

Abstract
Cytokines like interferons (IFNs) play a central role in regulating innate and specific immunities against the pathogens and neoplastic cells. A number of signaling pathways are induced in response to IFN in various cells. One classic mechanism employed by IFNs is the JAK-STAT signaling pathway for inducing cellular responses. Here we describe the non-STAT pathways that participate in IFN-induced responses. In particular, we will focus on the role played by transcription factor C/EBP-β in mediating these responses.
Keywords: cytokine, gene expression, signal transduction, antiviral, innate immunity
Introduction
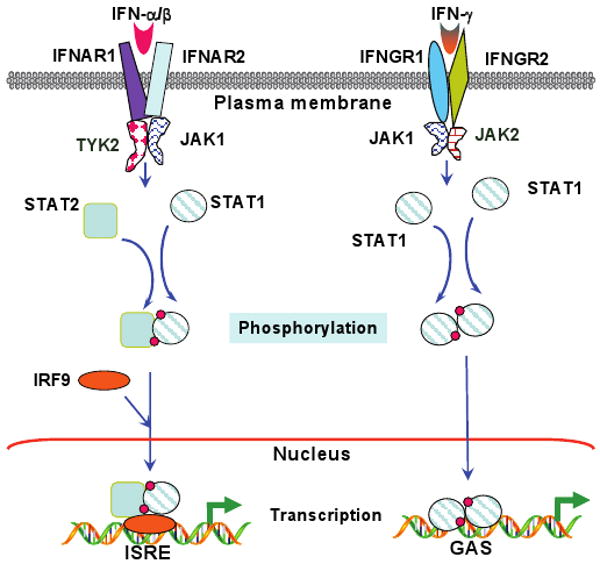
Interferons (IFNs) induce antiviral and antitumor effects; and promote the development of immune responses (1, 2). They regulate a broad range of physiologic processes, including cytokine and chemokine synthesis (3), mRNA translation (4), RNA and protein stability (5, 6), antigen presentation (7), nuclear trafficking (8), cell differentiation (9, 10), and cell division and apoptosis (11, 12). There are three main classes of interferons: type I (predominantly IFN-α, β and κ), type II (IFN-γ) (13) and type III (IFN-λ1 and -λ2 also known as IL-28 and IL-29). They transduce signals through related but distinct pathways. The Janus tyrosine kinase (JAK)-signal transducer and activator of transcription (STAT) pathway (Figure 1) is one of the best characterized IFN-signaling pathways (2). Most IFN-receptors are heteromers consisting of at least two different polypeptides. For example, the type I IFN-receptor is constituted by IFNAR1 and IFNAR2 (14); the type II IFN-receptor by IFNGR1 and IFNGR2 (15); and the type III-IFN receptor by IFNLR1 and IL-10R2 (16, 17). In all cases, receptor peptide 1 functions as the ligand binding protein while the receptor peptide 2 serves as the signaling chain. The STAT1 and STAT2 proteins have been reported to be activated by the IFN-α/β via tyrosyl phosphorylation at critical residues, using tyrosine kinases Tyk2 and JAK1 (2, 18, 19), although in some cells STAT3 is also activated (20, 21). After being activated (i.e. tyrosyl phosphorylated) STAT1 and STAT2 heterodimers associate with the non-STAT DNA-binding protein, IFN gene regulatory factor-9 (IRF-9/p48/ISGF3-γ) (22). The resultant trimeric transcription factor, ISGF3, binds to the IFN-stimulated regulatory elements (ISREs) to induce the expression of many IFN-α/β-regulated genes. Only the STAT1 protein is tyrosyl phosphorylated by JAK1 and JAK2 at the ligand-engaged type II IFN receptor. STAT1 dimers, thus formed, migrate to the nucleus, bind to the γ-IFN-activated sequence (GAS) to drive γ-IFN-induced gene expression (23). The ligand-bound type III IFN receptor, although activated in a manner similar to the type I receptor, activates multiple STATs-STAT1, STAT2, STAT3, STAT4 and STAT5 (24). However, receptor mutants that fail to activate STAT1 and STAT2, can still activate STAT3 and STAT4, indicating a novel aspect of the functioning of this receptor (24). The STAT proteins are rapidly activated by IFN-treatment usually in less than 10 sec, and reach a maximum by 15 min. Thereafter their nuclear activities decline owing to nuclear export (25-27), and/or desphosphorylation by TcPTPase (28), despite the presence of IFNs in the extracellular environment. Additionally, the suppressor of cytokine signaling-1 protein, a STAT-regulated inhibitory protein, turns off the activated JAKs (29). Although the JAK-STAT pathways may explain a lion's share of IFN-actions, there might be other signaling pathways given the observations that some IFNs, particularly IFN-γ, can regulate several genes through non-STAT binding promoter elements and at a time when the activated-STAT levels are barely detected.
Figure 1. IFN-induced JAK-STAT signaling pathways.
Additional IFN signaling pathways
Several other IFN-regulated signaling elements and pathways are required for the generation of diverse responses to IFNs. Some of these signaling pathways are STAT1-dependent and others are STAT1-independent (30, 31). Some of these additional IFN signaling cascades play key roles in optimizing the IFN-induced responses. For example, some studies have shown p38α is phosphorylated and activated in a type-I-IFN-dependent manner in several IFN-sensitive cell lines (32, 33) and a p38-inhibitor blocks IFN-α-dependent transcription of genes containing ISREs (34). However, inhibition of p38 activity did not block tyrosyl phosphorylation of STAT1 or STAT2, or formation of the mature ISGF3 complex and the binding of this complex to ISREs. These observations indicate that type-I-IFN-induced p38 activity acts independently of STAT1-activation (34). p38 is also required for type-I-IFN-driven gene transcription through GAS elements (32). Further studies have identified the upstream and downstream effectors of type-I-IFN-activated p38-signaling pathway (31). The biological function of this pathway accounts for the growth-inhibitory effects and antiviral responses of type I IFNs (31).
Other studies reported that type I IFNs activated a phosphatidylinositol 3-kinase (PI3K)-signaling pathway that occurred independently of STAT1 but was dependent on JAKs (35-37). The PI3K-signaling pathway can mediate either pro-apoptotic or anti-apoptotic signals in response to IFNs. The PI3K-signaling cascade also regulates IFN-inducible activation of the mammalian target of rapamycin (mTOR), via Akt, which mediates the initiation of mRNA translation (31).
A variety of IFN-γ response elements
Promoter analysis of a number of IFN-α/β-regulated genes and some IFN-γ-responsive genes revealed that a conserved ISRE is critical for IFN response (38). IFN-γ-stimulated genes contain a variety of response elements (39). The kinetics and requirements for de novo protein synthesis for the expression of several IFN-γ-responsive genes are quite distinct. Based on the temporal expression, they can be grouped into “early”- and “late”-induced genes. Most known early IFN-γ-stimulated genes possess a unique element known as the GAS (40) or the related palindromic IFN response element (pIRE), which binds a homodimer of STAT1 in response to IFN-γ (41).
As already said, the temporal and functional diversities in the IFN-γ induced responses suggest the activation of several other IFN-induced signaling pathways. Such regulatory signaling networks may be controlled by distinct transcription factors and their combinations. Such diversity in the biological response of IFNs may further be influenced by the post-translational modification of the transcription factors and/or by the interaction of these transcription factors with other transcription factors (42). In this context, IFN-γ-induced gene expression requires diverse regulatory factors such as the IRFs (43-45), the class II transactivator, and the X-box binding factor (46, 47). One recent study reported a novel IFN-γ-activated-c-Jun-dependent pathway for the induction of a subset of ISGs, such as ifi-205 and iNOS (48). IFN-γ activated DNA binding of AP1, constituted by c-Jun, occurred independently of JAK1 and STAT1. This pathway engages the MEK1/2-ERK1/2 module of the MAP kinase cascades. Surprisingly, the JNK1/2 and p38 MAPK pathways were dispensable for activating AP1 in these studies (48).
The IRF-9 protein plays a central role of IRF-9 in IFN-regulated pathways. IFN-γ augments IFN-α/β-induced gene expression via an up-regulation of the IRF-9 gene expression (49, 50). Certain oncogenic viruses down-regulate IRF-9 expression to evade the action of IFNs (51, 52); and its activity is inhibited in some human tumor cell lines (53-55). Induction of IRF-9 occurs in a temporally delayed manner, in contrast to that of other IFN-stimulated genes (ISGs) and some IRFs (50, 56), indicating that its expression is regulated via a novel element(s). Therefore, we studied the regulation of IRF-9 gene by IFN-γ and discovered a novel γ-IFN-activated transcriptional element (GATE) in IRF-9 promoter (57).
