

Abstract
Objective
To study the vitamin D receptor (VDR) gene in a young girl with severe rickets and clinical features of hereditary vitamin D resistant rickets, including hypocalcemia, hypophosphatemia, partial alopecia, and elevated serum levels of 1,25-dihydroxyvitamin D.
Study design
We amplified and sequenced DNA samples from blood for the patient, her mother, and the patient’s two siblings. We amplified and sequenced the VDR cDNA from RNA isolated from the patient’s blood.
Results
DNA sequence analyses of the VDR gene showed that the patient was homozygous for a novel guanine to thymine substitution in the 5′-splice site in the exon 8-intron J junction. Analysis of the VDR cDNA using reverse transcriptase-polymerase chain reaction showed that exons 7 and 9 were fused, and that exon 8 was skipped. The mother was heterozygous for the mutation and the two siblings were unaffected.
Conclusions
A novel splice site mutation was identified in the VDR gene that caused exon 8 to be skipped. The mutation deleted amino acids 303-341 in the VDR ligand-binding domain, which is expected to render the VDR non-functional. Nevertheless, successful outpatient treatment was achieved with frequent high doses of oral calcium. (190 words)
Keywords: Hereditary Vitamin D Resistant Rickets; Vitamin D Receptor; 1,25-dihydroxyvitamin D; Bone; Alopecia
INTRODUCTION
Active vitamin D, 1,25-dihydroxyvitamin D3 [calcitriol or 1,25(OH)2D], is crucial for normal calcium homeostasis. The vitamin D receptor (VDR) facilitates the downstream biological action of 1,25(OH)2D at target tissues. Mutations in the VDR cause hereditary vitamin D resistant rickets (HVDRR), also referred to as vitamin D dependent rickets type II.[1,2] Patients with HVDRR have rickets along with hypocalcemia, hypophosphatemia, secondary hyperparathyroidism, and elevated serum 1,25(OH)2D levels. Many patients also have alopecia. Multiple heterogeneous mutations in the VDR gene have been previously reported as the cause of HVDRR.[1,2] In this study, we identified a unique splice site mutation in the VDR gene as the molecular basis for a new case of HVDRR.
The treatment for patients with HVDRR is not standardized. Several reports describe intravenous calcium therapy to achieve normal serum calcium levels and healing of the rickets. [3-7] There are also limited publications regarding effective oral calcium therapy.[7-9] We describe the successful transition from intravenous to oral calcium therapy in a young child with HVDRR.
CASE DESCRIPTION
A small 24-month-old Hispanic female (length SDS -5.4; weight SDS -6.1) presented with acute respiratory distress, multiple painful fractures, generalized hypotonia, and severe gross motor delay. She was born at term without complications and the mother’s pregnancy was uneventful. The patient’s past medical history was significant for pneumonias and long bone fractures. Voluntary movements were restricted secondary to chronic bone pain, and she did not start to roll over until age 1 year. She was just beginning to sit with assistance. The child was the product of a consanguineous marriage, with the parents being distant cousins from a small town in Mexico. Both parents and two older siblings were healthy, and there was no family history of bone disease. Although the patient’s diet was low in calcium and vitamin D, her dentition did not appear abnormal. She had partial alopecia (including scalp and eyebrows) since birth (Fig 1A).
Figure 1.
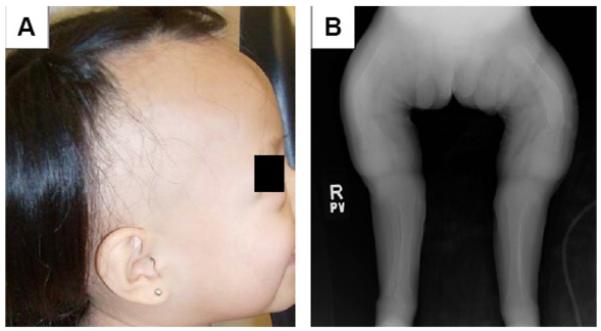
Clinical features of HVDRR. A. Photograph of the patient’s partial alopecia; and B. Radiograph of the patient’s severe rickets.
