

Abstract
Cancer propagating cells (CPCs) within primary central nervous system (CNS) tumors (glioblastoma multiforme (GBM), medulloblastoma (MB) and ependymoma) might be integral to tumor development and perpetuation. These cells, also known as brain cancer propagating cells (BCPCs), have the ability to self-renew and proliferate. BCPCs can initiate new tumors in mice with high efficiency and these exhibit many features that are characteristic of patient's brain tumors. Accumulating evidence suggests that BCPCs might originate from the transformation of neural stem cells (NSCs) and their progenitors. Furthermore, recent studies have shown that NSC surface markers also define BCPCs. Ultimately, treatments that include specific targeting of BCPCs might potentially be more effective at treating the entire tumor mass, translating to improved patient survival and quality of life.
Introduction to the cancer stem cell hypothesis
Recent evidence suggests that a subset of cancer cells, might underlie the growth of different types of cancer and be responsible for their resistance to therapy [1]. The terms CPC, cancer stem cells, cancer stem-like cells, or tumor initiating cells are variably used to describe tumor cells with stem-like properties [2] (Box 2). The new concept that a subset of cells within tumors might possess significant expansion capacity and the power to generate new tumors has been dubbed the cancer stem cell hypothesis [3]. This postulate also implies that the bulk of cancer cells within a solid tumor are progeny of CPCs, which cannot form new tumors and might represent a mix of partially differentiated cancer progenitor-like cells with limited proliferative capacity and terminally differentiated cancer cells (Box 1).
Box 2. Cancer propagating cells versus tumor-initiating cells (TICs).
CPCs are isolated from end-stage malignant tumors and those cells are highly tumorigenic in xenotransplantation assays. Some authors use the terms cancer stem cells or cancer stem-like cells. This terminology can be misleading as it may imply that the cell of origin was a stem cell, which is not necessarily the case.
TICs result from the transformation of the cell of origin by genetic alterations. TICs are identified in the early stages of tumor development and have not acquired full tumorigenic capacity.
Greater malignant properties of CPCs are due to their numerous mutations while TICs represent a cell population early in the transformation process.
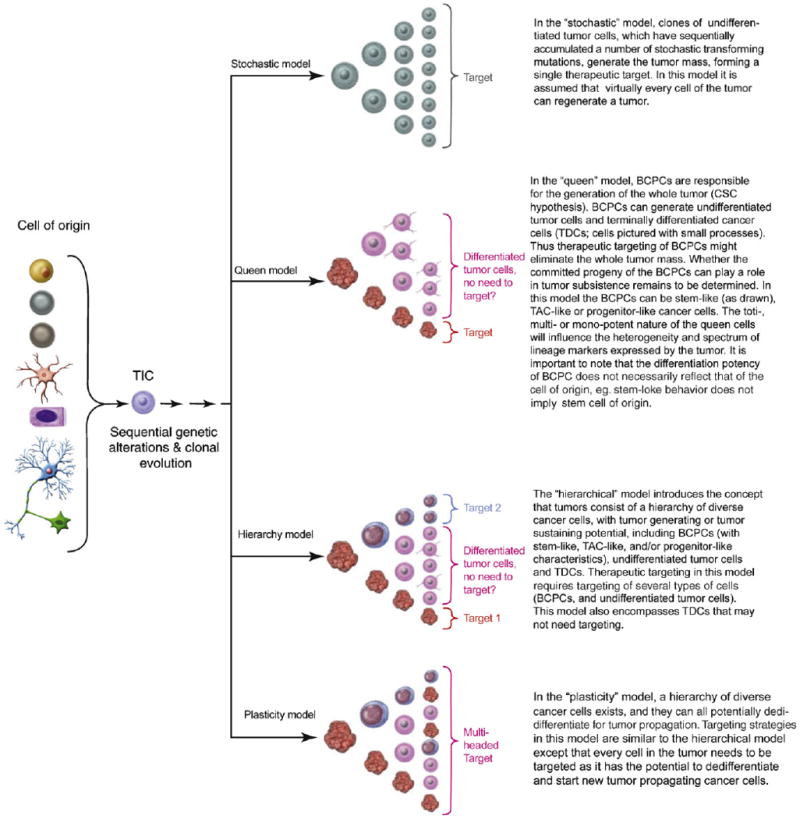
Box 1. Different models of tumor formation and therapeutic targeting.
A variety of normal cells in the CNS (left in Figure I) can potentially serve as cell of origin for the initiation of tumor development. A sequence of genetic and/or epigenetic alterations leads to the development of an early tumor cell population called tumor initiating cells (TICs). Further stochastic transforming mutations lead to progressively more malignant cell populations through an evolving “neoplastic ecosystem”. Various models are proposed below on how these transformed cells can propagate and generate the tumor mass. No single model needs to apply to all tumor types, different models may apply to different tumor types or stages of malignant progression.
The different cell types depicted are defined in Figure 2.
Figure I.
CPCs have been isolated from different types of tumors, including primary brain tumors such as GBM, MB and ependymoma [4–7] (Figure 1). These cells constitute a variable fraction of the total cell population within brain tumors, yet may be the drivers of their growth. They share characteristics similar to those of normal NSCs, including self-renewal (can divide and give rise to daughter stem cells with identical stem cell capabilities of the parent) and the proliferative ability for the generation of many progeny. Multipotency (the capability to differentiate into multiple cell types) is not a requirement for CPCs because some tumor types might have a single differentiated state (see Queen model in Box 1). The gold standard assay for the functional evaluation of both self-renewal and tumor propagation of CPCs is the ability to propagate serially in an undifferentiated state and form tumors in animals upon transplantation [8]. CPCs are isolated from dissociated tumors, propagated as neurospheres in specific neurobasal medium, and a subset express NSC surface markers such as Nestin and CD133 [9].
Figure 1.
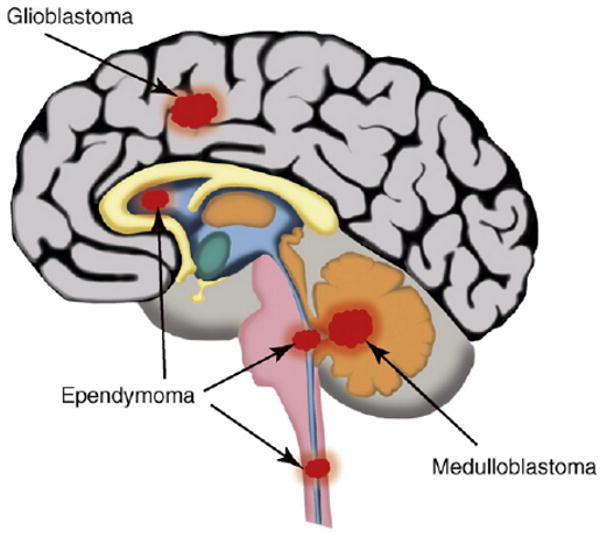
Brain tumors. Location of GBM, MB and ependymoma tumors in the CNS.
Definition and source of neural stem cells and progenitor cells in the brain
Current hypotheses postulate that BCPCs either originate from transformed NSC or neural progenitor cell populations in the brain or that they dedifferentiate from mature brain cells and reacquire phenotypic and functional similarities of NSCs [10] (Figure 2).