GATE was distinct from ISRE or GAS in terms of its sequence organization and bound to proteins, which were distinct from those bound to ISRE or GAS. GATE (24 bp) was also longer than ISRE (15 bp), and it exhibited a poor homology to ISRE or GAS. Similarly, other studies reported additional novel IFN response elements. For example, the MHC class I B gene promoter contains an atypical IFN-response element distinct from ISRE and GAS (58). The IFN-induced expression of a chemokine, RANTES, required an ISRE-like element, but none of the constituent proteins of the ISGF3 complex was required for gene induction through this element (59). An IFN response element from the IFP53/tryptophanyl tRNA synthase gene forms two distinct IFN-α-inducible complexes in an electrophoretic mobility shift assay (EMSA), one of which did not appear to be ISGF3 (60). Although an ISRE of MHC class I genes is crucial for the induction of gene expression, studies have shown that another distinct element, site α, is also required for their induction by IFN-γ (61, 62). In summary, a number of other regulatory elements respond to IFN-treatment. Our subsequent analyses discovered that CAAAT/enhancer binding protein-β (C/EBP-β) as a GATE-binding factor played a prominent role in IFN-action (63). This observation was also verified by another independent study (64).
Biology of C/EBP-β
C/EBPs belong to a superfamily constituted by transcription factors CREB, Fos, Jun/activator protein-1 (AP1), activating transcription factor (ATF), and musculoaponeurotic fibrosarcoma/nuclear factor E2-related factor (Maf/Nrf) (65). They participate in a number of physiologic activities, including energy metabolism, fat storage, tissue differentiation, hematopoiesis, immune responses, antibacterial defense, stress response, and the reproductive system (66-72). The C/EBP subfamily includes structurally similar but genetically and functionally distinct proteins - C/EBP-α, C/EBP-β, C/EBP-γ, C/EBP-δ, C/EBP-ε, and C/EBP-ζ. Most of these proteins possess a c-terminal bZIP domain (73, 74), which is essential for DNA binding; and homo- and hetero-dimeric interactions occur among various members of this family. Dimerization between different C/EBPs precisely modulates transcriptional activity of target genes (75). Their DNA binding specificity is determined by the DNA contact surface, the basic region of approximately 20 amino acids upstream of the leucine zipper (76). A less conserved, bi-partite transcription activation domain (TAD), located at the N-terminus, controls their transcriptional response. A constitutively high expression of C/EBP proteins is observed in human liver, intestine, lung, and adipose tissues. The pleiotropic transcriptional effects of C/EBP result from different mechanisms, such as their tissue and embryonic developmental stage-specific expression, leaky ribosomal reading, posttranscriptional modifications, and variable DNA binding specificities (77).
Unlike the other members of its family, C/EBP-β (also known as - nuclear factor induced by IL-6 (NF-IL-6)/IL-6 induced DNA binding protein (IL-6-DBP)/C-EBP related protein 2 (CRP2)/nuclear factor-myeloid (NF-M)) exhibits remarkable plasticity with respect to the range of transcriptional activities it takes part in. C/EBP-β has two central regulatory domains RD1 and RD2, which harbor sites that can be phosphorylated by several protein kinases (Figure 2). C/EBP-β also regulates IL-6 and IL-6-induced expression of the cytokines IL-1, IL-8, tumor necrosis factor-α (TNF-α), and granulocyte colony-stimulating factor (G-CSF), as well acute phase response proteins such as α1-acid glycoprotein, α2-microglobulin, and C-reactive protein (69).
Figure 2. Molecular organization of the C/EBP-β protein.

The ERK-phosphorylation site within the RD2 is shown.
Deletion of the C/EBP-β gene in mice causes death in utero, largely due to defective gluconeogenesis and adipogenesis (66). Rarely, some live pups are born, which develop severe defects in their immune system (70, 78). C/EBP-β-/- mice are highly susceptible to Candida albicans, Listeria monocytogenes, and Salmonella typhimurium infections (70, 78). These pathogens escape from the phagosome to the cytoplasm in C/EBP-β-/- macrophages (70, 78). The Th1 immune deficiencies include low IL-12 levels and a loss of delayed-type hypersensitivity in C/EBP-β-/- mice (78). Elevated serum IL-6 levels in C/EBP-β-/- mice coincide with splenomegaly, peripheral lymphadenopathy, plasmacytosis, and extramedullary hematopoiesis, as seen in Castleman's disease in humans (78). A number of defects in cytokine synthesis were also observed in C/EBP-β-/- macrophages (79). In the B cell lineage, C/EBP-γ is the predominant isoform in early cells, and it decreases with cellular maturation. C/EBP-β is highly expressed in mature B cells and induced further by LPS stimulation and regulates the expression of a number of genes involved in B-cell function (80). The other long-term effects of C/EBP-β deficiency in mice include the development of a lymphoproliferative disorder (78) and female infertility (72). Loss of C/EBP-β in mice also causes defective development and differentiation of hepatocytes (81), myelomonocytes (82), adipocytes (67), and neurons (83). Given the diverse effects of C/EBP-β, it is conceivable that its association with different cellular factors in a gene context and signal-specific manner may regulate its activity. Indeed, C/EBP-β can interact with transcription factors outside its family, such as NF-κB (84), retinoblastoma (pRb) tumor suppressor protein (85), Sp1 (86), STAT3 (87) to regulate cellular functions.
Regulation of C/EBP-β
Two C/EBP-β isoforms are expressed physiologically. They are generated from a single mRNA by a leaky ribosomal scanning mechanism. While the full-length C/EBP-β protein (Figure 2) has a complete modular organization (88), the truncated isoform, LIP, contains only the DNA-binding and leucine zipper domains (89). LIP dominantly inhibits the transcriptional activity of the full-length C/EBP-β by dimerizing with it (89). C/EBP-ζ is inducible only under the conditions of stress. It can form heterodimers with C/EBP-α and C/EBP-β, and attenuate their transcriptional activity (90). LPS, IL-6, IL-1, dexamethasone, and glucagon can strongly up-regulate the expression of C/EBP-β (91). We have shown that IFN-γ not only induces the expression of C/EBP-β but also enhances its transcriptional activity (63). Cytokine treatment further increases the transcriptional activity of C/EBP via enhanced DNA binding (92). Posttranscriptional modifications of C/EBP by protein kinases PKA, PKC, ribosomal S6 kinase (RSK), and extracellular signal-related kinase (ERK) appear to modulate its activity in different cells (93-96).
C/EBP-β binds to a variety of response elements and forms heteromeric complexes with other transcription factors, such as pRb, NF-κB, Sp1, Myb and PU.1 (85, 86, 97, 98). Indeed, synergistic activation by C/EBP-β and NF-κB members has been demonstrated for the genes encoding the acute-phase response proteins, serum amyloid A1, A2, A3, and α1-acid glycoprotein, as well as the cytokines IL-6, IL-8, and IL-12 and G-CSF (99-101). A cooperation between C/EBP-β and NF-κB has also been demonstrated in regulating transcription from the human immunodeficiency virus (HIV) long terminal repeat (102). In some instances, C/EBP-β and NF-κB interactions lead to antagonistic effects (84, 103), indicating that promoter organization and cell type specificities are likely to play a major role.
C/EBP-β-dependent transcription in response to IFN-γ
We have shown that C/EBP-β binds to GATE and stimulates transcription (63, 64). The b-Zip domain of C/EBP-β binds to a submotif within GATE. This motif exhibited homology with six of the eight conserved nucleotides of the consensus C/EBP binding sites, found in a number of C/EBP-regulated genes (63, 64). C/EBP-β not only induces the basal transcription from GATE but also stimulates it further following IFN-γ treatment. In contrast, the other members of the C/EBP family, such as C/EBP-α, -δ and -ζ, fail to stimulate GATE-dependent transcription (63). In fact, C/EBP-δ and -ζ suppressed IFN-γ-induced transcription through GATE probably by forming heteromeric complexes with C/EBP-β to inhibit transcription.
Similarly, IL-6 induces IRF-9 gene expression through GATE using C/EBP-β (104). Pretreatment with IL-6 potnetiated IFN-α induced responses. This is consistent with previous reports that IL-6 induces the expression of 2-5A synthetase and MHC class I genes (105). Neutralization of endogenous IFN-α using specific antibodies blocked the gene stimulatory effects of IL-6. IFN-γ and IL-6 have also been shown to synergistically up-regulate the expression of HLA class I and carcinoembryonic antigen in certain colorectal carcinomas (106). The HLA inductive effects of IL-6 are mediated indirectly because antibodies against type I IFNs neutralize them. Such cooperative effects of IL-6 and IFN on the tumor cells may allow the expression of tumor specific antigens or HLA simultaneously. Such synergy may be one underlying reason for an effective rejection of the tumor in vivo. These studies show that C/EBP-β may function as a bridge between IFN and IL-6 signaling.
