

Abstract
The enzyme aminoimidazole ribonucleotide (AIR) carboxylase catalyzes the synthesis of the purine intermediate, 4-carboxy-5-aminoimidazole ribonucleotide (CAIR). Previously, we have shown that the compound 4-nitro-5-aminoimidazole ribonucleotide (NAIR) is a slow, tight binding inhibitor of the enzyme with a Ki of 0.34 nM. The structural attributes and the slow, tight binding characteristics of NAIR implicated this compound as a transition state or reactive intermediate analog. However, it is unclear what molecular features of NAIR contribute to the mimetic properties for either of the two proposed mechanisms of AIR carboxylase. In order to gain additional information regarding the mechanism for the potent inhibition of AIR carboxylase by NAIR, a series of heterocyclic analogs were prepared and evaluated. We find that all compounds are weaker inhibitors than NAIR and that CAIR analogs are not alternative substrates for the enzyme. Surprisingly, rather subtle changes in the structure of NAIR can lead to profound changes in binding affinity. Computational investigations of enzyme intermediates and these inhibitors reveal that NAIR displays an electrostatic potential surface similar to a proposed reaction intermediate. The result indicates that AIR carboxylase is likely sensitive to the electrostatic surface of reaction intermediates and thus compounds which mimic these surfaces should possess tight binding characteristics. Given the evolutionary relationship between AIR carboxylase and N5-CAIR mutase, we believe that this concept extends to the mutase enzyme as well. The implications of this hypothesis for the design of selective inhibitors of the N5-CAIR mutase are discussed.
Introduction
The basic aspects of de novo purine biosynthesis were outlined in the 1950’s by Buchanan who studied the metabolism of labeled purine intermediates in chicken or pigeon liver extracts. From this work, a 10-step pathway for the conversion of PRPP into inosine monophosphate, the common branch point intermediate for both AMP and GMP biosynthesis was proposed. However, subsequent detailed biochemical studies of the purine biosynthetic enzymes from various organisms has revealed that differences in the pathway are observed. In vertebrates, purines are synthesized from phosphoribosyl pyrophosphate (PRPP) according to the 10 steps originally proposed by Buchanan; however, in bacteria, yeast and fungi, an extra step is required1–7. This extra step involves the synthesis of the intermediate 4-carboxy-5-aminoimidazole ribonucleotide (CAIR, figure 1)3–5.
Figure 1.

Differences in de novo purine biosynthesis between humans and microbes.
De novo purine biosynthesis contains only one carbon-carbon bond forming reaction, namely in the conversion of 5-aminoimidazole ribonucleotide (AIR) into 4-carboxy-5aminoimidazole ribonucleotide (CAIR). In vertebrates, AIR is converted directly to CAIR by the enzyme AIR carboxylase2,4. ATP is not required for this conversion and AIR carboxylase uses CO2, not bicarbonate, as the one-carbon substrate2,4. Biochemical studies on purine biosynthesis in bacteria, yeast and fungi, demonstrate that two enzymes are required for the synthesis of CAIR1,3–5. The first enzyme, N5-carboxyaminoimidazole ribonucleotide (N5-CAIR) synthetase, converts AIR to a previously unrecognized intermediate N5-CAIR. The second enzyme, N5-CAIR mutase, converts N5-CAIR to CAIR. Sequence alignments of AIR carboxylase and the microbial enzymes reveal that AIR carboxylase is evolutionarily related to N5-CAIR mutase and structural studies demonstrate that the architecture of the active sites of each enzyme are nearly superimposable. Despite these conserved features, biochemical studies have shown that both enzymes are highly specific. AIR carboxylase cannot use N5-CAIR as a substrate and N5-CAIR mutase cannot directly use AIR and CO22,4.
Our early efforts on these enzymes lead to the discovery of the most potent inhibitor of G. gallus (chicken) AIR carboxylase known to date. This compound, 4-nitro-5-aminoimidazole ribonucleotide (NAIR), is an analog of CAIR and has a Ki of 0.34 nM8. NAIR displays slow, tight-binding kinetics with a 10,000 fold tighter affinity for the enzyme than CAIR8. In contrast, NAIR is a steady-state competitive inhibitor of fungal and bacterial N5-CAIR mutase with a Ki value similar to the Km for CAIR3,4.
The slow, tight-binding characteristic of NAIR with AIR carboxylase implicates a possible reactive intermediate or transition state attribute to the inhibitor design. Two proposed mechanisms for AIR carboxylase are show in Figure 2A. In the first (path a), the N3 of AIR is protonated followed by deprotonation of C4 to generate a N-ylide. Previous studies have shown that N3 is the primary site of protonation for both AIR and CAIR9. Enzyme-mediated formation of ylides has been postulated for orotidylate decarboxylase and thiamine dependent enzymes10,11. Attack on carbon dioxide generates the desired product, CAIR. The second mechanism (path b) involves direct attack of CO2 with assistance of the electron-rich exocyclic amine. The resulting tetrahedral intermediate, previously termed iso-CAIR, is then converted into CAIR via deprotonation of the C4 carbon6. As shown in figure 2B, NAIR could (in principle) have characteristics of a transition state for either mechanism. NAIR could mimic the transition state for the N-ylide mechanism (path a) if NAIR was protonated at N3. However, previous studies have shown that the pKa value for N3 of NAIR is 0.3 reducing the possibility that protonation occurs when bound at the enzyme active site12. Protonation of NAIR at the C4 position could generate a compound which would be nearly identical to the transition state shown for path b. UV measurements of NAIR binding to AIR carboxylase showed a minimal 10 nm shift of the λmax for NAIR when the inhibitor was used to titrate the AIR carboxylase active sites8. Density functional calculations for the C4 protonated NAIR (figure 2b) have shown that the predicted UV properties of the C4 protonated NAIR differ from what is experimentally observed (see Supporting Information). Thus, existing data offers little support for NAIR to be a mimic of the tetrahedral intermediate in path b.
Figure 2.