Figure 2.
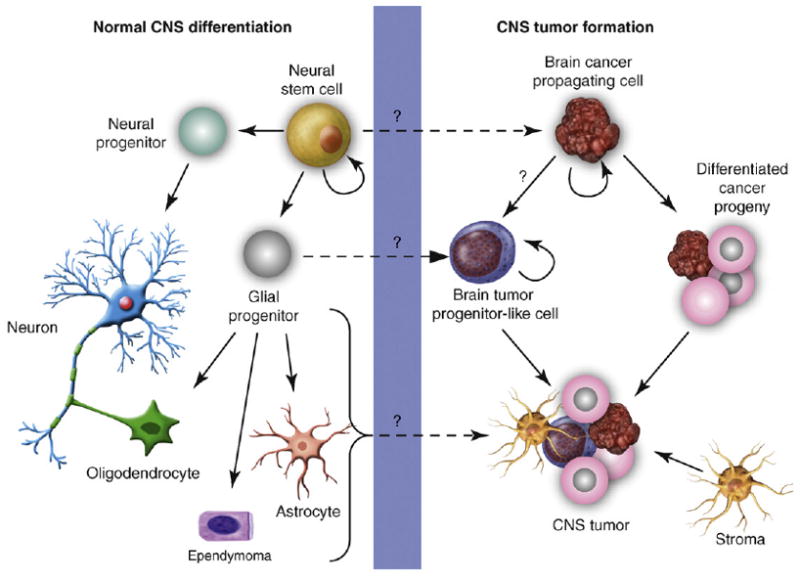
Normal CNS differentiation and transformation for CNS tumor formation. NSCs can differentiate into neural and glial progenitors. Neural progenitors differentiate into neurons, whereas glial progenitors are committed to oligodendrocytes, ependymal cells or astrocytes. CNS tumor formation might originate from the transformation of NSCs into BCPCs. Glial progenitors might transform into brain tumor progenitor-like cells that could engender CNS tumors (GBMs, MBs and ependymomas). The transformation of neurons, oligodendrocytes, ependymal cells and astrocytes has traditionally been thought to form CNS tumors. BCPCs can differentiate into brain tumor progenitor-like cells or more differentiated progeny, which can lead to CNS tumor formation. Stromal cells, either from the local brain microenvironment or recruited systemically, can be essential for tumor maintenance, progression and recurrence.
Individual adult NSCs were initially isolated from the adult striatum and shown to possess the ability to proliferate and generate clones of cells that showed multipotency, and grew as spheroids in defined medium supplemented with growth factors [epidermal growth factor (EGF) and fibroblast growth factor (FGF)-2], termed neurospheres [11]. In this neurobasal medium, NSCs can be propagated and expanded indefinitely, whereas most differentiating or differentiated cells rapidly die [12,13]. Upon growth factor removal or addition of serum, NSCs differentiate into neurons, astrocytes and oligodendrocytes.
NSCs are found in different areas of the brain, including around the ventricular system (subventricular zone (SVZ), subependymal zone, lining of the lateral ventricles and cerebellar ventricular zone), dentate gyrus, hippocampus and subcortical white matter [14–16]. During human development, the one important source of NSCs is the SVZ, a region localized between the lateral ventricle and parenchyma of the striatum. This region is widely viewed as the source of cells that can initiate GBM and ependymomas [17,18]. The SVZ contains astrocyte-like stem cells (also known as type B cells in mice) identified through their expression of the astroglial marker glial fibrillary acidic protein (GFAP). In contrast to mature astrocytes of the brain parenchyma, which also express GFAP, these cells can function as mature NSCs [15]. The murine postnatal cerebellum has been shown to contain multipotent NSCs that lack markers of neuronal and glial lineages (lin-) and can differentiate into glial cells (astrocytes and oligodendrocytes) and neurons in vitro and in vivo [19]. NSCs generate transit amplifying cells (TACs; also known as type C cells in mice) from which PGCs are derived. Prior evidence suggested that two types of PGCs are formed: those producing progeny along either the neuronal or glial lineages (Figure 2). New evidence suggests more plasticity, for example oligodendrocyte precursor cells, which are dispersed throughout the CNS, and initially thought to be lineage-restricted precursors that terminally differentiate to postmitotic oligodendrocytes seem to have the potential to form neuronal cells and might qualify as NSCs [20,21]. Although PGCs do replicate, their self-renewal capacity is finite in contrast to de facto stem cells.
In principle, any of these cell types might be subject to neoplastic transformation and engender brain tumors, although the rapidly expanding TACs are the most likely source [22]. In rodents, embryonic radial glial cells are believed to engender adult NSCs that share the functional and molecular characteristics of astroglial cells present in the SVZ of the lateral ventricles [23]. Radial glial cells are mitotically active, multipotent stem cells that can give rise through their progeny to the majority of neurons, astrocytes, oligodendrocytes and ependymal cells in the brain [24,25]. Radial glial cells can also give rise to adult NSCs, which could be the cell of origin of adult ependymomas [18].
The cerebellar ventricular zone, which consists of a band of NSCs and PGCs that line the fourth ventricle, gives rise to granule neuron precursors (GNPs) [14]. These are restricted PGCs that can give rise to MB [26]. Some subtypes of MBs can originate directly from NSCs [27,28].
Brain cancer stem cell formation and associated markers
How can we recognize and isolate BCPCs from the bulk of the tumors? Are there markers that can be used for their early detection and for tumor diagnosis, prognosis and response to therapy? Unfortunately, at this time no cell marker is absolute in identifying BCPCs. Not all tumor cells that are marker positive are BCPCs and not all BCPCs express known markers. Currently, functional assays of BCPC self-renewal and tumor propagation have defined this subpopulation of cells within tumors, and in vivo tumor generation remains the gold standard method to confirm the presence of BCPCs in neurospheres grown in serum-free media supplemented with growth factors.
CD133 (cluster of differentiation 133), also known as Prominin-1, is a 120 kD cell-surface 5-span transmembrane glycoprotein located on cellular protrusions originally described in mouse neuroepithelium and hematopoietic stem and progenitor cells [29] (Box 3). The loss of CD133 in the mouse results in the progressive degeneration of mature photoreceptors with complete loss of vision [30]. Embryonic NSCs, adult ependymal cells, functional endothelial precursor cells and colon CSCs have all been shown to express CD133 [10,31–33]. BCPCs that are CD133+ have been isolated from GBM, MB and ependymoma tumors and were shown to form aggressive tumors in the brain of mice at low inoculation numbers [4–6]. In patients with GBM, the percentage of tumor cells that express CD133 correlates with patient survival and risk of tumor regrowth [34]. The utility of CD133 in the isolation of cells with BCPC properties has been confirmed by multiple research groups [9,35,36]. It is unclear whether this marker identifies BCPC or whether it marks a subset of cells that can resist the immune system in partially immunodeficient mice strains such as nu/nu [37]. The CD133− cell subpopulation isolated from primary GBM tumors can also form orthotopic tumors, but is less proficient at tumor initiation [38,39]. This might reflect in part the presence of other types of BCPCs devoid of CD133, or that expression of CD133 by BCPCs is dynamically regulated and affected by different parameters including cell culture methods. Little is known about CD133 gene regulation and whether CD133 plays a role in the maintenance of the stemness phenotype.