Previous studies showed that mitogen-activated protein kinases (MAPKs) are activated by IFNs (107-109). The MAPK pathways (Figure 3) can be grossly grouped into two: one used by growth factors and the other by stress-activating signals (110, 111). In the growth factor-stimulated pathways, the Ras/Raf/MEK1/ERK cascade plays a key role in driving the responses. Ras activation is dependent on the recruitment of the adaptor proteins Grb and Sos to the growth factor receptors (112). Raf, a serine-threonine kinase, responds to Ras and phosphorylates MAP kinase kinase 1 (MEK1). MEK1, a dual specificity kinase, phosphorylates at the conserved threonine and tyrosine residues present in the activation loop of ERKs: ERK1 (p44) and ERK2 (p42). ERKs1/2 are known to phosphorylate members of the AP1 family, such as Elk1, Egr1, Fos, and Jun. The second type of MAPK pathway is initiated by stress-activating stimuli, such as IL-1, TNF-α, UV radiation and others. Rho and Rac, the G proteins similar to Ras, are activated first (113). These G-proteins then activate MAP kinase kinase kinases (MEKKs) 1-4. MEKKs then activate stress responsive kinases, such as the various isoforms of p38 MAPK (α, β, γ, δ) and c-Jun N-terminal kinases (JNK), using the intermediate enzymes SEK1/2. In these pathways, MEKKs and SEKs are equivalent to Raf and MEK1 of the growth factor induced pathways, respectively.
Figure 3. A simplified view of the mitogen-activated protein kinase (MAPK) pathways.
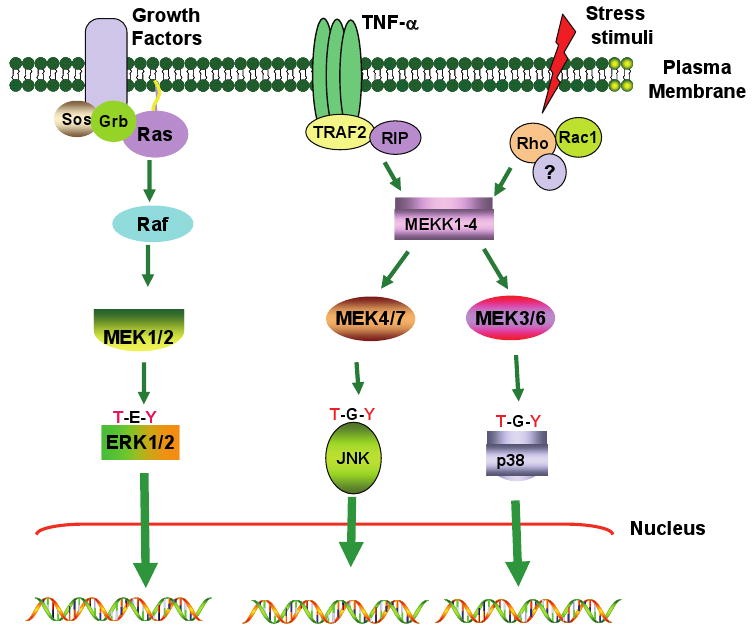
Three of the best studied modules, the ERK, JNK and p38 pathways are indicated. Each of them targets several cellular proteins including some transcription factors.
Although Raf activation by IFN-γ and IFN-β has been reported (107, 109), neither the catalytically inactive dominant negative Ras or Raf mutant, nor the loss of Raf in cells failed to block GATE-dependent transcription (114, 115). However, the inhibitors of ERK1/2 activation, PD98059 and U0126 (116); dominant negative forms of ERK1/2; and a catalytically inactive MEK1 potently ablated GATE-driven expression. C/EBP-β bears a consensus ERK phosphorylation site, GTPS, in its RD2 domain. A C/EBP-β mutant lacking the critical threonine residue (in the GTPS sequence) failed to promote GATE-dependent gene expression. These data indicate that C/EBP-β is regulated by ERK1/2, through MEK1, in response to IFN-γ.
Instead of Raf, MEKK1 was required for IFN-γ-induced ERK activation and the induction IRF-9 gene (115). Interestingly, IFN-γ-induced GAS-driven transcription was also inhibited in MEKK1-/- cells. Further analyses showed that GATE-driven transcription was inhibited by U0126 (an inhibitor of ERK1/2 pathway), whereas GAS-driven transcription was suppressed by SB202190 (an inhibitor of p38 MAPKs). These data indicate that MEKK1 acts as a common upstream effector for GAS- and GATE-dependent transcriptional responses. However, these signals seem to diverge thereafter. These results clearly differ from the existing paradigms wherein Raf1 is considered to be the upstream regulator of MEK1 and, therefore, ERK1/2.
Although the N-terminal half (367 amino acids) of the MEKK1 protein is dispensable for its catalytic activity (117-119), its deletion converts it into a constitutive enzyme. This domain acts as a scaffold for the binding of MEK1 and ERK1/2 (120), which permits the juxtaposition of the kinases and their activation. In fact, MEKK1 phosphorylates MEK1 on the same residue as does Raf (121). Thus, IFN-γ is the first known ligand to employ the MEKK1-MEK1-ERK1/2 signaling pathway to stimulate transcription using C/EBP-β. MEKK1 is also required for the activation of NF-κB (122, 123) and c-Jun N-terminal kinases (124). It also plays a major role in IFN-β gene induction (125), and double-stranded RNA-dependent responses (124). Thus, MEKK1 appears to play a broader role in innate immune responses that involve IFNs.
One consequence of ERK-induced phosphorylation at the GTPS motif of C/EBP-β is a conformational change that permits its interaction with other cellular proteins. One recent study showed that Ras induces phosphorylation of C/EBP-β and activated C/EBP-β interacts with the transcriptional mediator complex (126). GATE also binds another transcriptional activator, GBF1, in response to IFN-γ (127). However, it is an extremely poor DNA-binding protein. Our subsequent studies have shown that GBF1 recruitment to the IRF-9 promoter is dependent on C/EBP-β (128). Specifically, ERK1/2-dependent phosphorylation at T189 residue in the GTPS motif played a critical role. Mutation of this motif and/or interference with the ERK1/2 activation prevented the IFN-γ-induced association between GBF1 and C/EBP-β.
STAT1 is another important factor that controls IFN-γ-induced activation of ERKs. In the absence of STAT1, IFN-γ failed to promote activation of ERKs (114). Restoration of STAT1 into STAT1-/- cells rescued ERK1/2 activation. It is unlikely that STAT1 directly controls ERK activation. Rather, an unknown STAT1-dependent factor may regulate such a process. Although another study showed that Raf1 activation was JAK1-dependent (implying that ERK1/2 activation was also JAK1 dependent) and Ras-independent (108), we found that ERK1/2 were activated normally by IFN-γ in the absence of JAK1 (114). At present, we do not know the significance of Raf activation by IFNs. It is likely that such activation may be important for the stimulation of another transcription factor(s). It may also represent a cell type-specific effect of IFN-γ. With respect to GATE-dependent C/EBP-β-driven transcription, Ras and Raf are not required.
So, the question comes up what are the upstream effectors of this pathway? One of the effectors could possibly be Ras GTPase-activating protein 1 (RAP1), a ras like small GTPase (129). It is known that activation of MEKK1 or MEK1 usually occurs downstream of small GTPases (31). The function of small GTPases is regulated by guanine-nucleotide-exchange factors (GEFs). The GEF for RAP1, a G-protein-linked signaling molecules C3G, is linked by an adaptor protein CrkL (130). Studies showed that CRKL is tyrosine phosphorylated during IFN-treatment (131). It also has been shown that RAP1 regulates the activation of MAPK-signaling cascades (129, 132) in response to IFN. However, the precise mechanism that CrkL-RAP1 mediates IFN responses remains to be determined.
The role of mixed lineage kinases (MLK) in IFN-γ-induced C/EBP-β-dependent transcriptional response
Our recent studies show that mixed-lineage kinases (MLKs), a group of orphan MAPKs, also control another aspect of GATE-dependent gene regulation. MLKs are a subgroup of upstream kinases that regulate the MAPK signaling (133). The MLK family includes MLKs 1-4, the dual leucine zipper-bearing kinase (DLK), and the leucine zipper kinase (LZK). These kinases are sub-grouped based on the spacer motifs that divide two characteristic leucine zippers (Figure 4). Kinases in the MLK and DLK have a 13- and 31-amino acid spacers, respectively. The other difference between these proteins is the location of their catalytic domains.
Figure 4. The mixed lineage kinases.

Panel A shows the structure of MLK3. Panel B shows the groups of MLK family.