Proposed mechanisms for AIR carboxylase and the similarity between NAIR and the putative transition states for these enzymes. A. Two mechanisms proposed for AIR carboxylase. Path a. The ylide mechanism. Path b. The iso-CAIR mechanism. B. Comparison of transition states for two mechanisms outlined in A to NAIR.
To further understand the basic principles of NAIR binding to AIR carboxylase, we examined a series of azole nucleotide analogs of NAIR to establish the molecular features important for binding to AIR carboxylase6,7,13,14. In the studies presented here, we report the synthesis of a variety of heterocyclic analogs of NAIR and CAIR. We find that the inhibition of AIR carboxylase by NAIR is exquisitely sensitive to changes in the electronic structure of the compound suggesting that the electrostatic surface of the molecule plays a dominate role in inhibitor binding to AIR carboxylase.
Results
Compound Design
A structure-activity series for NAIR was designed to incorporate several key features. First, all compounds retained a phosphoribose since previous studies from our laboratory indicated that the phosphate group provided a substantial contribution to the binding (unpublished data). Second, only five member heterocyclic rings with ribosylation sites occurring at N1 were considered. This criterion was chosen to preserve the relative orientations of substituents at the C4 and C5 positions as is observed in both the product and substrate for the enzyme. This limited the heterocycle cores to a defined set of imidazoles, pyrroles, pyrazoles, and 1,2,3-triazoles (Figure 3). Our SAR also examined the role of a heteroatom at position 3 of the heterocycle. While this feature is inherited from the heterocycle choices listed above, the SAR allowed for probes of what role, if any, a basic nitrogen site plays in binding. The primary site of protonation for both the substrate and the product occurs at N3 of the heterocycle leading to the potential for this feature to be recognized by the enzyme9. If inhibitors could mimic or preserve this site, the potential for the recognition of protonation as a binding feature could be tested. The final feature explored was the nature of the substituents at C4 and C5. Here we limited the substituents at C4 to hydrogen, aldehyde, carboxylic acid, and nitro functional groups to mimic the product of the reaction or to mimic the parent inhibitor, NAIR. For C5, we used either hydrogen or an amino group (figure 3). By interchanging the substituents at C4 and C5, we could generate a number of possible combinations to explore the enzyme binding sites with these inhibitors. For example, replacement of a nitro group at C4 with hydrogen allowed us to address the possibility of a nitro binding pocket in the enzyme. If the placement of a nitro group at C4 was responsible for the majority of the binding energy of NAIR, then removal of the amino functionality at C5 should result in a compound with only a marginal decrease in Ki.
Figure 3.