The patient’s skeletal survey revealed severe rickets, multiple fractures, and osteopenia (Fig 1B) and the initial blood tests were consistent with vitamin D deficiency. However, despite significant improvement in her vitamin D levels following high-dose ergocalciferol and calcitriol therapy, the patient remained hypocalcemic and hypophosphatemic (Table). There was no apparent therapeutic response to pharmacological doses of vitamin D. This suggested resistance to the action of active vitamin D and the possible diagnosis of HVDRR.
Table.
The patient’s biochemical and treatment profiles at initial presentation and while hospitalized
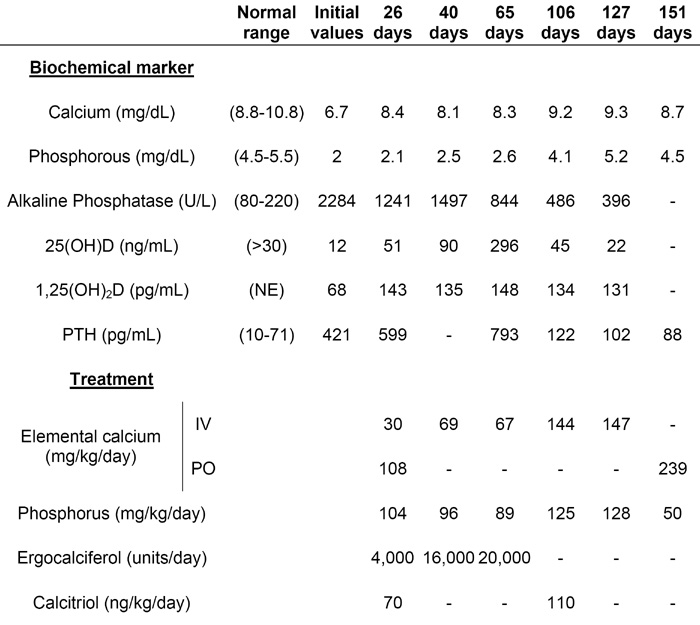 |
NE=Not established for our patient’s age (age 3-17 years: 27-71)
The patient had a prolonged hospitalization in which she was treated with large amounts of intravenous calcium and oral phosphorous (Table) after the diagnosis of HVDRR was confirmed (see below). Bone pain relief was observed within a few weeks of starting intravenous calcium therapy, but the alkaline phosphatase and parathyroid hormone (PTH) levels required several months to decrease to near-normal levels. After 4.5 months of intravenous calcium therapy, the patient demonstrated radiographic healing of the rickets and she was successfully transitioned to high-dose oral calcium therapy. Severe diarrhea secondary to anti-tuberculosis drugs given to treat a positive tuberculin skin test and a nosocomial Clostridium difficile infection precluded an earlier transition to oral therapy. The patient ultimately achieved serum calcium levels ≥8 mg/dL with frequent administration of oral calcium at high doses.
The patient demonstrated gradual improvement in muscle strength and range of motion and then started to sit up, bear weight, and cruise. During an intensive month of inpatient rehabilitation, the patient eventually started to walk. After a total of six months in the hospital, the patient was discharged home. She currently receives oral calcium (200 mg elemental calcium/kg/day) and ergocalciferol (800 units/day). The oral phosphorus supplementation was discontinued after her hyperparathyroidism resolved. The areas of partial hair loss remain unchanged.
RESULTS
After written informed consent was obtained from the family under Childrens Hospital Los Angeles and Stanford University institutional review board-approved protocols, genomic DNA was isolated from peripheral blood samples procured from the patient, her mother, and the patient’s two siblings. The VDR gene was amplified by polymerase chain reaction (PCR) and exons 2-9 were directly sequenced at the Stanford protein and nucleic acid facility.