MLKs contain several conserved structural motifs that are important for their function. These include a Src homology 3 (SH3) domain, two leucine zipper motifs, and a Cdc42/Rac interactive binding domain (CRIB). Signal-induced binding of Cdc42/Rac to the CRIB causes MLK dimerization, leading to their autophosphorylation (134). In the case of MLK3, autophosphorylation occurs at amino acids 277 and 281 of the activation loop, located in its catalytic domain (135). MLKs function as MKKKs and have been implicated in the activation of JNK and SEK1 and transcription factor NF-κB (136, 137). Some MLKs also activate p38-MAPK (133). One well-recognized function for the MLKs is regulation of apoptotic pathways in neuronal cells (138, 139). MLKs are expressed in a tissue-specific manner, with the exception of MLK3. Their role in regulation of neurodegeneration has been indicated by the observation that K252a, a metabolite found in the spent broths of the bacterium Narcodiopsis sp., can inhibit these kinases and prevent experimentally induced neurodegeneration in animal models (140).
We investigated the role of MLKs in IFN-γ-driven transcriptional responses through GATE/C/EBP-β (141). We observed that co-expression of MLK3, but not MLK2, significantly stimulated the IFN-γ-induced expression of IRF-9. Inhibition of MLKs did not significantly affect STAT1-driven and AP1-driven transcription. The fact that MLK-inhibitor CEP-11004 (an analog of K252a) inhibited neither the IFN-induced activation of ERK1/2 nor the phosphorylation of C/EBP-β at T189 indicated that MLK effects are exerted elsewhere on C/EBP-β. Indeed, constitutively phosphorylated serine residue (S64) in the transactivation domain of C/EBP-β was the target of MLK3-induced signals. IFN-γ treatment decreased S64 phosphorylation. Surprisingly, MLK3 caused a decrease in phosphorylation of C/EBP-β instead of its stimulation in an IFN-γ dependent manner. This observation suggests the requirement for a ligand-induced dephosphorylating activity to regulate GATE-driven transcription. Such dephosphorylation was required for the IFN-γ-induced recruitment of the transcriptional coactivator p300 to the IRF-9 promoter. Thus, IFN-induced GATE-driven transcription is dependent on MLK3 activity, which promotes a decrease in S64 phosphorylation on C/EBP-β.
Dephosphorylation plays a major role in other transcriptional events. For example, dephosphorylation of transcription factor NFAT by calcineurin (142) and homeodomain transcription factor Arix (143) have been shown to promote their transcription-activating function. The nature of the MLK3-driven dephosphorylating activity is unclear at present. Although we suggest a role for phosphatase in this process, our observations can also be explained by a MLK3-driven inactivation of a kinase that constitutively phosphorylates S64. One likely candidate is Cdk2, which phosphorylates C/EBP-β in vitro at S64 and T189 (144). IFN-γ treatment has been shown to cause a decrease in Cdk2 activity (145). One study showed that protein phosphatase PP2A activity was required for the IFN-γ-induced expression of the C1 inhibitor mRNA (146). It is unclear whether this phosphatase has any effect on S64 phosphorylation. Thus, further studies are needed to identify the relevant activity in this response.
One implication from these results is that signal-dependent phosphorylation (for growth promoters, such as Ras) and dephosphorylation (for growth inhibitors, such as IFN-γ) at S64 may act as a regulatory switch for routing C/EBP-β into specific promoter complexes. A post-induction re-phosphorylation at S64 following IFN-γ treatment may then reset this transcriptional switch. Consistent with our interpretation, cell cycle-dependent dual phosphorylation of C/EBP-β at S64 and T189 by Cdc2 or Cdk2 controls its activation by Ras signaling (144). In contrast, MEKK1 and MLK3 control the phosphorylation at these sites in response to IFN-γ. These results may also explain why IFN-γ circumvents Ras to activate ERK1/2 and, therefore, C/EBP-β (115). Otherwise, S64 might remain constitutively phosphorylated and would be unable to activate GATE-dependent transcription.
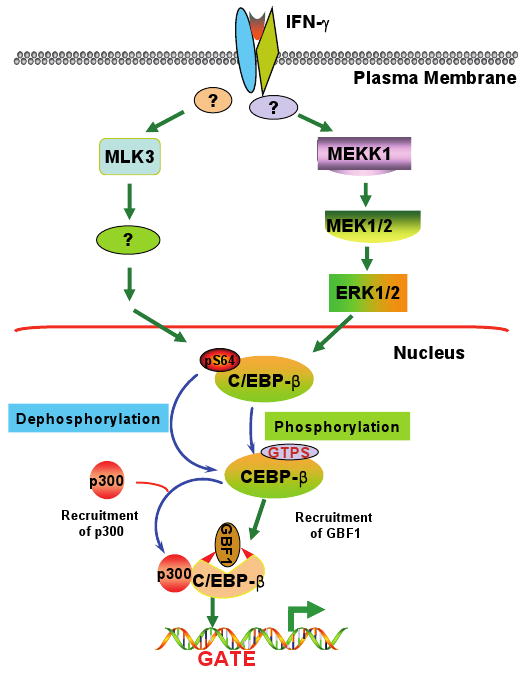
The IFN-γ-induced decrease in phosphorylation of S64 seems to occur independently of the ERK consensus motif present in RD2 of C/EBP-β. This conclusion is supported by three observations: (1) in cells lacking MEKK1, a normal dephosphorylation of S64 occurred in response to IFN-γ; (2) a C/EBP-β mutant lacking the ERK phosphorylation motif was dephosphorylated following IFN-γ treatment; and (3) CEP did not block the IFN-induced phosphorylation at T189 of C/EBP-β. These observations also rule out a potential IFN-γ-induced cascade-like relationship between MEKK1 and MLK3-driven signals. However, it is possible that these events can be coordinately regulated. Based on these results, we suggest that C/EBP-β is controlled by at least two independent IFN-γ-driven signaling pathways: one that promotes the phosphorylation at T189 through an MEKK1-MEK1-ERK1/2 cascade and the other that decreases phosphorylation at the S64 residue of the N-terminus through MLK3 (Figure 5).
Figure 5. The IFN-γ-induced MAPK-signaling pathways regulate C/EBP-β.
Conclusions
IFN-induced transcriptional control involves a multilayered and cross-regulating network. There is accumulating evidence that these signaling pathways may extend beyond the classical JAK-STAT pathways. The identification of GATE and C/EBP-β as its regulatory factor has uncovered hitherto undefined new regulatory processes controlled by IFNs. Such a highly synergistic and coordinate system provides an effective host defense against different pathogens and tumors. IFN-γ, using C/EBP-β, induces IRF-9 (63). IRF-9, in turn, promotes IFN-α/β responses (147). IFN-α/β and IFN-γ induce C/EBP-β (148). Some of this C/EBP-β is used for increasing IRF-9 synthesis in a positive feedback loop. A fraction of the C/EBP-β also participates in IL-6-, IL-1- and TNF-α-induced responses (68, 69). IL-6 also promotes IFN-α/β-inducible responses through C/EBP-β (104). IL-1 and TNF-α induce the synthesis of type I IFN, potentially amplifying this loop. IRF-9 also participates in IFN-β synthesis (149) in response to TNF-α (150). Thus, C/EBP-β connects multiple cytokine signaling pathways.
We have identified two C/EBP-β-dependent signaling pathways in response to IFN-γ. Both of them control the phosphorylation of C/EBP-β. At present, we only have a partial picture of the regulation of C/EBP-β by IFN-γ. It is unclear as to what and how specific regulatory proteins get recruited into the promoter complexes in response to IFN-γ and whether these factors are also subject to regulation by IFNs. IFN-induced control of C/EBP-β by more than one signal may recruit multi-proteins into specific transcriptional complexes. For example, the transcription coactivator, CBP/p300, is known to undergo phosphorylation at multiple sites (151-153) following its engagement with C/EBP-β (154, 155). Some proteins in the transcriptional mediator complex themselves appear to be targets for phosphorylation (126, 156). Whether IFNs or other cytokines control these events need further studies. Another important question is whether other genes are controlled by the IFN-γ-C/EBP-β regulatory pathways. In this connection, we have recently identified that ∼30 genes are dependent on this pathway for their expression (Gade P, et al., manuscript submitted). One such gene is the death-associated protein kinase, a protein required for suppressing metastasis (157, 158). The answers to these questions will provide us further insights about IFNs induced anti-tumor, antiviral and immunomodulatory effects.
Acknowledgments
DVK is supported by National Cancer Institute grant CA78282. We thank Shreeram Nallar for a critical reading of this manuscript.
