serine esterase motif; the multiple sequence alignment for BesA, IroE, IroD and Fes, as well as the corresponding dendrogram are shown in Figures S7-A and S7-B. Since BB is recognized by the same S. typhimurium receptor as DGE and both enzymes have been shown to participate in DGE hydrolysis,29 IroE and IroD are viable candidates for the hydrolysis of BB in yuiI-lacking bacterial strains.
Significantly, proteins involved in uptake and hydrolysis of trilactone-siderophores and their ferric complexes exhibit different recognition patterns: the binding of receptor proteins is determined by the coordination chemistry around the metal center (ferric catecholate complexes are not discriminated in this case) whereas identity and stereochemistry of the lactone-monomer are the selective features for backbone hydrolysis by the corresponding esterases. It remains unknown whether the divergent evolution and recognition of substrate-binding proteins and hydrolases is correlated to the production of specific siderophores. Moreover, the order of appearance of Ent and BB as bacterial metabolites has not been determined, and it is yet unexplained why only some BB-producing strains can produce adequate receptors and esterases to utilize exogenous Ent, and vice-versa. It is also remarkable that, when acidic solutions (6M HCl) of Ent and BB were stirred overnight and analyzed (Figure S8) by HPLC, Ent was completely degraded while BB remained essentially intact; hence the threonine macrocycle is much more resistant to acid hydrolysis than its serine counterpart. Methylation of the Ent backbone in BB could therefore be a bacterial strategy to produce a siderophore resistant to acidic environment.
Conclusion
While the triscatecholate ferric center found in [FeIII(Ent)]3- and [FeIII(BB)]3- serves as the primary recognition point for specific binding by cognate bacterial receptors or mammalian siderocalin, the trilactone scaffold incorporated in both siderophores is the crucial feature that determines substrate recognition by designated esterases and subsequent intracellular iron release. This is the source of the chiral selectivity of iron delivery by Ent in E. coli. For BB, insertion of glycine spacers between the macrocycle and the catecholate iron-binding units changes the chirality at the metal center of the ferric complex and results in a larger and more oblate shape; chirality at the metal center does not affect binding by receptor proteins, whereas the shape of the complex does. The use of a tri-threonine rather than a tri-l-serine backbone in BB is the seminal structural change that not only confers acid resistance to the siderophore but also prevents piracy of BB by Ent-producing bacteria. That is, inversion of the trilactone chiral configuration does not inhibit the transport of ferric complexes but does prevent enzymatic hydrolysis and intracellular iron release. Ent and BB are built on similar scaffolds and are the most powerful siderophores used in microbial iron uptake. Nevertheless, fine-tuned changes in the molecular structures of these ligands significantly affect their chemical properties and subsequent recognition in different biological systems. One consequence of understanding the siderophore-production strategies used by various bacterial populations in their arms race for efficient iron acquisition may be the rational design of antibiotics based on siderophores.
Methods
General
All chemicals were obtained from commercial suppliers and were used as received. The starting materials tris(N-hydrochloride-d-serine) trilactone (1)48 and 2,3-di(benzyloxy)benzoyl-glycinyl fluoride (2),37 as well as the ligands Ent,48 d-Ent48 and SGC37 were synthesized according to procedures described in the cited references. The siderophore BB was isolated from B. subtilis and purified as previously reported.18 Flash silica gel chromatography was performed using Merck 40-70 mesh silica gel. Melting points were taken on a Büchi melting apparatus and are uncorrected. All NMR spectra were recorded at ambient temperature on Bruker FT-NMR spectrometers at the NMR Laboratory, College of Chemistry, UC Berkeley. Microanalyses were performed by the Microanalytical Services Laboratory, College of Chemistry, UC Berkeley. Mass spectra were recorded at the Mass Spectrometry Laboratory, College of Chemistry, UC Berkeley. UV-Visible absorption spectra were taken on a Varian Cary 300 UV-Vis spectrometer. CD spectra were recorded on a Jasco J-810 spectropolarimeter.