A unique guanine (G) to thymine (T) substitution was found in the patient’s 5′-splice site in the exon 8-intron J junction in the VDR gene (Fig 2A). The mother was heterozygous for this substitution (Fig 2B) and two siblings were homozygous for the wild-type VDR sequence (data not shown). The wild-type VDR exon 8-intron J sequence is CAG|gtatggggcc (Fig 2C). Substitution of G with T generated the sequence CAG|ttatggggcc that altered the 5′-splice site. The mutation was predicted to cause exon 8 to be skipped during the processing of the mature mRNA (Fig 3A). The VDR cDNA was then amplified from RNA isolated from the patient’s blood using reverse transcription-PCR. Sequence analysis of the VDR cDNA showed that the sequence from exon 7 was followed by the sequence from exon 9 (Fig 3B) confirming that exon 8 was skipped. The fusion of exons 7-9 did not cause a shift in the reading frame and the remaining amino acid sequence encoded by exon 9 was normal (Fig 3B). The skipping of exon 8 deleted amino acids 303-341 in the VDR ligand-binding domain (LBD) including H305, the residue that contacts the 25-hydroxyl group of 1,25(OH)2D.
Figure 2.
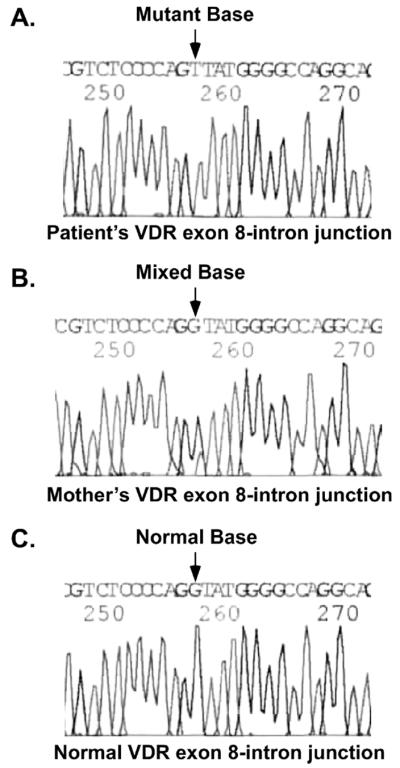
DNA sequence of the patient’s VDR gene. A. The patient’s exon 8-intron J sequence with the homozygous G to T substitution; B. The mother’s exon 8 sequence is heterozygous with the normal sequence (large peak) and the G to T substitution (smaller peak); and C. Normal sequence.
Figure 3.

Exon 8 is skipped in the patient’s VDR cDNA. A. Model showing normal exon splicing of the VDR. Exons are indicated by boxes with the exon number above each box and introns by lines between the boxes. The G to T mutation is shown and its location is indicated. The lower model shows the resultant mutant cDNA with exon 8 skipped. B. Sequence of VDR cDNA showing the fusion between exon 7 and exon 9 and the resultant amino acid sequence. The nucleotide sequence remains in frame. The translated amino acid sequence is shown above the nucleotide sequence. Amino acids 303-341 were deleted due to exon 8 skipping.
An additional finding was that the patient was homozygous (f/f) for the FokI polymorphism that is present at the translation start site.[10-11] The mother was heterozygous (F/f) (data not shown) indicating that the f allele contained the mutation and that the F allele was normal. The father’s DNA was not available for study. The two siblings were unaffected and genotyping revealed that both were homozygous for the wild-type VDR sequence and F/F polymorphism. They, therefore, received an F allele from both the mother and father coding for the wild-type VDR.
DISCUSSION
There are three splice site mutations in the VDR gene that previously have been described in patients with HVDRR.[12-14] The mutations in these cases lead to frame shifts and premature stops in the mRNA. In our patient with the classical features of HVDRR, a unique single base substitution in a 5′-splice site in the VDR gene was found. It caused exon 8 to be skipped during RNA processing and the deletion of 39 amino acids in the LBD of the VDR including the H305 residue that contacts the 25-hydroxyl group of 1,25(OH)2D. This patient’s mutation is expected to alter the conformation of the LBD distal to the deletion including the location of helix 12 and, thus, disrupt 1,25(OH)2D binding and transactivation, and render the VDR non-functional.[15,16] The mutation also likely alters amino acid positions critical for retinoid X receptor binding contributing to the development of alopecia.
An additional feature of this patient’s clinical presentation was concomitant vitamin D deficiency. Her nutritional intake was poor secondary to anorexia from multiple painful fractures. The initial 25-hydroxyvitamin D level was low and, despite secondary hyperparathyroidism, the 1,25(OH)2D level was not significantly elevated. Only after the vitamin D status was improved with high-dose ergocalciferol and calcitriol therapy did the patient’s 1,25(OH)2D level increase to concentrations more typically seen in patients with HVDRR. This helped to elucidate the patient’s resistance to active vitamin D and sustained requirement for calcium therapy.