Box 3. Cell surface markers in brain cancer propagating cells.
CD133, also known as Prominin-1, is the most studied cell surface marker for BCPCs. It comprises a 120 kDa cell surface transmembrane glycoprotein located on cellular protrusions. BCPCs that are CD133+ have been isolated from GBMs, MBs and ependymoma tumors. The figure shows confocal imaging of a single neurosphere derived from a fresh human glioblastoma tumor specimen depicting the cell population with green fluorescently labeled anti-CD133 antibody (a), nuclear DAPI blue staining (b) and costaining (c). (Figure I)
Figure I.
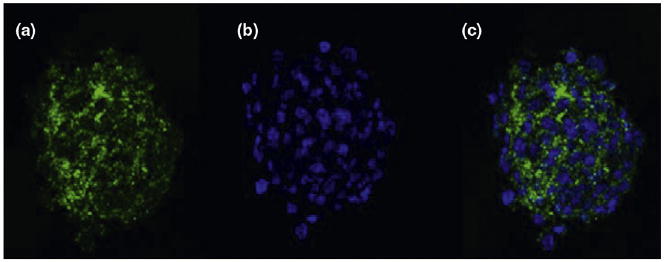
Photographs courtesy of D. Brat and C. Tucker-Burden.
The intermediate filament protein Nestin has been recognized as a NSC marker and is expressed by brain tumor cells [11,40,41]. Nestin is expressed in all CNS lineage-restricted PGCs and in astrocytes. The use of the Nestin gene promoter to drive the expression of oncogenes can allow for brain tumor formation in mice [42]. Nestin expression has also been shown to increase after the activation of the NOTCH signaling pathway in BCPCs [43]. However, the expression of Nestin is variable and not specific for BCPCs.
Other markers have been identified in BCPCs that are RNA-binding proteins, oncogenes, transcription factors, cell cycle modulators, signaling pathway proteins and ganglioside antigens. Musashi1, an RNA-binding protein expressed in NSCs/PGCs, is also expressed by BCPCs [6,9,44]. Bmi1 is an oncogene that is expressed in MBs and astrocytomas [45,46] and BCPCs [6]. Sox2 (sex determining region Y–box 2) is a transcription factor expressed by NSCs as well as BCPCs [6,9]. Maternal embryonic leucine zipper kinase (MELK), a cell cycle modulator, is expressed by NSCs and found to be elevated in BCPCs [47]. Notch proteins involved in cell signaling have been shown to be frequently expressed by BCPCs [43]. The ganglioside antigen A2B5 has been shown to be present in a population of BCPCs that are CD133− and able to propagate tumors [48]. A2B5 is a marker of all of the glial-restricted precursor cells from developing and mature animals. CD15, also known as Lewis X (LeX) or stage-specific embryonic antigen-1 (SSEA-1), has been shown to be a marker for type B and C cells in the adult SVZ, normal granule precursor cells and MB cells [49,50].
Glioblastoma multiforme
GBM, also called astrocytoma WHO grade IV, is the most frequent and lethal primary cancer of the CNS with patient survival at five years being less than 5% [51]. These tumors are highly cellular and vascularized, very resistant to radio- and chemotherapy and can diffusely and deeply infiltrate the normal brain parenchyma precluding total surgical resection [52]. The pathology of GBM is that of a highly heterogeneous tumor (as indicated by the old descriptor multiforme), which includes a variety of cancer cell types. Some of these cell types have differentiated stellar morphology and expression of markers such as GFAP and S100, whereas others have small and rounded undifferentiated cells mixed with stromal elements such as microvascular cell proliferation, reactive astrocytes, microglia and other immune infiltrates.
Discovery and characterization of GBM propagating cells
The heterogeneity of GBM was previously understood as a heterogeneous mix of highly motile tumor cells at different stages in their genetic progression towards malignancy, admixed with stromal elements. Genetic analyses confirmed that a single astrocytoma could contain different cell populations that were hierarchically organized in matters of the number of genetic alterations they contained, the earliest being remnants of a precursor cell population from which the rest of the tumor derived [53]. Alternatively, these early cells could be actively generating the bulk of the tumor. Addressing such questions became possible once dissociated cells from fresh tumor specimens could be cultured in media developed for the culture of human NSCs that would preserve their original stage of differentiation. These studies evidenced the existence in GBM of clonogenic, neurosphere-forming precursor cells that harbored stem-like characteristics, and could be induced to differentiate into cells expressing neuronal and astroglial markers (Figure 3) [5,7,54]. The identification of CD133 as a stem cell marker in normal CNS tissue prompted its use for the isolation of CD133-positive cells from human GBM using cell sorting technology. These cells were shown to possess the characteristics of BCPCs, for example they harbored genetic alterations and had the key hallmarks of stem cells, namely self-renewal, propagation as neurospheres and the ability to differentiate into neurons, astrocytes and oligodendrocytes [55]. These studies evidenced a remarkable difference in tumorigenic capacity between CD133-positive and -negative GBM cells As little as 100 transplanted CD133+ GBM stem cells were able to form tumors in vivo into the brains of immunocompromised mice, whereas >105 CD133− cells failed to form orthotopic tumors. Moreover, the tumors recapitulated the key pathognomonic features of GBM, including extensive infiltration in the brain parenchyma, pseudopalisading micronecrosis and extensive microvascular proliferation – features usually absent with the transplantation of traditional cell lines (Figure 4).
Figure 3.
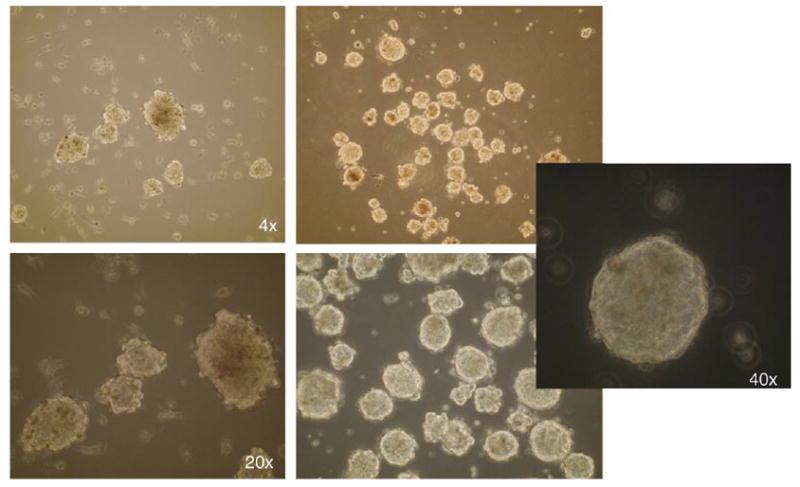
Human neurospheres cultured from glioblastoma tumor. The figure shows different magnifications of cultured neurospheres (4×, 20× and 40×).
Figure 4.