N,N′,N″-Tris[2,3-di(benzyloxy)benzoyl-glycinyl]cyclotri-d-seryl trilactone, Bn6-d-SERglyCAM (3)
Tris(N-hydrochloride-d-serine) trilactone (0.176 g, 0.48 mmol) was suspended in 40 mL of dry and degassed THF and cooled in an ice/water bath. Solutions of 2,3-di(benzyloxy)benzoyl-glycinyl fluoride (2.5 mmol) in 10 mL of THF and triethylamine (0.49 g, 4.8 mmol) were added simultaneously dropwise via syringes over 10 minutes into this suspension while stirring under nitrogen. The mixture was allowed to warm to room temperature and stirred over night. It was then filtered, concentrated, applied to a silica gel column and eluted with 98:2 CH2Cl2:MeOH. Fractions were combined and evaporated to a red solid. Yield: 0.17 g (26%). 1H NMR (400 MHz, CDCl3): δ 3.66 (dd, J = 11.2 Hz, J′ = 5.6 Hz, 3H), 3.80 (dd, J = 11.2 Hz, J′ = 5.6 Hz, 3H), 4.12 (d, J = 11.6 Hz, 3H), 4.80 (d, J = 7.6 Hz, 3H), 4.90 (d, J = 8.4 Hz, 3H), 5.00-5.13 (m, 12H), 7.00-7.35 (m, 36H), 7.72 (dd, J = 3.2 Hz, J′ = 2.8 Hz, 3H), 8.21 (d, J = 7.6 Hz, 3H), 8.61 (br t, 3H) ppm. 13C NMR (400 MHz, CDCl3): δ 43.4, 65.0, 71.2, 117.6, 123.1, 124.5, 126.6, 127.7, 128.3, 128.4, 128.5, 128.6, 129.3, 136.3, 147.3, 152.0, 165.7, 169.0, 169.8 ppm. Mp: 79-80 °C. (+)-FABMS: m/z 1381.5 (MH+).
N,N′,N″-Tris[2,3-dihydroxybenzoyl-glycinyl]cyclotri-d-seryl trilactone, d-SERglyCAM (4)
Absolute ethanol (14 mL) was added to a suspension of Bn6-d-SERglyCAM (170 mg, 0.12 mmol) in 100 mL of ethyl acetate. The solution was hydrogenated over 10% Pd-C (34.0 mg) at room temperature and atmospheric hydrogen pressure for 24 hours. The reaction mixture was filtered over celite, washed with acetone and evaporated under vacuum. The product was collected as a light beige powder. Yield: 99.0 mg (88%). 1H NMR (500 MHz, DMSO-d6): δ 4.01 (d, J = 5.5 Hz, 6H), 4.27-4.35 (m, 6H), 4.71 (dd, J = 8.0 Hz, J′ = 4.5 Hz, 3H), 6.69 (t, J = 8.0 Hz, 3H), 6.91 (d, J = 6.5 Hz, 3H), 7.30 (d, J = 8.0 Hz, 3H), 8.76 (d, J = 4.5 Hz, 3H), 9.04 (s, 3H), 9.23 (s, 3H), 12.22 (s, 3H) ppm. Mp: 132-133 °C. (+)-FABMS: m/z 841 (MH+). Anal. Calcd (Found) for C36H36N6O18.2H2O.MeOH: C, 48.90 (48.86); H, 4.88 (5.03); N, 9.25 (9.04).
Solution Thermodynamics
Protonation and ferric complex formation constants for SGC were determined using procedures and equipment described previously.49-51
Titration Solutions and Equipment
Corning high performance combination glass electrodes (response to [H+] was calibrated before each titration)52 were used together with either an Accumet pH meter or a Metrohm Titrino to measure the pH of the experimental solutions. Metrohm autoburets (Dosimat or Titrino) were used for incremental addition of acid or base standard solutions to the titration cell. The titration instruments were fully automated and controlled using LabView software.53 Titrations were performed in 0.1 M KCl supporting electrolyte under positive Ar gas pressure. The temperature of the experimental solution was maintained at 25 °C by an external circulating water bath. UV-Visible spectra for incremental titrations were recorded on a Hewlett-Packard 8452a spectrophotometer (diode array). Solid reagents were weighed on a Metrohm analytical balance accurate to 0.01 mg. All titrant solutions were prepared using distilled water that was further purified by passing through a Millipore Milli-Q reverse osmosis cartridge system. Titrants were degassed by boiling for 1 h while being purged under Ar. Carbonate-free 0.1 M KOH was prepared from Baker Dilut-It concentrate and was standardized by titrating against potassium hydrogen phthalate using phenolphthalein as an indicator. Solutions of 0.1 M HCl were similarly prepared and were standardized by titrating against sodium tetraborate to Methyl Red endpoint. Stock solutions of ferric ion were obtained by dissolving solid FeCl3.6H2O in 0.1 M HCl and were standardized by titrations with EDTA according to the methods of Welcher.54
Incremental Titrations
Initial titration solutions were assembled from the constituent reagents in ratios determined previously by modeling using estimated formation constants and the modeling program Hyss.55,56 The solutions were incrementally perturbed by the addition of either acid (HCl) or base (KOH) titrant, followed by a time delay for equilibration (90 seconds for protonation studies; 2 hours for EDTA competition titrations). Potentiometric and EDTA competition titrations were conducted in pairs: first a forward titration from low to high pH, then a reverse titration back to low pH. The data for the two titrations comprising each experiment were pooled for calculation of formation constants when reversibility was achieved. All absorbance measurements used for calculation of formation constants were less than 1.05 absorbance units.