Some patients with HVDRR have been previously shown to be effectively treated with intravenous calcium therapy.[1-7] Our patient was initially treated with 30-69 mg elemental calcium/kg/day intravenously and, during that time, her alkaline phosphatase levels decreased, her bone pain resolved, and she started to demonstrate improved musculoskeletal strength. Her hyperparathyroidism also eventually improved, although this was not observed until treatment day 106 after the intravenous calcium dose had been increased to almost 150 mg elemental calcium/kg/day. It is unclear whether our patient’s hyperparathyroidism improved secondary to treatment duration, the higher intravenous calcium dose, and/or a combination of these two factors. However, the acute decrease in PTH levels between 65 and 106 days of treatment in our patient is more convincing of a dose-related response. Large amounts of calcium are necessary to suppress PTH secretion in children with HVDRR. It is also possible that our patient’s oral phosphorus therapy contributed to a delayed improvement in her secondary hyperparathyroidism. Patients with HVDRR do not usually require phosphorous supplementation. The hypophosphatemia eventually normalizes after the hypocalcemia is corrected and the hyperparathyroidism improves.[1,2] However, secondary to very low serum phosphorus levels, our child required phosphorus treatment. Repetitive transient elevations in serum phosphorus presumably bound the ionized calcium in the circulation and transiently worsened our patient’s hypocalcemia and stimulated PTH secretion. In addition, multiple acute decreases in serum calcium may induce a more robust reflexive hyperparathyroidism compared to chronic hypocalcemia.
Our patient successfully transitioned to oral calcium therapy after treatment with intravenous calcium for 4.5 months and demonstrated radiographic signs of healing. It is reasonable to believe that our patient’s severe diarrhea precluded an earlier transition to oral calcium therapy. Currently, frequent administration of oral calcium allows sufficient passive transport through the intestinal wall to achieve serum calcium levels ≥8 mg/dL. This and other cases[7-9] illustrate that HVDRR can be treated with high-dose oral calcium therapy.
On the other hand, alopecia in children with HVDRR generally remains unchanged despite treatment and improvement of rickets.[1,2] It remains unclear how the VDR influences the regulation of normal hair growth and why some patients have some scalp hair and others with HVDRR exhibit total alopecia. Our patient’s pattern of alopecia is quite unusual as there are areas of full scalp hair adjacent to areas of total baldness. We have no explanation for the ability of some parts of the scalp to grow abundant hair in this setting.
In conclusion, we identified a young girl with HVDRR, vitamin D deficiency, and partial alopecia who had a novel splice site mutation in the VDR gene. After 4.5 months of intravenous calcium therapy, she was successfully transitioned to oral calcium therapy. With on-going physical therapy, the patient continues to gain musculoskeletal strength and flexibility.
Acknowledgments
Supported by NIH Grant DK42482 (to D.F.).