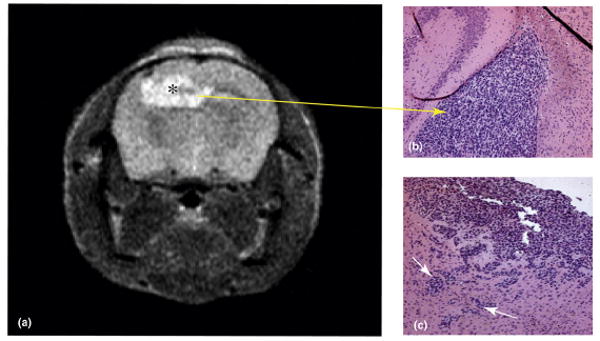
Human glioblastoma xenograft generated in an athymic nu/nu mouse eight weeks after the intracranial implantation of neurospheres harvested from a resected glioblastoma specimen. a) T2-weighted MRI of mouse brain showing xenograft (marked by *). b) Hematoxylin/eosin stained section of mouse brain showing xenograft (marked by arrow). c) Hematoxylin/eosin stained section of mouse brain demonstrating the invasion of human glioblastoma xenograft into surrounding brain (marked by arrows).
The purification of BCPCs in malignant gliomas can also be achieved based upon their ability to extrude the Hoechst dye 33342, a technique called side population (SP) [56]. GBM or rat C6 glioma cell line-derived SP cells form aggressive tumors in immunocompromised mice and can be propagated in vitro as neurospheres. SP cells express various ATP-binding cassette (ABC) transporters, probably explaining why they do not stain with the Hoechst dye, but more importantly conferring them with drug resistance because these transporters pump drugs outside the cells. The SP technique is controversial because the assay can be detrimental to non-dye excluding cells.
Signaling pathways leading to the genesis of glioblastoma
There has been a longstanding debate as to whether the cell of origin for the formation of GBM results from the transformation of NSCs/PGCs or through the dedifferentiation of a mature glial cell in the brain [57,58]. Genetic studies have demonstrated that a number of signaling pathways are commonly altered in human GBM, namely the p53/mdm2/ARF, pRb/p16Ink4a, PTEN/PI3kinase and HIF/IDH1/2 pathways. Concurrently, oncogenic signals are activated such as those deriving from EGF, c-Met and platelet-derived growth factor (PDGF) tyrosine kinase receptors [59]. The alteration of the same pathways in various NSC and PGC populations in the brain using genetically engineered mice with multiple gene combinations (EGF receptor/ARF loss, PDGF overexpression, Akt/Ras activation, NF1/p53 losses and PTEN/p53 losses) has demonstrated that they can lead to transformation in rodents [42,60,61] (Table 1). These models confirm that NSCs and PGCs initiate the tumorigenic process. Accordingly, the gene expression profiles of GBM resemble those of NSCs and PGCs of the developing forebrain [62]. OLIG2, a transcription factor that can promote the proliferation of neural progenitors by repressing the p21 tumor suppressor, has similar effects on GBM stem cells [60]. MELK, a cell cycle modulator, regulates NSC self-renewal and has been shown to regulate BCPC proliferation [6,47]. The commonality of signaling pathways activated in NSCs and GBM stem cells [54] has important implications for potential differentiation therapy. For example, blockage of the Sonic hedgehog (SHH) signaling pathway depletes BCPC cell populations in GBM [63].
Table 1. BCPC signaling pathways implicated in CNS tumor formation.
| CNS tumor type | Signaling pathway(s) |
|---|---|
| GBM | Ras, Myc, EGF receptor, PDGF receptor, Akt, PI(3)-kinase (loss of PTEN), SHH, TP53, OLIG2, Rb, MELK, NOTCH |
| MB | SHH, Wnt, MELK, NOTCH |
| Ependymoma | NOTCH |
The evidence for the formation of gliomas from mature differentiated glial cells is not as extensive, perhaps because it is a rare event. Mature, differentiated astrocytes or oligodendrocytes might be less prone to transformation. They could be intrinsically more resistant to carcinogenesis and thereby necessitate more stochastic transformation steps. The activation of two oncogenic pathways, such as ras and Akt in conjunction with p53 and Rb inactivation and expression of hTERT, can lead to the transformation of human astrocytes in vitro [64]. The inactivation of the pRb and p53 pathways through the combined genetic loss of p16INK4a and p19ARF in mouse astrocytes leads to dedifferentiation in response to EGF receptor activation, and these cells can induce high-grade gliomas [65]. The dedifferentiation of astrocytes into glial progenitors or stem cells can produce gliomas when infected by PDGF-encoding or EGF-encoding retroviruses [66].
Medulloblastoma
MB is the most common malignant brain tumor in children and is an embryonal neuroepithelial tumor of the cerebellum [26]. Approximately one-third of patients with MB tumors remain incurable and current treatments have associated toxicities that can cause significant disabilities in long-term survivors. While the tumors form relatively well-circumscribed masses that can be excised surgically, morbidity stems from their metastatic spread via the cerebrospinal fluid to the leptomeningeal spaces. MB tumors are characterized by particular morphologic and genetic attributes [26]. The morphologic differentiation of MB tumors occurs principally along neuroepithelial lines, whereas glial differentiation is uncommon. Like embryonal CNS tumors, MBs can express markers of neuronal and glial cell lineage such as synaptophysin and GFAP.
Discovery and characterization of MB cancer propagating cells
MB CPCs were isolated by their expression of CD133 and were found in 1–21% of surgically resected tumors from both classic and desmoplastic variants of MB [5]. As few as 1,000–5,000 CD133+ cells could form MB tumors in nu/nu mice, whereas 10-fold more CD133− cells could not [5,36].
Signaling pathways leading to the genesis of medulloblastoma
The cell of origin of MBs remains nondefinitive [26]. The morphology of tumor cells and their location in the cerebellum have led to the thought that tumors might arise from GNPs present in the external germinal layer (EGL) of the cerebellum. These are restricted PGCs that give rise to only granule neurons. The cerebellar ventricular zone, which consists of a band of stem and progenitor cells that line the fourth ventricle, gives rise to the GNPs [14]. The GNPs proliferate in the EGL and then migrate to form the internal granule layer after differentiation to neurons. MB cells have been shown to express markers that commonly are associated with GNPs, such as p75NTR, TrkC, Zic1 and Math1 [67,68]. MBs have also been shown to express stem cell markers and possess the ability to differentiate into neurons and glia, suggesting their origin might be from NSCs [6,55]. Recently, it has been suggested that subtypes of MB (desmoplastic, anaplastic and large-cell MBs and MB with extensive nodularity) arise from GNPs and others from NSCs [26–28].
The activation of the SHH pathway plays a critical role in MB formation (Table 1). The SHH pathway regulates NSC development and can induce GNP proliferation [69]. The Patched (Ptc) receptor is an antagonist of the SHH signaling pathway, and mutations of Ptc will activate the pathway [70]. Ptc mutations have been observed in many human MBs including sporadic tumors and in tumors arising in patients with Gorlin's syndrome (an autosomal dominant disorder in which patients present with various neoplasms including basal cell carcinoma and MB) [71,72]. Use of the ptc mutant mouse has become a widely studied model of MB [73]. The blockade of the SHH pathway using a small molecule inhibitor can eliminate MB tumors in mice harboring a ptc1 mutation [74].