References
- 1.Kalvakolanu DV, Borden EC. Interferons: cellular and molecular biology of their actions. In: Bertion JR, editor. Encyclopedia Cancer. Vol. 2. 2002. pp. 511–521. [Google Scholar]
- 2.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to Interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 3.Biron CA. Initial and innate responses to viral infections-pattern setting in immunity or disease. Curr Opin Microbiol. 1999;2:374–381. doi: 10.1016/s1369-5274(99)80066-6. [DOI] [PubMed] [Google Scholar]
- 4.Williams BR. Signal integration via PKR. Sci STKE. 2001;2001:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- 5.Silverman R. 2-5A Dependent RNase L: a regulated endoribonuclease in the interferon system. In: D'Alessio GRJ, editor. Rionucleases: Structure and Functions. 1997. pp. 515–551. [Google Scholar]
- 6.Nyman TA, Matikainen S, Sareneva T, Julkunen I, Kalkkinen N. Proteome analysis reveals ubiquitin-conjugating enzymes to be a new family of interferon-α-regulated genes. Eur J Biochem. 2000;267:4011–4019. doi: 10.1046/j.1432-1327.2000.01433.x. [DOI] [PubMed] [Google Scholar]
- 7.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 8.Enninga J, Levy DE, Blobel G, Fontoura BM. Role of nucleoporin induction in releasing an mRNA nuclear export block. Science. 2002;295:1523–1525. doi: 10.1126/science.1067861. [DOI] [PubMed] [Google Scholar]
- 9.Takayanagi H, Kim S, Matsuo K, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 10.Horiuchi M, Hayashida W, Akishita M, et al. Interferon-γ induces AT(2) receptor expression in fibroblasts by Jak/STAT pathway and interferon regulatory factor-1. Circ Res. 2000;86:233–240. doi: 10.1161/01.res.86.2.233. [DOI] [PubMed] [Google Scholar]
- 11.Kalvakolanu DV. The GRIMs: a new interface between cell death regulation and interferon/retinoid induced growth suppression. Cytokine Growth Factor Rev. 2004;15:169–194. doi: 10.1016/j.cytogfr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Levy-Strumpf N, Kimchi A. Death-associated proteins (DAPs): from gene identification to the analysis of their apoptotic and turmor suppressive functions. Oncogene. 1998;17:3331–3340. doi: 10.1038/sj.onc.1202588. [DOI] [PubMed] [Google Scholar]
- 13.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 14.de Weerd NA, Samarajiwa SA, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282:20053–20057. doi: 10.1074/jbc.R700006200. [DOI] [PubMed] [Google Scholar]
- 15.Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem. 2007;282:20047–20051. doi: 10.1074/jbc.R700004200. [DOI] [PubMed] [Google Scholar]
- 16.Kotenko SV, Gallagher G, Baurin VV, et al. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 17.Uze G, Monneron D. IL-28 and IL-29: newcomers to the interferon family. Biochimie. 2007;89:729–734. doi: 10.1016/j.biochi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Leung S, Qureshi SA, Kerr IM, Darnell JE, Jr, Stark GR. Role of STAT2 in the α interferon signaling pathway. Mol Cell Biol. 1995;15:1312–1317. doi: 10.1128/mcb.15.3.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darnell JE., Jr Studies of IFN-induced transcriptional activation uncover the Jak-Stat pathway. J Interferon Cytokine Res. 1998;18:549–554. doi: 10.1089/jir.1998.18.549. [DOI] [PubMed] [Google Scholar]
- 20.Pfeffer LM, Mullersman JE, Pfeffer SR, Murti A, Shi W, Yang CH. STAT3 as an adapter to couple phosphatidylinositol 3-kinase to the IFNAR1 chain of the type I interferon receptor. Science. 1997;276:1418–1420. doi: 10.1126/science.276.5317.1418. [DOI] [PubMed] [Google Scholar]
- 21.Yang CH, Murti A, Pfeffer LM. STAT3 complements defects in an interferon-resistant cell line: evidence for an essential role for STAT3 in interferon signaling and biological activities. Proc Natl Acad Sci U S A. 1998;95:5568–5572. doi: 10.1073/pnas.95.10.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veals SA, Santa Maria T, Levy DE. Two domains of ISGF3 γ that mediate protein-DNA and protein-protein interactions during transcription factor assembly contribute to DNA-binding specificity. Mol Cell Biol. 1993;13:196–206. doi: 10.1128/mcb.13.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shuai K, Stark GR, Kerr IM, Darnell JE., Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-γ. Science. 1993;261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- 24.Dumoutier L, Tounsi A, Michiels T, Sommereyns C, Kotenko SV, Renauld JC. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-λ1: similarities with type I interferon signaling. J Biol Chem. 2004;279:32269–32274. doi: 10.1074/jbc.M404789200. [DOI] [PubMed] [Google Scholar]
- 25.McBride KM, McDonald C, Reich NC. Nuclear export signal located within theDNA-binding domain of the STAT1 transcription factor. EMBO J. 2000;19:6196–6206. doi: 10.1093/emboj/19.22.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McBride KM, Reich NC. The ins and outs of STAT1 nuclear transport. Sci STKE. 2003;2003:RE13. doi: 10.1126/stke.2003.195.re13. [DOI] [PubMed] [Google Scholar]
- 27.Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J Cell Biol. 2004;165:823–833. doi: 10.1083/jcb.200403057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexander WS, Starr R, Fenner JE, et al. SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 30.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-γ-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 31.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 32.Uddin S, Lekmine F, Sharma N, et al. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon α-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem. 2000;275:27634–27640. doi: 10.1074/jbc.M003170200. [DOI] [PubMed] [Google Scholar]
- 33.Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999;18:5601–5608. doi: 10.1093/emboj/18.20.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uddin S, Majchrzak B, Woodson J, et al. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274:30127–30131. doi: 10.1074/jbc.274.42.30127. [DOI] [PubMed] [Google Scholar]
- 35.Uddin S, Yenush L, Sun XJ, Sweet ME, White MF, Platanias LC. Interferon-α engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3′-kinase. J Biol Chem. 1995;270:15938–15941. doi: 10.1074/jbc.270.27.15938. [DOI] [PubMed] [Google Scholar]
- 36.Uddin S, Fish EN, Sher D, et al. The IRS-pathway operates distinctively from the Stat-pathway in hematopoietic cells and transduces common and distinct signals during engagement of the insulin or interferon-α receptors. Blood. 1997;90:2574–2582. [PubMed] [Google Scholar]
- 37.Uddin S, Majchrzak B, Wang PC, et al. Interferon-dependent activation of the serine kinase PI 3′-kinase requires engagement of the IRS pathway but not the Stat pathway. Biochem Biophys Res Commun. 2000;270:158–162. doi: 10.1006/bbrc.2000.2402. [DOI] [PubMed] [Google Scholar]
- 38.Friedman RL, Stark GR. α-Interferon-induced transcription of HLA and metallothionein genes containing homologous upstream sequences. Nature. 1985;314:637–639. doi: 10.1038/314637a0. [DOI] [PubMed] [Google Scholar]
- 39.Sen GC, Ransohoff RM. Transcriptional regulation in the interferon system. Austin, TX: Chapman & Hall and Landes Bioscience; 1997. [Google Scholar]
- 40.Decker T, Lew DJ, Mirkovitch J, Darnell JE., Jr Cytoplasmic activation of GAF, an IFN-γ-regulated DNA-binding factor. EMBO J. 1991;10:927–932. doi: 10.1002/j.1460-2075.1991.tb08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanno Y, Kozak CA, Schindler C, et al. The genomic structure of the murine ICSBP gene reveals the presence of the γ interferon-responsive element, to which an ISGF3 α subunit (or similar) molecule binds. Mol Cell Biol. 1993;13:3951–3963. doi: 10.1128/mcb.13.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tjian R, Maniatis T. Transcriptional activation: a complex puzzle with few easy pieces. Cell. 1994;77:5–8. doi: 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 43.Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. 2000;13:155–165. doi: 10.1016/s1074-7613(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 44.Kamijo R, Harada H, Matsuyama T, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 45.Briken V, Ruffner H, Schultz U, et al. Interferon regulatory factor 1 is required for mouse Gbp gene activation by γ interferon. Mol Cell Biol. 1995;15:975–982. doi: 10.1128/mcb.15.2.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109(Suppl):S21–33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 47.Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol. 2001;19:331–373. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- 48.Gough DJ, Sabapathy K, Ko EY, et al. A novel c-Jun-dependent signal transduction pathway necessary for the transcriptional activation of interferon γ response genes. J Biol Chem. 2007;282:938–946. doi: 10.1074/jbc.M607674200. [DOI] [PubMed] [Google Scholar]