Protonation Constants: Potentiometric Titrations
Solutions were assembled from a weighed portion of ligand and the supporting electrolyte solution, with resulting ligand concentrations between 0.2 and 0.5 mM. An average of 60 – 90 data points were collected in each pair of titrations (forward and back), each data point consisting of a volume increment and a pH reading over the pH range 4 to 9.5. Refinement of the protonation constants was accomplished using the program Hyperquad,57 which allows simultaneous nonlinear least squares refinement of the data from multiple titration curves.
Formation Constants: EDTA Competition Spectrophotometric Titrations
Each EDTA competition experiment consisted of a pair of titrations: forward vs. KOH and reverse vs. HCl. At the lower pH end of the titration, the colorless iron-EDTA complex dominated, but as the pH was raised, the solution turned pink to red, corresponding to the gradual formation of the iron-experimental ligand complex. Solutions were assembled from a weighed portion of ligand and the supporting electrolyte solution, with resulting ligand concentrations of about 0.1 mM. The EDTA was present in a ten fold excess and the iron in a 0.9 fold default over the ligand in order to prevent formation of insoluble iron species. In each incremental experiment, an average of 24 points was collected over a period of 48 hours (∼2 hr equilibration time per point). Each data point consisted of a pH measurement and an absorbance spectrum over at least 80 different wavelengths between 300 and 700 nm over the pH range 5 to 7.5. The data were imported into the refinement program pHab58 and analyzed by non-linear least-squares refinement.
Data Treatment
All equilibrium constants were defined as cumulative formation constants, βmlh according to eq. 1, where the ligand is designated as L. Stepwise protonation constants, Kan, may be derived from these cumulative constants according to eq. 2. In the case of the competition experiments, the equilibration of iron between SGC and EDTA was calculated by including the proton association and iron formation constants for EDTA59 as fixed parameters in the refinements. Because of the hexacoordinate nature of the ligand, it was assumed that ternary (i.e., mixed EDTA-Fe-SGC) complexes were not formed. This assumption was supported by spectral data showing that the band shapes and λmax values of the LMCT transitions of the Fe-SGC complex were unchanged in the presence and absence of EDTA. Each pair of titrations (i.e., forward titration against KOH and reverse titration against HCl) was combined for simultaneous refinement. For potentiometric titrations, both the proton and ligand concentrations were refined, only the proton concentration was allowed to vary in the spectrophotometric studies, and all other concentrations were held at estimated values determined from the volume of standardized stock or the weight of ligand (measured to 0.01 mg). Refined concentrations were within 5% of the analytical values. For spectral titrations, all species formed with the ligand were considered to have significant absorbance to be observed in the UV-Vis spectra.
Circular Dichroism Spectroscopic Measurements
Solutions for CD were made in situ. The ferric complexes were prepared from DMSO stock solutions of the free ligands (4 mM, 25 μL) with iron trichloride (27 mM, 3.7 μL). Sodium phosphate (pH = 7.4, 100 μL) was added to form the tris-catecholate complex, and the solutions were diluted with water to yield a final FeL concentration of 0.1 mM. The spectra were obtained with cuvettes of 1 cm path length and recorded on a Jasco J-810 spectropolarimeter.