Recently, the issue of which CNS cells are capable of being transformed into MB tumors by SHH activation has been addressed by showing that MB tumors can be initiated by the deletion of ptc in lineage-restricted progenitors (GNPs) or in neural stem cells [27]. These studies confirm both GNPs and NSCs can respond to the SHH signaling pathway and serve as cells of origin for MB. Furthermore, genetic changes induced in either multipotent ventricular zone stem cells or lineage-committed cells result in MB tumors in the EGL, thereby acquiring a GNP identity with SHH activation [28].
A second signaling pathway involved in MBs is the Wnt pathway, which is associated with MBs of the classic subtype [73]. Wnt pathway mutations have been identified in MB associated with Turcot's type 2 syndrome (caused by the loss of the Adenomatous Polyposis Coli gene function). The Wnt signaling pathway does not seem to regulate EGL precursors, providing further evidence that MB tumors arise from NSCs/PGCs outside the EGL [75].
Ependymomas
Ependymomas are tumors in the CNS that originate from the wall of the ventricular system and can occur along the entire craniospinal axis in both children and adults (Figure 1). They display characteristic ultrastuctural and immunohistochemical features of cells of ependymal lineage. CPCs can be isolated from fresh samples of ependymomas and cultured as neurospheres [4]. Ependymoma CPCs are believed to derive from embryonic radial glial cells and share similar cell surface markers with NSCs such as CD133, Nestin, RC2 and brain lipid-binding protein (BLBP). These cells exhibit the ability to self-renew and differentiate along divergent neuronal lines. Immunocompromised nu/nu mice injected with as few as 10 000 CD133+/RC2+/BLBP+ ependymoma cells develop tumors that arise within the cerebral SVZ and fill the lateral or third ventricles [4]. These tumors have morphologic features of ependymomas. CD133− ependymoma cells have not been able to develop tumors in mice.
Ependymomas from supratentorial, posterior fossa or spine locations exhibit distinct patterns of gene expression [4,18]. Many genes expressed by these tumors are known regulators of neural precursor cells in the corresponding region of the CNS. Recently, it has been shown that embryonic radial glial cells are likely to be a source of ependymomas independent of patient age [4]. Embryonic radial glial cells are believed to be NSCs that share the functional and molecular characteristics of astroglial cells present in the SVZ of the lateral ventricles. Radial glial cells are mitotically active, multipotent stem cells that can give rise to the majority of neurons, astrocytes, oligodendrocytes and ependymal cells in the brain, and are believed to be the cell of origin of adult ependymomas [24,25].
Signaling pathways leading to the genesis of ependymomas
The initiation of ependymomas is thought to occur from the transformation of radial glial cells and might involve aberrant cell division mediated by certain transcription factors such as EMX2, the disruption of adherens junctions and the deregulation of cell signaling pathways such as NOTCH [18] (Table 1). The NOTCH ligands JAGGED 1/2 and the NOTCH signal targets HES1/5 and ERBB2 are upregulated in supratentorial ependymomas [4,76]. The concurrent activation of NOTCH cell signaling and deletion of the INK4A/ARF gene locus were found to be present in supratentorial ependymomas.
Role of the brain cancer stem cell niche
Normal NSCs are concentrated in regions of the brain that are rich in blood vessels, called “vascular niches”, where they are sheltered from apoptotic stimuli and maintain a proper balance between self-renewal and differentiation [77,78]. The endothelial cells (ECs) in the vascular niche secrete paracrine factors that can promote stem cell survival and self-renewal. Microvascular proliferation and high vascularity are the pathologic hallmarks of GBM tumors. Therefore, tumors might be an alternative harbor for NSCs and BCPCs requiring paracrine factors for their maintenance and survival. In fact, this might provide a role for the exuberant glomeruloid microvascular proliferations seen in GBM, which are devoid of lumen and blood flow. NSCs are also known to thrive in hypoxic regions, and the tumor might provide a hypoxic niche [79]. BCPCs have been found to be closely associated with the vascular niche in GBM [36], and sorted CD133+ tumor cells selectively associate with ECs in co-cultures, which augments their self-renewal capacity. Intracranially transplanted CD133+ tumor cells form tumors more rapidly when co-transplanted with ECs, and these contain many more CD133+ CPCs. Reciprocally, CPCs might also sustain vascular development, as CD133+ GBM propagating cells have a higher level of production of vascular endothelial growth factor (VEGF) than CD133− cells [80], which is consistent with CD133+ GBM cells forming highly vascular and hemorrhagic tumors in vivo.
Impact of brain cancer stem cells on therapy
Individual parameters that characterize primary brain tumors (proliferation, apoptosis, invasion, angiogenesis, immune resistance, etc.) have all been obtained through the analysis of the bulk of the tumor or cell lines in culture, which in light of the CPC hypothesis might not accurately represent the tumor-propagating cell population. Such studies have been useful for identifying the critical oncogenic pathways that can be targeted therapeutically. Current studies are determining whether BCPCs use the same pathways, to validate the impact of existing therapies on this cell population and possibly develop novel ones that will be more effective at targeting BCPCs and causing their demise (Table 2). This also suggests that eliminating a tumor might need combination therapies against multiple targets (Box 1).
Table 2. BCPC therapeutic targeting in CNS tumors.
| CNS tumor type | Mechanism |
|---|---|
| GBM | Radiosensitivity enhancement (e.g. Chk1 and Chk2 inhibitors) |
| GBM, MB and ependymoma | Blockade of signaling pathways (e.g. EGF receptor, AKT, PI(3)-kinase, SHH, NOTCH pathways) |
| GBM | Differentiation (e.g. BMPs) |
| GBM | Dendritic cell vaccine |
| GBM | Disruption of vascular niche (e.g. bevacizumab, AZD2171) |
New therapeutic strategies targeting brain cancer propagating cells
Conventional therapies, such as radiation therapy and chemotherapy, have been used to treat primary brain tumors with modest efficacy, and recent evidence suggests BCPCs are involved in radio-/chemoresistance [81,82]. CD133-expressing glioma cells survive ionizing radiation in increased proportions relative to most tumor cells, which lack CD133, both in vitro and in nu/nu mice. These cells were found to more efficiently repair damaged DNA in response to ionizing radiation compared with the bulk of more differentiated tumor cells [9], and the preferential phosphorylation of the DNA repair checkpoint proteins Chk1/2 might underlie this resistance. Therapeutic targeting of the checkpoint kinases Chk1 and Chk2 with small molecules can reverse the radioresistance of CD133+ GBM stem cells in vitro, providing potential targeted therapy to BCPCs.
Primary brain tumor progression might also result from chemoresistant BCPCs. GBM stem cells have recently been shown to be resistant to the currently used chemotherapy agent temozolomide (TMZ) [82]. The elevated expression of transporters that pump out chemotherapeutic agents might be one important mechanism for chemoresistance [83]. The ABC drug transporter genes ABCG2 and ABCA3 have been shown to be highly expressed in side population cells and have also been found in GBM cell lines [84].