Glossary
ABBREVIATIONS
- (VDR)
Vitamin D receptor
- [1,25(OH)2D]
1,25-dihydroxyvitamin D3
- (HVDRR)
hereditary vitamin D resistant rickets
- (PTH)
parathyroid hormone
- (PCR)
polymerase chain reaction
- (G)
guanine
- (T)
thymine
- (LBD)
ligand-binding domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr Rev. 1999;20:156–88. doi: 10.1210/edrv.20.2.0359. [DOI] [PubMed] [Google Scholar]
- [2].Malloy PJ, Pike JW, Feldman D. Hereditary 1,25-dihydroxyvitamin D resistant rickets. In: Feldman D, Glorieux F, Pike JW, editors. Vitamin D. Second Edition Elsevier; San Diego, CA: 2005. pp. 1207–38. [Google Scholar]
- [3].Balsan S, Garabedian M, Liberman UA, Eil C, Bourdeau A, Guillozo H, et al. Rickets and alopecia with resistance to 1,25-dihydroxyvitamin D: two different clinical courses with two different cellular defects. J Clin Endocrinol Metab. 1983;57:803–11. doi: 10.1210/jcem-57-4-803. [DOI] [PubMed] [Google Scholar]
- [4].Bliziotes M, Yergey AL, Nanes MS, Muenzer J, Begley MG, Viera NE, et al. Absent intestinal response to calciferols in hereditary resistance to 1,25-dihydroxyvitamin D: documentation and effective therapy with high dose intravenous calcium infusions. J Clin Endocrinol Metab. 1988;66:294–300. doi: 10.1210/jcem-66-2-294. [DOI] [PubMed] [Google Scholar]
- [5].Balsan S, Garabedian M, Larchet M, Gorski AM, Cournot G, Tau C, et al. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77:1661–7. doi: 10.1172/JCI112483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Weisman Y, Bab I, Gazit D, Spirer Z, Jaffe M, Hochberg Z. Long-term intracaval calcium infusion therapy in end-organ resistance to 1,25-dihydroxyvitamin D. Am J Med. 1987;83:984–90. doi: 10.1016/0002-9343(87)90666-8. [DOI] [PubMed] [Google Scholar]
- [7].Hochberg Z, Tiosano D, Even L. Calcium therapy for calcitriol-resistant rickets. J Pediatr. 1992;121:803–8. doi: 10.1016/s0022-3476(05)81919-5. [DOI] [PubMed] [Google Scholar]
- [8].Sakati N, Woodhouse NJY, Niles N, Harfi H, de Grange DA, Marx S. Hereditary resistance to 1,25-dihydroxyvitamin D: clinical and radiological improvement during high-dose oral calcium therapy. Hormone Res. 1986;24:280–7. doi: 10.1159/000180568. [DOI] [PubMed] [Google Scholar]
- [9].Wong GW, Leung SS, Law WY, Cheung NK, Oppenheimer SJ. Oral calcium treatment in vitamin D-dependent rickets type II. J Paediatr Child Health. 1994;30:444–6. doi: 10.1111/j.1440-1754.1994.tb00699.x. [DOI] [PubMed] [Google Scholar]
- [10].Saijo T, Ito M, Takeda E, Huq AH, Naito E, Yokota I, et al. A unique mutation in the vitamin D receptor gene in three Japanese patients with vitamin D-dependent rickets type II: utility of single-strand conformation polymorphism analysis for heterozygous carrier detection. Amer J Hum Gen. 1991;49:668–73. [PMC free article] [PubMed] [Google Scholar]
- [11].Gross C, Eccleshall TR, Malloy PJ, Villa ML, Marcus R, Feldman D. The presence of a polymorphism at the translation initiation site of the vitamin D receptor gene is associated with low bone mineral density in postmenopausal Mexican-American women. J Bone Miner Res. 1996;11:1850–5. doi: 10.1002/jbmr.5650111204. [DOI] [PubMed] [Google Scholar]
- [12].Hawa NS, Cockerill FJ, Vadher S, Hewison M, Rut AR, Pike JW, et al. Identification of a novel mutation in hereditary vitamin D resistant rickets causing exon skipping. Clin Endocrinol. 1996;45:85–92. [PubMed] [Google Scholar]
- [13].Cockerill FJ, Hawa NS, Yousaf N, Hewison M, O’Riordan JL, Farrow SM. Mutations in the vitamin D receptor gene in three kindreds associated with hereditary vitamin D resistant rickets. J Clin Endocrinol Metab. 1997;82:3156–60. doi: 10.1210/jcem.82.9.4243. [DOI] [PubMed] [Google Scholar]
- [14].Katavetin P, Katavetin P, Wacharasindhu S, Shotelersuk V. A girl with a novel splice site mutation in VDR supports the role of a ligand-independent VDR function on hair cycling. Horm Res. 2006;66:273–6. doi: 10.1159/000095546. [DOI] [PubMed] [Google Scholar]
- [15].Malloy PJ, Eccleshall TR, Gross C, Van Maldergem L, Bouillon R, Feldman D. Hereditary vitamin D resistant rickets caused by a novel mutation in the vitamin D receptor that results in decreased affinity for hormone and cellular hyporesponsiveness. J Clin Invest. 1997;99:297–304. doi: 10.1172/JCI119158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–9. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]