Protein Cloning, Expression and Purification
The esterase Fes from the human pathogen Shigella flexneri 2a str. 2457T was purified by Dr. Natalia Maltseva (Argonne National Laboratory), as described elsewhere.45 The substrate-binding protein FeuA from B. subtilis 168 and the esterase BesA from B. cereus ATCC 14579 were cloned, expressed and purified as follows. The besA gene (iroE, BC_3734) was amplified by PCR from the chromosomal DNA using the following primers: besF (5′-CACCGTGAATACTACAGTTGAAAAACAGCA-3′) and besR (5′-TCCCTGAAAATACAGGTTTTCTACGTAACTAATAAACCTCAATCCTTT-3′). These primers were designed to enable directional cloning into a pET101/D-TOPO expression vector introducing a C-terminal His6-tag; a TEV protease cleavage site was incorporated into the besR primer to remove the tag after protein purification (underlined sequences). The feuA gene was amplified by PCR from the chromosomal DNA using the following primers: feuF (5′-CACCATGGGCAGCAAAAATGAATCAACT-3′) and feuR (5′-TCAATGGTGATGGTGATGATGGTTTTGTGTCAATTTTTCAGCAG-3′). The His6 sequence followed by the stop codon was introduced into the feuR primer (underlined) so that the recombinant protein has His6 residues added immediately at the C-terminus. The PCR products were cloned into a pET101/D-TOPO vector using a Champion pET Directional TOPO expression kit (Invitrogen) and used to transform One Shot TOP10 chemically competent Escherichia coli cells. The correct sequence of the insert was confirmed by sequencing and the construct was transformed into chemically competent E. coli BL21(DE3) (Invitrogen). Expression of the recombinant BesA-His6 or FeuA-His6 was induced with 0.5 mM of IPTG when the cultures reached an OD600 of 0.6-0.8. After induction, cultures were incubated an additional 4-5 h at 37 °C. Cells were harvested by centrifugation at 4 °C and the pellets frozen at -20 °C. For protein purification, the harvested cells were suspended in BugBuster Protein Extraction Reagent Master Mix (5 mL per 1 g of wet cells) (EMD Biosciences) and the protease inhibitor cocktail for purification of His-tagged proteins (Sigma) was added. After 10 min of incubation at room temperature, glycerol (5%), NaCl (300 mM) and imidazole (10 mM) were added and the mixture was incubated for an additional 10 min. The cell debris were removed by centrifugation for 20 min at 16,000 × g at 4 °C. Recombinant BesA-His6 and FeuA-His6 were purified from the clarified cell extract using HIS-Select nickel affinity agarose gel (Sigma-Aldrich) to >95% purity (Figure S9). The equilibration and wash buffer consisted of 50 mM sodium phosphate, pH 8.0, 0.5 M NaCl, 10% glycerol, and 10 mM imidazole, for both proteins. The proteins were eluted with a buffer containing 50 mM sodium phosphate, pH 8.0, 0.5 M NaCl, 10% glycerol, and 250 mM imidazole. The purified BesA-His6 (Yield from culture of transformed E. coli: 9.0 mg.L-1) and FeuA-His6 (Yield from culture of transformed E. coli: 2.8 mg.L-1) were stored frozen at −20 °C in the elution buffer. The C-terminal His6-tag was removed from the recombinant BesA-His6 using AcTEV protease (Invitrogen). Prior to tag removal the buffer was exchanged into Tris-buffered saline (TBS), pH 7.4 by gel filtration (Sephadex G-25). BesA was purified using nickel affinity chromatography. The elution buffer for BesA and FeuA-His6 was exchanged into TBS, pH 7.4, the working buffer for fluorescence experiments. The concentration of eluted proteins was measured by bicinchoninic acid (BCA) protein assay. SDS-PAGE analysis was performed on polyacrylamide gels and the gels were stained with Coomasie Brilliant Blue R250 (Figure S9). Molecular weight and homogeneity of BesA and FeuA-His6 were examined using electrospray ionization-mass spectrometry.
Fluorescence Quenching Binding Assay
Fluorescence quenching of recombinant FeuA-His6 was measured on a Cary Eclipse fluorescence spectrometer with 10 nm slit band pass and a high voltage detector, using the characteristic excitation and emission wavelengths λexc = 281 nm and λem = 340 nm. Measurements were made at a protein concentration of 100 nM in buffered aqueous solutions, plus 32 μg/mL ubiquitin (Sigma). Fluorescence values were corrected for dilution upon addition of substrate. Fluorescence data were analyzed by nonlinear regression analysis of fluorescence response versus ligand concentration using a one-site binding model as implemented in DYNAFIT.60 The Kd values are the results of three independent titrations. All binding experiments were done at pH 7.4 using TBS buffer. Control experiments were performed to ensure the stability of the protein in these experimental conditions, and to account for dilution and photobleaching. Substrate solutions were freshly prepared in situ. An aliquot of a DMSO stock solution of the free ligand (4 mM, 25 μL) and FeCl3 salt (27 mM, 1 eq., solution standardized by EDTA titration according to the methods of Welcher54) were combined, vigorously shaken and diluted with TBS buffer to form the tris-catecholate complexes at a concentration of 0.1 mM (concentrations were checked by UV-Visible spectroscopy; for [FeIII(d-SGC)]3-, ε(λ = 494 nm) = 4,900 M-1cm-1 and ε(λ = 338 nm) = 15,100 M-1cm-1). The solutions were equilibrated for 2 h and diluted to a final concentration of 6 μM in aqueous TBS buffer (pH 7.4).