The vulnerability of CNS PGCs to chemotherapeutic agents has recently been shown and raises the concern of toxicity to the normal CNS stem cell lineage [85]. Clinically relevant concentrations of BCNU, cisplatin or cytarabine were associated with increased cell death and decreased cell division in the SVZ of the CNS, the dentate gyrus of the hippocampus and the corpus callosum. The potential vulnerability of areas in the brain rich in NSC to chemotherapeutics might result in untoward effects. Targeting BCPCs and sparing NSCs and their progenitors is an important challenge that needs to be addressed with novel therapies.
Signaling pathway inhibition of brain cancer propagating cells
Key signaling pathways (PI(3)-kinase, OLIG2, SHH, NOTCH and Wnt) essential for the development and regulation of NSCs have been shown to be active in BCPCs of GBMs, MBs and ependymomas [4,14,62] and need to be considered candidate targets. The blockade of the SHH pathway in brain tumors can decrease neurosphere formation, deplete CD133+ cells, interfere with xenograft tumor growth and cure mice with MB tumors [74]. The inhibition of the NOTCH signaling pathway by gamma secretase inhibitors (e.g. GSI-18) can attenuate NOTCH activity and interfere with BCPC function in vitro and xenograft tumor formation in vivo [43]. The use of AKT inhibitors and targeting of the PI(3)-kinase pathway have shown activity against BCPCs with apoptosis induction and inhibition of invasion [86]. The inhibition of the PDGF signaling pathway can inhibit BCPC neurosphere growth in culture [87].
Differentiation of brain cancer propagating cells
Promoting the differentiation of BCPCs might be a new therapeutic mechanism for targeting BCPCs and primary brain tumors. The induction of differentiation by bone morphogenetic proteins (BMPs), specifically BMP4, can trigger a significant reduction in GBM CPCs in vitro and extended mouse survival and reduced tumor growth [35]. Of note, a small number of animals with tumors did not respond to BMP and died three months after treatment. In those animals with a treatment response, CD133+ cells could not be recovered from the xenograft tumors.
Further evidence has confirmed BMPs promote the glial differentiation of GBM CPCs [88]. However, in some cases BMPs can paradoxically cause BCPC proliferation and tumorigenesis. In these cells, BMPs did not induce STAT3-dependent glial differentiation because of the epigenetic silencing of BMP1RB by an EZH2-dependent mechanism. In fact, a significant number (20%) of GBM tumor samples were found to have low levels of the BMP1 receptor B (BMP1RB), and the majority of these samples had hypermethylation of the BMP1RB gene promoter. Epigenetic resistance mechanisms to differentiation therapy suggest that BMPs in combination therapy with epigenetic modulators might have to be tailored to patients with BMP1RB silencing.
Brain cancer propagating cell niche
The vascular niche has been shown to be important for providing a nurturing milieu to BCPCs, suggesting that its disruption might be therapeutic [77,78]. The inhibition of blood vessel growth might be an effective method for eliminating GBM CPCs. Bevacizumab (an anti-VEGF antibody) treatment of CD133+ GBM CPCs can block their ability to induce EC migration and tube formation in culture, and initiate tumors in vivo. The treatment of mice carrying GBM with bevacizumab can result in a large reduction in the number of GBM CPCs and in the growth of tumors [36]. Human clinical trials of bevacizumab and the pan-VEGF receptor tyrosine kinase inhibitor cediranib (AZD2171) have demonstrated efficacy in GBM patients [89,90], and it will be interesting to examine whether this was linked to BCPC depletion by disruption of the vascular niche.
Perspectives
The discovery of BCPCs has provided a potential new paradigm shift in our understanding of the growth of primary brain tumors and the development of targeted therapeutics against these cells [9,36,84]. It is unclear at this time whether the growth of primary brain tumors is driven exclusively by BCPCs [91] or the majority of the cells within the tumor (Box 1). Furthermore, the overlap between normal NSC lineage and BCPCs needs further characterization. Exactly when transformation of a NSC or PGC occurs remains to be determined. Progenitors that might be implicated in the transformation process to form a BCPC could be TACs, which share similar signaling pathways to BCPC and GBM tumor cells [22]. Nearly half of all high-grade astrocytomas demonstrate EGF receptor amplification, and EGF receptor mutation is a classic step in the development of primary glioblastomas. EGF-responsive TACs within the adult SVZ constitute a large population of dividing progenitors [92]. The EGF-mediated stimulation of this population prevents TAC differentiation and induces cell migration into an infiltrative phenotype comparable to that seen in GBM tumors.
Other cellular relationships in the heterogeneous tumor remain to be explored. One might speculate that the coevolution of different cell populations within tumors with different terminal genetic alterations might occur and coexist, and might even be codependent. It is likely that normal NSCs might be recruited to become part of the tumor stroma, and stromal cells can all potentially be modified by adjacent tumor cells through the transfer of oncogenic proteins through microvesicles or exosomes [93] (Figure 2). Heterotypic cell–cell communications between all the tumor elements need to be better understood.
Current markers used to identify BCPCs are suboptimal. Furthermore, additional studies on BCPCs might possibly reveal subsets of brain tumor progenitor-like cells with more limited self-renewal potential that could also contribute to the tumor mass (Box 1). Also, a significant overlap currently exists between cell markers of BCPCs and the NSC lineage. Because of the limited markers available for characterizing BCPCs, the isolation of BCPCs for study and therapeutic targeting relies on culturing clonogenic neurospheres with growth factors in a serum-free culture method [54].
Clear differences exist in the biological properties of BCPCs in comparison with cells derived from the bulk of the tumor [9,54]. Yet, the clonal transformation process that engendered BCPCs implies that the more differentiated tumor cell populations that comprise the bulk of tumors carry the same genetic alterations [53]. A clear understanding needs to be developed of the different cell populations in the tumor to guide the development of new therapeutic strategies. Based on the current cancer stem cell hypothesis, the bulk of the tumor is comprised of the differentiated progeny from BCPCs. It is reasonable to speculate that some tumors might consist of three broad categories of cells: BCPCs (tumor-seeding), brain tumor progenitor-like cells (with limited capacity for progeny generation) and terminally differentiated cancer cells (TDCs, with no regenerative potential) [94,95] (see Hierarchy model in Box 1).
New technology needs to be developed to track BCPCs in patients, because their fate in response to therapy might become a new guiding principle for judging the efficacy of various agents. Magnetic resonance imaging (MRI) or positron emission tomography (PET) imaging might permit a better understanding of the relationship between BCPCs and tumor response to therapy. Recently, magnetic resonance spectroscopy has been shown to identify neural progenitor cells in vivo in the human brain [96]. Emerging nanotechnologies might permit the simultaneous imaging of BCPCs as well as targeted therapy with conjugated antitumor agents and/or the ability, for example, to generate local hyperthermia through their magnetic properties [97].
Vaccine peptides and immunotherapy that can elicit a natural immune response to BCPCs might be important. Newer BCPC surface markers might permit the development of peptides derived from BCPCs that are specific and able to generate an immune response. Dendritic cells pulsed with BCPCs might form the basis of selective immunotherapy against these cells [98].
Therapeutic strategies against BCPCs in the form of oncolytic viruses [herpes simplex virus 1 (HSV-1), adenoviruses, etc.] might be designed to selectively infect and replicate within BCPCs [99]. The selective uptake of these viruses by BCPCs would need to be based on viral constructs that can express a ligand specific for BCPCs. Alternatively, viral replication can be made conditional to transcription factors specifically activated in BCPCs [100].