- 49.Lewis JA, Huq A, Shan B. β and γ interferons act synergistically to produce an antiviral state in cells resistant to both interferons individually. J Virol. 1989;63:4569–4578. doi: 10.1128/jvi.63.11.4569-4578.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bandyopadhyay SK, Kalvakolanu DV, Sen GC. Gene induction by interferons: functional complementation between transacting factors induced by α interferon and γ interferon. Mol Cell Biol. 1990;10:5055–5063. doi: 10.1128/mcb.10.10.5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kalvakolanu DV. Virus interception of cytokine-regulated pathways. Trends Microbiol. 1999;7:166–171. doi: 10.1016/s0966-842x(99)01476-6. [DOI] [PubMed] [Google Scholar]
- 52.Barnard P, McMillan NA. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-α. Virology. 1999;259:305–313. doi: 10.1006/viro.1999.9771. [DOI] [PubMed] [Google Scholar]
- 53.Wong LH, Krauer KG, Hatzinisiriou I, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3γ. J Biol Chem. 1997;272:28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 54.Clifford JL, Walch E, Yang X, et al. Suppression of type I interferon signaling proteins is an early event in squamous skin carcinogenesis. Clin Cancer Res. 2002;8:2067–2072. [PubMed] [Google Scholar]
- 55.Wu WZ, Sun HC, Gao YQ, et al. Reduction in p48-ISGFγ levels confers resistance to interferon-α2a in MHCC97 cells. Oncology. 2004;67:428–440. doi: 10.1159/000082928. [DOI] [PubMed] [Google Scholar]
- 56.Levy DE, Lew DJ, Decker T, Kessler DS, Darnell JEJ. Synergistic interaction between interferon- α and interferon-γ through induced synthesis of one subunit of the transcription factor ISGF3. EMBO J. 1990;9:1105–1111. doi: 10.1002/j.1460-2075.1990.tb08216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weihua X, Kolla V, Kalvakolanu DV. Interferon γ-induced transcription of the murine ISGF3γ (p48) gene is mediated by novel factors. Proc Natl Acad Sci U S A. 1997;94:103–108. doi: 10.1073/pnas.94.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vallejo AN, Pease LR. The locus-specific enhancer activity of the class I major histocompatibility complex interferon-responsive element is associated with a γ-interferon (IFN)-inducible factor distinct from STAT1α, p48, and IFN regulatory factor-1. J Biol Chem. 1996;271:29813–29821. doi: 10.1074/jbc.271.47.29813. [DOI] [PubMed] [Google Scholar]
- 59.Cremer I, Ghysdael J, Vieillard V. A non-classical ISRE/ISGF3 pathway mediates induction of RANTES gene transcription by type I IFNs. FEBS Lett. 2002;511:41–45. doi: 10.1016/s0014-5793(01)03276-8. [DOI] [PubMed] [Google Scholar]
- 60.Lehmann J, Seegert D, Strehlow I, Schindler C, Lohmann-Matthes ML, Decker T. IL-10-induced factors belonging to the p91 family of proteins bind to IFN-γ-responsive promoter elements. J Immunol. 1994;153:165–172. [PubMed] [Google Scholar]
- 61.Gobin SJ, Peijnenburg A, Keijsers V, van den Elsen PJ. Site α is crucial for two routes of IFN γ-induced MHC class I transactivation: the ISRE-mediated route and a novel pathway involving CIITA. Immunity. 1997;6:601–611. doi: 10.1016/s1074-7613(00)80348-9. [DOI] [PubMed] [Google Scholar]
- 62.Martin BK, Chin KC, Olsen JC, et al. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity. 1997;6:591–600. doi: 10.1016/s1074-7613(00)80347-7. [DOI] [PubMed] [Google Scholar]
- 63.Roy SK, Wachira SJ, Weihua X, Hu J, Kalvakolanu DV. CCAAT/enhancer-binding protein-β regulates interferon-induced transcription through a novel element. J Biol Chem. 2000;275:12626–12632. doi: 10.1074/jbc.275.17.12626. [DOI] [PubMed] [Google Scholar]
- 64.Xiao W, Wang L, Yang X, et al. CCAAT/enhancer-binding protein β mediates interferon-γ-induced p48 (ISGF3-γ) gene transcription in human monocytic cells. J Biol Chem. 2001;276:23275–23281. doi: 10.1074/jbc.M010047200. [DOI] [PubMed] [Google Scholar]
- 65.Kalvakolanu DV. Alternate interferon signaling pathways. Pharmacol Ther. 2003;100:1–29. doi: 10.1016/s0163-7258(03)00070-6. [DOI] [PubMed] [Google Scholar]
- 66.Croniger C, Trus M, Lysek-Stupp K, et al. Role of the isoforms of CCAAT/enhancer-binding protein in the initiation of phosphoenolpyruvate carboxykinase (GTP) gene transcription at birth. J Biol Chem. 1997;272:26306–26312. doi: 10.1074/jbc.272.42.26306. [DOI] [PubMed] [Google Scholar]
- 67.Darlington GJ, Ross SE, MacDougald OA. The role of C/EBP genes in adipocyte differentiation. J Biol Chem. 1998;273:30057–30060. doi: 10.1074/jbc.273.46.30057. [DOI] [PubMed] [Google Scholar]
- 68.Poli V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem. 1998;273:29279–29282. doi: 10.1074/jbc.273.45.29279. [DOI] [PubMed] [Google Scholar]
- 69.Akira S, Kishimoto T. NF-IL6 and NF-κB in cytokine gene regulation. Adv Immunol. 1997;65:1–46. [PubMed] [Google Scholar]
- 70.Tanaka T, Akira S, Yoshida K, et al. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell. 1995;80:353–361. doi: 10.1016/0092-8674(95)90418-2. [DOI] [PubMed] [Google Scholar]
- 71.Wang XZ, Lawson B, Brewer JW, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sterneck E, Tessarollo L, Johnson PF. An essential role for C/EBPβ in female reproduction. Genes Dev. 1997;11:2153–2162. doi: 10.1101/gad.11.17.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 74.Williams SC, Baer M, Dillner AJ, Johnson PF. CRP2 (C/EBP β) contains a bipartite regulatory domain that controls transcriptional activation, DNA binding and cell specificity. EMBO J. 1995;14:3170–3183. doi: 10.1002/j.1460-2075.1995.tb07319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams SC, Cantwell CA, Johnson PF. A family of C/EBP-related proteins capable of forming covalently linked leucine zipper dimers in vitro. Genes Dev. 1991;5:1553–1567. doi: 10.1101/gad.5.9.1553. [DOI] [PubMed] [Google Scholar]
- 76.Agre P, Johnson PF, McKnight SL. Cognate DNA binding specificity retained after leucine zipper exchange between GCN4 and C/EBP. Science. 1989;246:922–926. doi: 10.1126/science.2530632. [DOI] [PubMed] [Google Scholar]
- 77.Lekstrom-Himes J, Xanthopoulos KG. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J Biol Chem. 1998;273:28545–28548. doi: 10.1074/jbc.273.44.28545. [DOI] [PubMed] [Google Scholar]
- 78.Screpanti I, Romani L, Musiani P, et al. Lymphoproliferative disorder and imbalanced T-helper response in C/EBP β-deficient mice. EMBO J. 1995;14:1932–1941. doi: 10.1002/j.1460-2075.1995.tb07185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gorgoni B, Maritano D, Marthyn P, Righi M, Poli V. C/EBP β gene inactivation causes both impaired and enhanced gene expression and inverse regulation of IL-12 p40 and p35 mRNAs in macrophages. J Immunol. 2002;168:4055–4062. doi: 10.4049/jimmunol.168.8.4055. [DOI] [PubMed] [Google Scholar]
- 80.Cooper CL, Berrier AL, Roman C, Calame KL. Limited expression of C/EBP family proteins during B lymphocyte development. Negative regulator Ig/EBP predominates early and activator NF-IL-6 is induced later. J Immunol. 1994;153:5049–5058. [PubMed] [Google Scholar]
- 81.Diehl AM. Roles of CCAAT/enhancer-binding proteins in regulation of liver regenerative growth. J Biol Chem. 1998;273:30843–30846. doi: 10.1074/jbc.273.47.30843. [DOI] [PubMed] [Google Scholar]
- 82.Natsuka S, Akira S, Nishio Y, et al. Macrophage differentiation-specific expression of NF-IL6, a transcription factor for interleukin-6. Blood. 1992;79:460–466. [PubMed] [Google Scholar]