HPLC Separation Assay
All High Pressure Liquid Chromatography runs were performed in the same conditions, by injection of 20 μL of filtered solutions (0.22 μM) through a reverse-phase analytical C-18 column. A gradient from 5% CH3CN in ddH2O/0.1%TFA to 50% CH3CN in ddH2O/0.1% TFA over 20 min at 1 mL/min was used to elute the siderophores and their derivatives (detection by UV-Vis absorption at 316 nm and identification by electrospray mass spectrometry). All enzymatic reactions were carried out in 75 mM HEPES buffer pH 7.5, as described previously,29 with 64 μM substrates and 72 nM enzymes. Fes and BesA frozen stocks were thawed on ice, diluted to 1 μM with cold buffer (25 mM Tris pH 8, 50 mM NaCl, 1 mM DTT, 10% v glycerol) and then added to the reaction mixture. The reactions were quenched at 2, 8, 25 min and 2 h with 0.5 volumes of 2.5 N HCl in methanol and then frozen on dry ice. For HPLC analysis, the samples were thawed just before injection. For strong acid hydrolysis reactions, acidic solutions of Ent and BB (1 mg/mL, 6 M HCl) were stirred for 12 h before HPLC injection.
Enzymatic Hydrolyses Followed by UV-Vis Spectroscopy
All reactions were carried out in 75 mM HEPES buffer pH 7.5, as described previously,29 with 100 μM substrates and 1 μM enzymes. The ferric-siderophore complexes were prepared from DMSO stock solutions of the free ligands (4 mM, 6.76 μL) with iron trichloride (27 mM, 1.0 μL), and diluted to 270 μL. Fes and BesA frozen stocks were thawed on ice, diluted to 1 μM with cold buffer (25 mM Tris pH 8, 50 mM NaCl, 1 mM DTT, 10% v glycerol) and then added to the reaction mixture. UV-Visible absorption spectra were recorded in a 100 μL quartz cuvette (1 cm path length), over 12 h, on a Varian Cary 300 UV-Vis spectrometer.
Disc Diffusion Assays
Growth assays were performed as described in the literature61 with B. subtilis strains HB5600 (dhbA∷spc) and HB5625 (feuA∷spc dhbA∷mls) obtained from Prof. John Helmann (Cornell University).35 Both strains were grown to late exponential phase in LB medium supplemented with spectinomycin (100 μg mL-1); erythromycin (1 μg mL-1) and linkomycin (25 μg mL-1) were added in the case of HB5625. Cells were washed twice with TE buffer at pH 8, resuspended in sterile water and diluted 1:100 in 0.7% noble agar cooled to 45 °C. Aliquots of 3 mL of these suspensions were overlaid onto LB agar plates supplemented with 0.5 mM 2,2′-dipyridyl. Sterile paper discs infused with 20 nmoles of tested siderophore were placed on the agar overlays, incubated at 37 °C overnight and examined for the presence of zones of growth around the discs. The bacterial growth can only be supported by the siderophores that have higher or comparable affinity for iron than 2,2′-dipyridyl.