Currently, all therapies designed against BCPCs run the risk of targeting NSCs and their progenitor lineage. Morphologic and phenotypic differences need to be defined for each set of cells for proper targeting and for minimizing normal neurologic deficit. Recent evidence suggests that hypoxia inducible factor HIF2α is preferentially upregulated in glioma BCPCs, but absent in normal neural progenitors [101]. This warrants the evaluation of HIF-targeted therapies on BCPCs [102,103].
Concluding remarks
In summary, and in spite of some controversies as to their nature or existence [37,91] (Box 4), the discovery of CPCs is strongly impacting the field of neuro-oncology [52]. A major rethinking of the basic concepts on which neuro-oncology has been built is occurring. The genetics, biology, imaging and therapeutics (chemo- and radiotherapy) of primary CNS tumors have been affected. Studies of clonal evolution have to account for the novel concept of possibly only a small subset of cells in the cancer driving tumorigenesis [53,104]. The characterization of tumor pathology needs to develop better markers to trace and characterize BCPCs among a much larger population of more differentiated cancer cells. Studies of cell signaling and modeling in animal systems need to be performed with novel neurosphere cultures, and prior findings with traditionally grown cell lines should be revisited. Most importantly, the response of this new target cell population to different traditional and newly developed therapeutic modalities needs to be reassessed in light of their unique biology. This will also require the development of novel imaging tracers that can track the response of BCPCs in the background of the bulk of the tumor. This revolution in neuro-oncology research opens up novel opportunities for great discoveries and promises to accelerate the development of new therapies, which are an absolute priority for these devastating diseases.
Box 4. Brain cancer propagating cells: current limitations and controversies.
Limited cell surface markers for BCPCs.
Transplantation assays have limitations in recognizing BCPCs.
Potential hierarchy of cells in CNS tumors including BCPCs, undifferentiated and more differentiated tumor cells.
Need for therapeutic targeting of various tumor cell populations.
Potential problem of affecting normal NSC populations when targeting BCPCs.
Acknowledgments
Our work is supported in part by grants from the NIH (CA86335, CA116804 to EGVM, NS053454 to CGH), American Brain Tumor Association (to CGH), Goldhirsh Foundation (to EGVM), Southeastern Brain Tumor Foundation (to CGH and EGVM), the Brain Tumor Funders Collaborative (to EGVM) and the Georgia Cancer Coalition, Distinguished Cancer Clinicians and Scientists Program (to CGH).
References
- 1.Jordan CT, et al. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 2.Reilly KM, et al. Rethinking brain tumors: the fourth mouse models of human cancers consortium nervous system tumors workshop. Cancer Res. 2008;68:5508–5511. doi: 10.1158/0008-5472.CAN-08-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reya T, et al. Stem cells, cancer and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 4.Taylor MD, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Singh SK, et al. Identification of human brain tumor initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 6.Hemmati HD, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galli R, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 8.Clarke MF, et al. Cancer stem cells – perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 9.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 10.Uchida N, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 12.Vescovi AL, et al. Isolation and cloning of multipotential stem cells from the embryonic human CNS and establishment of transplantable human neural stem cell lines by epigenetic stimulation. Exp Neurol. 1999;156:71–83. doi: 10.1006/exnr.1998.6998. [DOI] [PubMed] [Google Scholar]
- 13.Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- 14.Eberhart CG. Even cancers want commitment: lineage identity and medulloblastoma formation. Cancer Cell. 2008;14:105–107. doi: 10.1016/j.ccr.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanai N, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature. 2004;427:740–744. doi: 10.1038/nature02301. [DOI] [PubMed] [Google Scholar]
- 16.Nunes MC, et al. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439–447. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- 17.Sanai N, et al. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- 18.Poppleton H, Gilbertson RJ. Stem cells of ependymoma. Br J Cancer. 2007;96:6–10. doi: 10.1038/sj.bjc.6603519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee A, et al. Isolation of neural stem cells from the postnatal cerebellum. Nat Neurosci. 2005;8:723–729. doi: 10.1038/nn1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo T, Raff M. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science. 2000;289:1754–1757. doi: 10.1126/science.289.5485.1754. [DOI] [PubMed] [Google Scholar]
- 21.Webber DJ, et al. Minimally manipulated oligodendrocyte precursor cells retain exclusive commitment to the oligodendrocyte lineage following transplantation into intact and injured hippocampus. Eur J Neurosci. 2007;26:1791–1800. doi: 10.1111/j.1460-9568.2007.05823.x. [DOI] [PubMed] [Google Scholar]
- 22.Doetsch F, et al. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36:1021–1034. doi: 10.1016/s0896-6273(02)01133-9. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez-Buylla A, et al. Identification of neural stem cells in the adult vertebrate brain. Brain Res Bull. 2002;57:751–758. doi: 10.1016/s0361-9230(01)00770-5. [DOI] [PubMed] [Google Scholar]
- 24.Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 25.Spassky N, et al. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci. 2005;25:10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annu Rev Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- 27.Yang ZJ, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuller U, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form SHH-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weigmann A, et al. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci U S A. 1997;94:12425–12430. doi: 10.1073/pnas.94.23.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zacchigna S, et al. Loss of the cholesterol-binding protein Prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J Neurosci. 2009;29:2297–2308. doi: 10.1523/JNEUROSCI.2034-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Brien CA, et al. A human colon cancer cell capable of initiating tumor growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 32.Peichev M, et al. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–958. [PubMed] [Google Scholar]
- 33.Pfenninger CV, et al. CD133 is not present on neurogenic astrocytes in the adult subventricular zone, but on embryonic neural stem cells, ependymal cells and glioblastoma cells. Cancer Res. 2007;67:5727–5736. doi: 10.1158/0008-5472.CAN-07-0183. [DOI] [PubMed] [Google Scholar]
- 34.Zeppernick F, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14:123–129. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 35.Piccirillo SG, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumor-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 36.Calabrese C, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 37.Quintana E, et al. Efficient tumor formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beier D, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 39.Sakariassen PO, et al. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc Natl Acad Sci U S A. 2006;103:16466–16471. doi: 10.1073/pnas.0607668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dahlstrand J, et al. Expression of the class VI intermediate filament Nestin in human central nervous system tumors. Cancer Res. 1992;52:5334–5341. [PubMed] [Google Scholar]
- 41.Lendahl U, et al. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 42.Holland EC, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 43.Fan X, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 44.Kaneko Y, et al. Musashi1: an evolutionally conserved marker for CNS progenitor cells including neural stem cells. Dev Neurosci. 2000;22:139–153. doi: 10.1159/000017435. [DOI] [PubMed] [Google Scholar]