- 83.Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPβ. Nat Neurosci. 2001;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- 84.Stein B, Cogswell PC, Baldwin AS., Jr Functional and physical associations between NF-κB and C/EBP family members: a Rel domain-bZIP interaction. Mol Cell Biol. 1993;13:3964–3974. doi: 10.1128/mcb.13.7.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen PL, Riley DJ, Chen-Kiang S, Lee WH. Retinoblastoma protein directly interacts with and activates the transcription factor NF-IL6. Proc Natl Acad Sci U S A. 1996;93:465–469. doi: 10.1073/pnas.93.1.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee YH, Williams SC, Baer M, Sterneck E, Gonzalez FJ, Johnson PF. The ability of C/EBP β but not C/EBP α to synergize with an Sp1 protein is specified by the leucine zipper and activation domain. Mol Cell Biol. 1997;17:2038–2047. doi: 10.1128/mcb.17.4.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alonzi T, Maritano D, Gorgoni B, Rizzuto G, Libert C, Poli V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol Cell Biol. 2001;21:1621–1632. doi: 10.1128/MCB.21.5.1621-1632.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Descombes P, Chojkier M, Lichtsteiner S, Falvey E, Schibler U. LAP, a novel member of the C/EBP gene family, encodes a liver-enriched transcriptional activator protein. Genes Dev. 1990;4:1541–1551. doi: 10.1101/gad.4.9.1541. [DOI] [PubMed] [Google Scholar]
- 89.Descombes P, Schibler U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 90.Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- 91.Akira S, Isshiki H, Sugita T, et al. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990;9:1897–1906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Poli V, Mancini FP, Cortese R. IL-6DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related to C/EBP. Cell. 1990;63:643–653. doi: 10.1016/0092-8674(90)90459-r. [DOI] [PubMed] [Google Scholar]
- 93.Trautwein C, Caelles C, van der Geer P, Hunter T, Karin M, Chojkier M. Transactivation by NF-IL6/LAP is enhanced by phosphorylation of its activation domain. Nature. 1993;364:544–547. doi: 10.1038/364544a0. [DOI] [PubMed] [Google Scholar]
- 94.Nakajima T, Kinoshita S, Sasagawa T, et al. Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc Natl Acad Sci U S A. 1993;90:2207–2211. doi: 10.1073/pnas.90.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Buck M, Poli V, van der Geer P, Chojkier M, Hunter T. Phosphorylation of rat serine 105 or mouse threonine 217 in C/EBP β is required for hepatocyte proliferation induced by TGF α. Mol Cell. 1999;4:1087–1092. doi: 10.1016/s1097-2765(00)80237-3. [DOI] [PubMed] [Google Scholar]
- 96.Buck M, Poli V, Hunter T, Chojkier M. C/EBP-β phosphorylation by RSK creates a functional XEXD caspase inhibitory box critical for cell survival. Mol Cell. 2001;8:807–816. doi: 10.1016/s1097-2765(01)00374-4. [DOI] [PubMed] [Google Scholar]
- 97.Shimizu H, Yamamoto K. NF-κB and C/EBP transcription factor families synergistically function in mouse serum amyloid A gene expression induced by inflammatory cytokines. Gene. 1994;149:305–310. doi: 10.1016/0378-1119(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 98.Oelgeschlager M, Nuchprayoon I, Luscher B, Friedman AD. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol Cell Biol. 1996;16:4717–4725. doi: 10.1128/mcb.16.9.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ray A, Hannink M, Ray BK. Concerted participation of NF-κB and C/EBP heteromer in lipopolysaccharide induction of serum amyloid A gene expression in liver. J Biol Chem. 1995;270:7365–7374. doi: 10.1074/jbc.270.13.7365. [DOI] [PubMed] [Google Scholar]
- 100.Plevy SE, Gemberling JH, Hsu S, Dorner AJ, Smale ST. Multiple control elements mediate activation of the murine and human interleukin 12 p40 promoters: evidence of functional synergy between C/EBP and Rel proteins. Mol Cell Biol. 1997;17:4572–4588. doi: 10.1128/mcb.17.8.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee YM, Miau LH, Chang CJ, Lee SC. Transcriptional induction of the α-1 acid glycoprotein (AGP) gene by synergistic interaction of two alternative activator forms of AGP/enhancer-binding protein (C/EBP β) and NF-κB or Nopp140. Mol Cell Biol. 1996;16:4257–4263. doi: 10.1128/mcb.16.8.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ruocco MR, Chen X, Ambrosino C, et al. Regulation of HIV-1 long terminal repeats by interaction of C/EBP(NF-IL6) and NF-κB/Rel transcription factors. J Biol Chem. 1996;271:22479–22486. doi: 10.1074/jbc.271.37.22479. [DOI] [PubMed] [Google Scholar]
- 103.Brasier AR, Ron D, Tate JE, Habener JF. A family of constitutive C/EBP-like DNA binding proteins attenuate the IL-1α induced, NFκB mediated trans-activation of the angiotensinogen gene acute-phase response element. EMBO J. 1990;9:3933–3944. doi: 10.1002/j.1460-2075.1990.tb07614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Weihua X, Hu J, Roy SK, Mannino SB, Kalvakolanu DV. Interleukin-6 modulates interferon-regulated gene expression by inducing the ISGF3γ gene using CCAAT/enhancer binding protein-β (C/EBP-β) Biochim Biophys Acta. 2000;1492:163–171. doi: 10.1016/s0167-4781(00)00111-1. [DOI] [PubMed] [Google Scholar]
- 105.Cohen B, Gothelf Y, Vaiman D, Chen L, Revel M, Chebath J. Interleukin-6 induces the (2′-5′) oligoadenylate synthetase gene in M1 cells through an effect on the interferon-responsive enhancer. Cytokine. 1991;3:83–91. doi: 10.1016/1043-4666(91)90027-b. [DOI] [PubMed] [Google Scholar]
- 106.Dansky-Ullmann C, Salgaller M, Adams S, Schlom J, Greiner JW. Synergistic effects of IL-6 and IFN-γ on carcinoembryonic antigen (CEA) and HLA expression by human colorectal carcinoma cells: role for endogenous IFN-β. Cytokine. 1995;7:118–129. doi: 10.1006/cyto.1995.1016. [DOI] [PubMed] [Google Scholar]
- 107.Stancato LF, Sakatsume M, David M, et al. B interferon and oncostatin M activate Raf-1 and mitogen-activated protein kinase through a JAK1-dependent pathway. Mol Cell Biol. 1997;17:3833–3840. doi: 10.1128/mcb.17.7.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sakatsume M, Stancato LF, David M, et al. Interferon γ activation of Raf-1 is Jak1-dependent and p21ras-independent. J Biol Chem. 1998;273:3021–3026. doi: 10.1074/jbc.273.5.3021. [DOI] [PubMed] [Google Scholar]
- 109.Stancato LF, Yu CR, Petricoin EF, 3rd, Larner AC. Activation of Raf-1 by interferon γ and oncostatin M requires expression of the Stat1 transcription factor. J Biol Chem. 1998;273:18701–18704. doi: 10.1074/jbc.273.30.18701. [DOI] [PubMed] [Google Scholar]
- 110.Lewis TL, Shapiro PS, Ahn NG. Cell Signaling transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 111.Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 112.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 113.Fanger GR, Johnson NL, Johnson GL. MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J. 1997;16:4961–4972. doi: 10.1093/emboj/16.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hu J, Roy SK, Shapiro PS, et al. ERK1 and ERK2 activate CCAAAT/enhancer-binding protein-β-dependent gene transcription in response to interferon-γ. J Biol Chem. 2001;276:287–297. doi: 10.1074/jbc.M004885200. [DOI] [PubMed] [Google Scholar]
- 115.Roy SK, Hu J, Meng Q, et al. MEKK1 plays a critical role in activating the transcription factor C/EBP-β-dependent gene expression in response to IFN-γ. Proc Natl Acad Sci U S A. 2002;99:7945–7950. doi: 10.1073/pnas.122075799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 117.Yan M, Dai T, Deak JC, et al. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- 118.Deak JC, Templeton DJ. Regulation of the activity of MEK kinase 1 (MEKK1) by autophosphorylation within the kinase activation domain. Biochem J. 1997;322(Pt 1):185–192. doi: 10.1042/bj3220185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yujiri T, Sather S, Fanger GR, Johnson GL. Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science. 1998;282:1911–1914. doi: 10.1126/science.282.5395.1911. [DOI] [PubMed] [Google Scholar]