Ferric complex transport assays in B. subtilis ATCC 6051
These assays were performed following modified described procedures and using radio-labeled ferric-siderophore complexes.43 We chose to perform these experiments with the wild-type BB producer B. subtilis ATCC 6051 rather than with the mutant strain B. subtilis HB5600 (dhbA∷spc) to be consistent with previous siderophore-mediated iron uptake studies in this model organism.12,43 The iron complexes were formed by mixing 55FeCl3 (2.8 Ci/mmol, 6.6 μL, 19 nmol) and FeCl3 (2.7 μL, 72 nmol) with the corresponding ligand (25 μL, 100 nmol) in a ratio of 0.9:1 and ddH2O (165.7 μL). These solutions were then incubated at room temperature for 2 hours and diluted with sodium phosphate (100 μL, 1 M, pH 7.4) and ddH2O (700 μL). Free iron was removed by centrifugation at 14,000 rpm for 1 min. An aliquot of each concentrated stock solution (100 μL, 91 μM Fe) was diluted with 9.9 mL of the iron-limited medium and filtered twice through membrane filters (HAWP Millipore, 0.45 μm pore size). The concentrations of the ferric complexes in the working solutions were quantified by determining the specific radioactivity. B. subtilis ATCC 6051, acquired from the American Type Culture Collection and cultured on nutrient agar plates, was grown in iron-free medium to the late exponential phase. The cells were then washed, suspended in the iron-limited medium, and kept on ice until the transport assay. The resuspension was set to an OD600 of 0.61 ± 0.02. After incubation of the cells (9 mL), for 10 min at 37 °C (for cold transport assays, cells were kept on ice throughout the experiment), the assay was started by addition of 55Fe-siderophore (1 mL at 0.9 μM). Aliquots (1 mL) were removed at appropriate times, filtered through membrane filters (HAWP Millipore, 0.45 μm pore size), and washed with 10 mL of cold 0.1 M sodium citrate. Filters were dried and 6 mL of liquid scintillation Ecolume (ICN) was added. The vials were shaken, stored for 12 hours, and the radioactivity determined using a Perkin Elmer Tri-Carb 2800 TR liquid scintillation analyzer.
Supplementary Material
Acknowledgments
We thank Dr. Youngchang Kim for many helpful discussions, as well as Prof. John Helmann and Dr. Natalia Maltseva for kindly providing us with bacterial strains and purified Fes samples, respectively. This research was supported by the National Institutes of Health (Grant AI11744).
Footnotes
The pM value is a direct measure of the chemical free energy of the metal ion at equilibrium with the chelating siderophore. It is defined as the negative logarithm of the free iron concentration in equilibrium with complexed and free ligand, at physiological pH (pH 7.4) with 1 μM total iron concentration and 10 μM total ligand concentration.
Supporting Information Available: Additional figures and spectral data obtained for both enzymatic and acidic hydrolysis experiments; Synthetic scheme for d-SGC. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Coordination Chemistry of Microbial Iron Transport. 86. Part 85. Zawadzka AM, Abergel RJ, Nichiporuk R, Andersen UN, Raymond KN. Biochemistry. 2009;48:3645–3657. doi: 10.1021/bi8018674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris WR. In: Molecular and Cellular Iron Transport. Templeton DM, editor. Marcel Dekker, Inc.; New York: 2002. pp. 1–40. [Google Scholar]
- 3.Braun V, Hantke K, Koster W. In: Metal ions in biological systems. Sigel A, Sigel H, editors. Vol. 35. Marcel Dekker; New York: 1998. pp. 67–145. [PubMed] [Google Scholar]
- 4.Raymond KN, Dertz EA. In: Iron transport in bacteria. Crosa JH, Mey AR, Payne SM, editors. ASM Press; Washington, D. C.: 2004. pp. 3–17. [Google Scholar]
- 5.Boukhalfa H, Crumbliss AL. BioMetals. 2002;15:325–339. doi: 10.1023/a:1020218608266. [DOI] [PubMed] [Google Scholar]