- 45.Tirabosco R, et al. Expression of the Polycomb-Group protein BMI1 and correlation with p16 in astrocytomas: an immunohistochemical study on 80 cases. Pathol Res Pract. 2008;204:625–631. doi: 10.1016/j.prp.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 46.Leung C, et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 47.Nakano I, et al. Maternal embryonic leucine zipper kinase is a key regulator of the proliferation of malignant brain tumors, including brain tumor stem cells. J Neurosci Res. 2008;86:48–60. doi: 10.1002/jnr.21471. [DOI] [PubMed] [Google Scholar]
- 48.Ogden AT, et al. Identification of A2B5+ and CD133− tumor-initiating cells in adult human gliomas. Neurosurgery. 2008;62:505–514. doi: 10.1227/01.neu.0000316019.28421.95. discussion 514-505. [DOI] [PubMed] [Google Scholar]
- 49.Read TA, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15:135–147. doi: 10.1016/j.ccr.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Capela A, Temple S. LeX/SSEA-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron. 2002;35:865–875. doi: 10.1016/s0896-6273(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 51.Legler JM, et al. Cancer surveillance series: brain and other central nervous system cancers: recent trends in incidence and mortality. J Natl Cancer Inst. 1999;91:1382–1390. doi: 10.1093/jnci/91.16.1382. [DOI] [PubMed] [Google Scholar]
- 52.Van Meir EG, editor. CNS Cancer: Models, Markers, Prognostic Factors, Targets and Therapeutic Approaches. Humana Press (Springer); 2009. p. 1284. [Google Scholar]
- 53.Fulci G, et al. Initiation of human astrocytoma by clonal evolution of cells with progressive loss of p53 functions in a patient with a 283H TP53 germline mutation: evidence for a precursor lesion. Cancer Res. 2002;62:2897–2906. [PubMed] [Google Scholar]
- 54.Lee J, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 55.Singh SK, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 56.Kondo T, et al. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A. 2004;101:781–786. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Emmenegger BA, Wechsler-Reya RJ. Stem cells and the origin and propagation of brain tumors. J Child Neurol. 2008;23:1172–1178. doi: 10.1177/0883073808321062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alcantara Llaguno S, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Y, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stiles CD, Rowitch DH. Glioma stem cells: a midterm exam. Neuron. 2008;58:832–846. doi: 10.1016/j.neuron.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 62.Phillips HS, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 63.Bar EE, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pieper RO. Transformed human brain cells in culture as a model for brain tumors. In: Van Meir EG, editor. CNS Cancer: Models, Markers, Prognostic Factors, Targets and Therapeutic Approaches. Humana Press (Springer); 2009. pp. 163–180. Chapter 9. [Google Scholar]
- 65.Bachoo RM, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 66.Dai C, et al. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pomeroy SL, et al. Neurotrophins in cerebellar granule cell development and medulloblastoma. J Neurooncol. 1997;35:347–352. doi: 10.1023/a:1005841206252. [DOI] [PubMed] [Google Scholar]
- 68.Salsano E, et al. Expression of MATH1, a marker of cerebellar granule cell progenitors, identifies different medulloblastoma subtypes. Neurosci Lett. 2004;370:180–185. doi: 10.1016/j.neulet.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 69.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 70.Ahn S, Joyner AL. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- 71.Hahn H, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 72.Johnson RL, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 73.Fan X, Eberhart CG. Medulloblastoma stem cells. J Clin Oncol. 2008;26:2821–2827. doi: 10.1200/JCO.2007.15.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Romer JT, et al. Suppression of the SHH pathway using a small molecule inhibitor eliminates medulloblastoma in ptc1(+/−)p53(−/−) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 75.Thomas KR, Capecchi MR. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature. 1990;346:847–850. doi: 10.1038/346847a0. [DOI] [PubMed] [Google Scholar]
- 76.Gilbertson RJ, et al. ERBB receptor signaling promotes ependymoma cell proliferation and represents a potential novel therapeutic target for this disease. Clin Cancer Res. 2002;8:3054–3064. [PubMed] [Google Scholar]
- 77.Yang ZJ, Wechsler-Reya RJ. Hit'em where they live: targeting the cancer stem cell niche. Cancer Cell. 2007;11:3–5. doi: 10.1016/j.ccr.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 78.Gilbertson RJ, Rich JN. Making a tumor's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 79.Keith B, Simon MC. Hypoxia-inducible factors, stem cells and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bao S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 81.Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980–8984. doi: 10.1158/0008-5472.CAN-07-0895. [DOI] [PubMed] [Google Scholar]
- 82.Liu G, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 84.Hirschmann-Jax C, et al. A distinct side population of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dietrich J, et al. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J Biol. 2006;5:22. doi: 10.1186/jbiol50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eyler CE, et al. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells. 2008;26:3027–3036. doi: 10.1634/stemcells.2007-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gal H, et al. MIR-451 and imatinib mesylate inhibit tumor growth of glioblastoma stem cells. Biochem Biophys Res Commun. 2008;376:86–90. doi: 10.1016/j.bbrc.2008.08.107. [DOI] [PubMed] [Google Scholar]
- 88.Lee J, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vredenburgh JJ, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 90.Batchelor TT, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Adams JM, Strasser A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. 2008;68:4018–4021. doi: 10.1158/0008-5472.CAN-07-6334. [DOI] [PubMed] [Google Scholar]
- 92.Doetsch F, et al. Lack of the cell cycle inhibitor p27Kip1 results in selective increase of transit amplifying cells for adult neurogenesis. J Neurosci. 2002;22:2255–2264. doi: 10.1523/JNEUROSCI.22-06-02255.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Al-Nedawi K, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumor cells. Nat Cell Biol. 2008;10:619–624. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 94.Hadjipanayis CG, Van Meir EG. Tumor initiating cells in malignant gliomas: biology and implications for therapy. J Mol Medicine. 2009;87:363–374. doi: 10.1007/s00109-009-0440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Canoll P, Goldman JE. The interface between glial progenitors and gliomas. Acta Neuropathol. 2008;116:465–477. doi: 10.1007/s00401-008-0432-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Manganas LN, et al. Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain. Science. 2007;318:980–985. doi: 10.1126/science.1147851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hadjipanayis CG, et al. Metallic iron nanoparticles for MRI contrast enhancement and local hyperthermia. Small. 2008;4:1925–1929. doi: 10.1002/smll.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pellegatta S, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006;66:10247–10252. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]
- 99.Nandi S, et al. Low-dose radiation enhances survivin-mediated virotherapy against malignant glioma stem cells. Cancer Res. 2008;68:5778–5784. doi: 10.1158/0008-5472.CAN-07-6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Post DE, et al. Replicative oncolytic adenoviruses in multimodal cancer regimens. Hum Gene Ther. 2003;14:933–946. doi: 10.1089/104303403766682205. [DOI] [PubMed] [Google Scholar]
- 101.Li Z, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Post DE, et al. Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin Cancer Res. 2004;10:8603–8612. doi: 10.1158/1078-0432.CCR-04-1432. [DOI] [PubMed] [Google Scholar]
- 103.Narita T, et al. Identification of a novel small molecule HIF-1a translation inhibitor. Clin Cancer Res. 2009;15:6128–6136. doi: 10.1158/1078-0432.CCR-08-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ishii N, et al. Cells with TP53 mutations in low grade astrocytic tumors evolve clonally to malignancy and are an unfavorable prognostic factor. Oncogene. 1999;18:5870–5878. doi: 10.1038/sj.onc.1203241. [DOI] [PubMed] [Google Scholar]