- 120.Karandikar M, Xu S, Cobb MH. MEKK1 binds raf-1 and the ERK2 cascade components. J Biol Chem. 2000;275:40120–40127. doi: 10.1074/jbc.M005926200. [DOI] [PubMed] [Google Scholar]
- 121.Xu S, Robbins D, Frost J, Dang A, Lange-Carter C, Cobb MH. MEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen-activated protein kinase. Proc Natl Acad Sci U S A. 1995;92:6808–6812. doi: 10.1073/pnas.92.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee FS, Peters RT, Dang LC, Maniatis T. MEKK1 activates both IκB kinase α and IκB kinase β. Proc Natl Acad Sci U S A. 1998;95:9319–9324. doi: 10.1073/pnas.95.16.9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xia Y, Makris C, Su B, et al. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc Natl Acad Sci U S A. 2000;97:5243–5248. doi: 10.1073/pnas.97.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim T, Kim TY, Lee WG, Yim J, Kim TK. Signaling pathways to the assembly of an interferon-β enhanceosome. Chemical genetic studies with a small molecule. J Biol Chem. 2000;275:16910–16917. doi: 10.1074/jbc.M000524200. [DOI] [PubMed] [Google Scholar]
- 126.Mo X, Kowenz-Leutz E, Xu H, Leutz A. Ras induces mediator complex exchange on C/EBP β. Mol Cell. 2004;13:241–250. doi: 10.1016/s1097-2765(03)00521-5. [DOI] [PubMed] [Google Scholar]
- 127.Hu J, Meng Q, Roy SK, et al. A novel transactivating factor that regulates interferon-γ-dependent gene expression. J Biol Chem. 2002;277:30253–30263. doi: 10.1074/jbc.M202679200. [DOI] [PubMed] [Google Scholar]
- 128.Meng Q, Raha A, Roy S, Hu J, Kalvakolanu DV. IFN-γ-stimulated transcriptional activation by IFN-γ-activated transcriptional element-binding factor 1 occurs via an inducible interaction with CAAAT/enhancer-binding protein-β. J Immunol. 2005;174:6203–6211. doi: 10.4049/jimmunol.174.10.6203. [DOI] [PubMed] [Google Scholar]
- 129.Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- 130.Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–6371. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- 131.Alsayed Y, Uddin S, Ahmad S, et al. IFN-γ activates the C3G/Rap1 signaling pathway. J Immunol. 2000;164:1800–1806. doi: 10.4049/jimmunol.164.4.1800. [DOI] [PubMed] [Google Scholar]
- 132.Huang CC, You JL, Wu MY, Hsu KS. Rap1-induced p38 mitogen-activated protein kinase activation facilitates AMPA receptor trafficking via the GDI.Rab5 complex. Potential role in (S)-3,5-dihydroxyphenylglycene-induced long term depression. J Biol Chem. 2004;279:12286–12292. doi: 10.1074/jbc.M312868200. [DOI] [PubMed] [Google Scholar]
- 133.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 134.Bock BC, Vacratsis PO, Qamirani E, Gallo KA. Cdc42-induced activation of the mixed-lineage kinase SPRK in vivo. Requirement of the Cdc42/Rac interactive binding motif and changes in phosphorylation. J Biol Chem. 2000;275:14231–14241. doi: 10.1074/jbc.275.19.14231. [DOI] [PubMed] [Google Scholar]
- 135.Leung IW, Lassam N. The kinase activation loop is the key to mixed lineage kinase-3 activation via both autophosphorylation and hematopoietic progenitor kinase 1 phosphorylation. J Biol Chem. 2001;276:1961–1967. doi: 10.1074/jbc.M004092200. [DOI] [PubMed] [Google Scholar]
- 136.Tibbles LA, Ing YL, Kiefer F, et al. MLK-3 activates the SAPK/JNK and p38/RK pathways via SEK1 and MKK3/6. EMBO J. 1996;15:7026–7035. [PMC free article] [PubMed] [Google Scholar]
- 137.Nihalani D, Meyer D, Pajni S, Holzman LB. Mixed lineage kinase-dependent JNK activation is governed by interactions of scaffold protein JIP with MAPK module components. EMBO J. 2001;20:3447–3458. doi: 10.1093/emboj/20.13.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mota M, Reeder M, Chernoff J, Bazenet CE. Evidence for a role of mixed lineage kinases in neuronal apoptosis. J Neurosci. 2001;21:4949–4957. doi: 10.1523/JNEUROSCI.21-14-04949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Roux PP, Dorval G, Boudreau M, et al. K252a and CEP1347 are neuroprotective compounds that inhibit mixed-lineage kinase-3 and induce activation of Akt and ERK. J Biol Chem. 2002;277:49473–49480. doi: 10.1074/jbc.M203428200. [DOI] [PubMed] [Google Scholar]
- 141.Roy SK, Shuman JD, Platanias LC, et al. A role for mixed lineage kinases in regulating transcription factor CCAAT/enhancer-binding protein-β-dependent gene expression in response to interferon-γ. J Biol Chem. 2005;280:24462–24471. doi: 10.1074/jbc.M413661200. [DOI] [PubMed] [Google Scholar]
- 142.Okamura H, Aramburu J, Garcia-Rodriguez C, et al. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol Cell. 2000;6:539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 143.Adachi M, Lewis EJ. The paired-like homeodomain protein, Arix, mediates protein kinase A-stimulated dopamine β-hydroxylase gene transcription through its phosphorylation status. J Biol Chem. 2002;277:22915–22924. doi: 10.1074/jbc.M201695200. [DOI] [PubMed] [Google Scholar]
- 144.Shuman JD, Sebastian T, Kaldis P, et al. Cell cycle-dependent phosphorylation of C/EBPβ mediates oncogenic cooperativity between C/EBPβ and H-RasV12. Mol Cell Biol. 2004;24:7380–7391. doi: 10.1128/MCB.24.17.7380-7391.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bromberg JF, Horvath CM, Wen Z, Schreiber RD, Darnell JE., Jr Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon α and interferon γ. Proc Natl Acad Sci U S A. 1996;93:7673–7678. doi: 10.1073/pnas.93.15.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Heda GD, Kehoe KJ, Mahdi F, Schmaier AH. Phosphatase 2A participates in interferon-γ's induced upregulation of C1 inhibitor mRNA expression. Blood. 1996;87:2831–2838. [PubMed] [Google Scholar]
- 147.Bluyssen AR, Durbin JE, Levy DE. ISGF3γ p48, a specificity switch for interferon activated transcription factors. Cytokine Growth Factor Rev. 1996;7:11–17. doi: 10.1016/1359-6101(96)00005-6. [DOI] [PubMed] [Google Scholar]
- 148.Der SD, Zhou A, Williams BRG, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kawakami T, Matsumoto M, Sato M, Harada H, Taniguchi T, Kitagawa M. Possible involvement of the transcription factor ISGF3 γ in virus-induced expression of the IFN-β gene. FEBS Letters. 1995;358:225–229. doi: 10.1016/0014-5793(94)01426-2. [DOI] [PubMed] [Google Scholar]
- 150.Bachmann A, Hanke B, Zawatzky R, et al. Disturbance of tumor necrosis factor α-mediated β interferon signaling in cervical carcinoma cells. J Virol. 2002;76:280–291. doi: 10.1128/JVI.76.1.280-291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ait-Si-Ali S, Ramirez S, Barre FX, et al. Histone acetyl-transferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature. 1998;396:184–186. doi: 10.1038/24190. [DOI] [PubMed] [Google Scholar]
- 152.Impey S, Fong AL, Wang Y, et al. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 153.Ait-Si-Ali S, Carlisi D, Ramirez S, et al. Phosphorylation by p44 MAP Kinase/ERK1 stimulates CBP histone acetyl transferase activity in vitro. Biochem Biophys Res Commun. 1999;262:157–162. doi: 10.1006/bbrc.1999.1132. [DOI] [PubMed] [Google Scholar]
- 154.Kovacs KA, Steinmann M, Magistretti PJ, Halfon O, Cardinaux JR. CCAAT/enhancer-binding protein family members recruit the coactivator CREB-binding protein and trigger its phosphorylation. J Biol Chem. 2003;278:36959–36965. doi: 10.1074/jbc.M303147200. [DOI] [PubMed] [Google Scholar]
- 155.Schwartz C, Beck K, Mink S, et al. Recruitment of p300 by C/EBPβ triggers phosphorylation of p300 and modulates coactivator activity. EMBO J. 2003;22:882–892. doi: 10.1093/emboj/cdg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pandey PK, Udayakumar TS, Lin X, Sharma D, Shapiro PS, Fondell JD. Activation of TRAP/mediator subunit TRAP220/Med1 is regulated by mitogen-activated protein kinase-dependent phosphorylation. Mol Cell Biol. 2005;25:10695–10710. doi: 10.1128/MCB.25.24.10695-10710.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the γ interferon-induced cell death. Genes Dev. 1995;9:15–30. doi: 10.1101/gad.9.1.15. [DOI] [PubMed] [Google Scholar]
- 158.Inbal B, Cohen O, Polak-Charcon S, et al. DAP kinase links the control of apoptosis to metastasis. Nature. 1997;390:180–184. doi: 10.1038/36599. [DOI] [PubMed] [Google Scholar]