- 6.Andrews SC, Robinson AK, Rodriguez-Quinones F. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 7.Winkelmann G. Microbial Transport Systems. Wiley-VCH; Weinheim: 2001. [Google Scholar]
- 8.Brown JS, Holden DW. Micr Infec. 2002;4:1149–1156. doi: 10.1016/s1286-4579(02)01640-4. [DOI] [PubMed] [Google Scholar]
- 9.Weinberg ED. BioMetals. 2000;13:85–89. doi: 10.1023/a:1009293500209. [DOI] [PubMed] [Google Scholar]
- 10.Raymond KN, Dertz EA, Kim SS. Proc Natl Acad Sci USA. 2003;100:3584–3588. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loomis LD, Raymond KN. Inorg Chem. 1991;30:906–911. [Google Scholar]
- 12.Dertz EA, Xu J, Stintzi A, Raymond KN. J Am Chem Soc. 2006;128:22–23. doi: 10.1021/ja055898c. [DOI] [PubMed] [Google Scholar]
- 13.O'Brien IG, Gibson F. Biochim Biophys Acta. 1970;215:393–397. doi: 10.1016/0304-4165(70)90038-3. [DOI] [PubMed] [Google Scholar]
- 14.Pollack JR, Neilands JB. Biochem Biophys Res Comm. 1970;38:989–992. doi: 10.1016/0006-291x(70)90819-3. [DOI] [PubMed] [Google Scholar]
- 15.May JJ, Wendrich TM, Marahiel MA. J Biol Chem. 2001;276:7209–7217. doi: 10.1074/jbc.M009140200. [DOI] [PubMed] [Google Scholar]
- 16.Koppisch AT, Browder CC, Moe AM, Shelley JT, Kinkel BA, Hersman LE, Iyer S, Ruggiero CE. BioMetals. 2005;18:577–585. doi: 10.1007/s10534-005-1782-6. [DOI] [PubMed] [Google Scholar]
- 17.Koppisch AT, Dhungana S, Hill KK, Boukhalfa H, Heine HS, Colip LA, Romero RB, Shou Y, Ticknor LO, Marrone BL, Hersman LE, Iyer S, Ruggiero CE. BioMetals. 2008;21 doi: 10.1007/s10534-008-9144-9. epub. [DOI] [PubMed] [Google Scholar]
- 18.Wilson MK, Abergel RJ, Raymond KN, Arceneaux JEL, Byers BR. Biochem Biophys Res Comm. 2006;348:320–325. doi: 10.1016/j.bbrc.2006.07.055. [DOI] [PubMed] [Google Scholar]
- 19.Temirov YV, Esikova TZ, Kashparov IA, Balshova TA, Vinokurov LM, Alakhov YB. Russ J Bioorg Chem (Engl Transl) 2003;29:542–549. doi: 10.1023/b:rubi.0000008894.80972.2e. [DOI] [PubMed] [Google Scholar]
- 20.Chen X, Koumoutsi A, Scholz R, Borriss R. J Mol Microbiol Biotechnol. 2009;16:14–24. doi: 10.1159/000142891. [DOI] [PubMed] [Google Scholar]
- 21.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 22.Abergel RJ, Wilson MK, Arceneaux JEL, Hoette TM, Strong RK, Byers BR, Raymond KN. Proc Natl Acad Sci USA. 2006;103:18499–18503. doi: 10.1073/pnas.0607055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 24.Bister B, Bischoff D, Nicholson G, Valdebenito M, Schneider K, Winkelmann G, Hantke K, Sussmuth RD. BioMetals. 2004;17:471–481. doi: 10.1023/b:biom.0000029432.69418.6a. [DOI] [PubMed] [Google Scholar]
- 25.Fischbach MA, Lin H, Zhou L, Yu Y, Abergel RJ, Liu DR, Raymond KN, Wanner BL, Strong RK, Walsh CT, Aderem A, Smith KD. Proc Natl Acad Sci USA. 2006;103:16502–16507. doi: 10.1073/pnas.0604636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischbach MA, Lin HN, Liu DR, Walsh CT. Nature Chem Biol. 2006;2:132–138. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- 27.Rabsch W, Methner U, Voigt W, Tschape H, Reissbrodt R, Williams PH. Infect Immun. 2003;71:6953–6961. doi: 10.1128/IAI.71.12.6953-6961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miethke M, Marahiel MA. Microbiol Mol Biol Rev. 2007;71:413–451. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin H, Fischbach MA, Liu DR, Walsh CT. J Am Chem Soc. 2005;127:11075–11084. doi: 10.1021/ja0522027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miethke M, Klotz O, Linne U, May JJ, Beckering CL, Marahiel MA. Mol Microbiol. 2006;61:1413–1427. doi: 10.1111/j.1365-2958.2006.05321.x. [DOI] [PubMed] [Google Scholar]
- 31.Cooper SR, McArdle JV, Raymond KN. Proc Natl Acad Sci USA. 1978;75:3551–3554. doi: 10.1073/pnas.75.8.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neilands JB, Erickson TJ, Rastetter WH. J Biol Chem. 1981;256:3831–3832. [PubMed] [Google Scholar]
- 33.Thulasiraman P, Newton SM, Xu J, Raymond KN, Mai C, Hall A, Montague MA, Klebba PE. J Bacteriol. 1998;180:6689–6696. doi: 10.1128/jb.180.24.6689-6696.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rabsch W, Voigt W, Reissbrodt R, Tsolis RM, Baumler AJ. J Bacteriol. 1999;181:3610–3612. doi: 10.1128/jb.181.11.3610-3612.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ollinger J, Song K, Antelmann H, Hecker M, Helmann JD. J Bacteriol. 2006;188:3664–3673. doi: 10.1128/JB.188.10.3664-3673.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bluhm ME, Kim SS, Dertz EA, Raymond KN. J Am Chem Soc. 2002;124:2436–2437. doi: 10.1021/ja016651s. [DOI] [PubMed] [Google Scholar]
- 37.Bluhm ME, Hay BP, Kim SS, Dertz EA, Raymond KN. Inorg Chem. 2002;41:5475–5478. doi: 10.1021/ic025531y. [DOI] [PubMed] [Google Scholar]
- 38.Ramirez RJA, Karamanukyan L, Ortiz S, Gutierrez CG. Tetrahed Lett. 1997;38:749–752. [Google Scholar]
- 39.Dertz EA, Xu J, Raymond KN. Inorg Chem. 2006;45:5465–5478. doi: 10.1021/ic060321x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karpishin TB, Dewey TM, Raymond KN. J Am Chem Soc. 1993;115:1842–1851. [Google Scholar]
- 41.Karpishin TB, Stack TDP, Raymond KN. J Am Chem Soc. 1993;115:3052–3055. [Google Scholar]
- 42.Moore CM, Helmann JD. Curr Opin Microbiol. 2005;8:188–195. doi: 10.1016/j.mib.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 43.Dertz EA, Stintzi A, Raymond KN. J Biol Inorg Chem. 2006;11:1087–1097. doi: 10.1007/s00775-006-0151-4. [DOI] [PubMed] [Google Scholar]
- 44.Sprencel C, Cao Z, Qi Z, Scott DC, Montague MA, Ivanoff N, Xu J, Raymond KN, Newton SMC, Klebba PE. J Bacteriol. 2000;182:5359–5364. doi: 10.1128/jb.182.19.5359-5364.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y, Maltseva NI, Abergel RJ, Binkowski TA, Quartey P, Dementieva I, Holzle D, Collart F, Raymond KN, Joachimiak A. Submitted for publication. 2009 [Google Scholar]
- 46.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu Y. FEBS J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer M, Telford JR, Cohen SM, White DJ, Xu J, Raymond KN. J Am Chem Soc. 1997;119:10093–10103. [Google Scholar]
- 49.Cohen SM, O'Sullivan B, Raymond KN. Inorg Chem. 2000;39:4339–4346. doi: 10.1021/ic000239g. [DOI] [PubMed] [Google Scholar]
- 50.Johnson AR, O'Sullivan B, Raymond KN. Inorg Chem. 2000;39:2652–2660. doi: 10.1021/ic991471t. [DOI] [PubMed] [Google Scholar]
- 51.Xu J, O'Sullivan B, Raymond KN. Inorg Chem. 2002;41:6731–6742. doi: 10.1021/ic025610+. [DOI] [PubMed] [Google Scholar]
- 52.Gans P, O'Sullivan B. Talanta. 2000;51:33–37. doi: 10.1016/s0039-9140(99)00245-3. [DOI] [PubMed] [Google Scholar]
- 53.Wandersman C, Delepaire P. Annu Rev Microbiol. 2004;58:611–647. doi: 10.1146/annurev.micro.58.030603.123811. [DOI] [PubMed] [Google Scholar]
- 54.Welcher FJ. The Analytical Uses of Ethylenediamine Tetraacetic Acid. D van Nostrand Co.; Princeton, NJ: 1958. [Google Scholar]
- 55.Alderighi L, Gans P, Ienco A, Peters D, Sabatini A, Vacca A. HYSS. Leeds; U.K. and Florence, Italy: 1999. [Google Scholar]
- 56.Alderighi L, Gans P, Ienco A, Peters D, Sabatini A, Vacca A. Coord Chem Rev. 1999;184:311–318. [Google Scholar]
- 57.Gans P, Sabatini A, Vacca A. HYPERQUAD2000. Leeds; U. K. and Florence, Italy: [Google Scholar]
- 58.Gans P, Sabatini A, Vacca A. Ann Chim (Rome) 1999;89:45–49. [Google Scholar]
- 59.Smith RM, Martell AE. Critical Stability Constants. 1-4 Plenum; New York: 1977. [Google Scholar]
- 60.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 61.Lee JY, Janes BK, Passalacqua KD, Pfleger B, Bergman NH, Liu H, Hakansson K, Somu RV, Aldrich CC, Cendrowski S, Hanna PC, Sherman DH. J Bacteriol. 2007;189:1698–1710. doi: 10.1128/JB.01526-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.