Nucleoside and nucleotides involved in this project. Compounds 1 and 2 are the substrate and product of AIR carboxylase. Compound 3 is the most potent known inhibitor of the enzyme. Compounds 4–10 are the targets for this study.
Chemistry
We anticipated that the required ribonucleotides could be prepared from the corresponding ribonucleosides by phosphorylation (scheme I). The nucleoside precursors, 4-NIRs (11)15, 3-NPyRs (12)16, 4-NPzRs (13)17, and CAPzRs (14)18 are either known compounds or could be readily prepared from known compounds. Thus, their syntheses were carried out following the reported procedure. However, FAIRs (15), NAPzRs (16) and CATRs (17) have not been described previously and their syntheses are discussed here.
Scheme I.
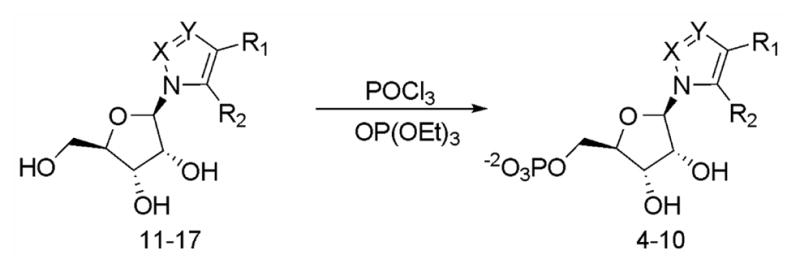
Phosphorylation of nucleosides
Synthesis of FAIRs (15)
Ribosylation of commercially available ethyl 4(5)-nitroimidazole-5(4)-carboxylate was accomplished using a stereospecific chlororibofuranose, 1719 to generate both regioisomers (Scheme II). The regioisomers were separated by silica gel chromatography and the 5-nitro regioisomer (18) was verified by conversion to AICAR, a known purine intermediate (see supplementary information). Reduction of 18 with DIBAL, followed by conversion of the nitro functionality to the free amine generated compound 20, the latter gave the free nucleoside 15 after deprotection.
Scheme II.

Synthesis of FAIRs (15) and NAPzRs (16).
Synthesis of NAPzRs (16)
Ribosylation of 5-amino-4-nitropyrazole or 5-acetylamino-4-nitropyrazole using standard Lewis acid mediated conditions failed to generate the desired compound. Reaction of the mercury salt of 5-amino-4-nitropyrazole and tribenzoylribfuranose chloride20 in heated toluene gave exclusively the 3-amino-4-nitropyrazole nucleoside.
The same reaction was done at lower temperature with the stereospecifically prepared ribosyl chloride 17 (scheme II). NMR analysis of the reaction products revealed that all four possible products had been formed in the reaction. The α- and β-isomers of the reaction could be separated by column chromatography and the regioisomeric mixture of β-isomers were deprotected using standard conditions. The desired compound, 16, could be isolated by selective crystallization of the nucleosides.
Synthesis of Triazole Nucleotides
Synthesis of 1,2,3-triazole nucleoside focused on a 1,3-dipolar cycloaddition reaction involving an active methylene compound and 1-azidoribofuranoside as reported for other triazole analogs of purine intermediates21 (Scheme III). t-Butyl cyanoacetate was chosen as the active methylene compound since the t-butyl group could be removed under acidic conditions which would prevent the facile Dimroth rearrangements observed with 5-amino-1,2,3-triazoles22. Condensation of 2,3-O-isopropylidene-5-O-tert-butylsilyl-ribofuranose azide, 2223, with t-butyl cyanoacetate under basic conditions failed to give the desired product. However, if the anion of the cyanoacetate was preformed in THF and then added to a solution of the ribofuranosyl azide in t-butyl alcohol under refluxing conditions, the required nucleoside 23 could be prepared in good yield. Compound 23 was deprotected smoothly using TFA to give desired nucleoside 17.
Scheme III.
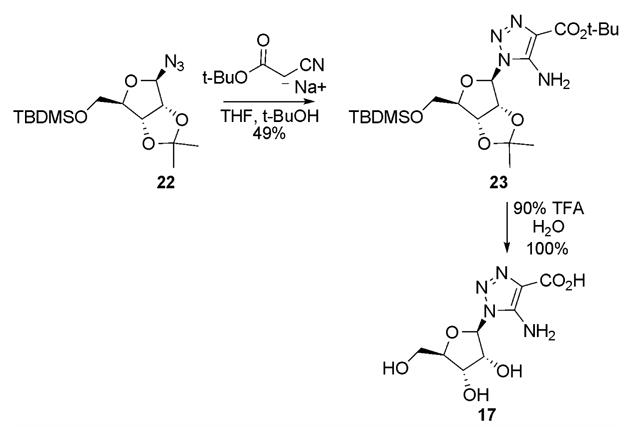
Synthesis of CATRs (17)
Phosphorylation
Phosphorylation of the azole ribonucleosdies with Yoshikawa procedure gave desired ribonucleoside monophosphates in various yields for each specific nucleoside and they were generally purified by anion exchange chromatography (scheme I)24–26. An efficient purification method was developed during the course of the work which used a boronate affinity gel. Since the main byproduct of the Yoshikawa reaction is phosphorylation of either the 2′ or 3′ hydroxyl, the boronate column can easily separate these from the desired 5′-monophosphates27.
Biochemistry
Inhibition of G. gallus AIR carboxylase
The nucleotides were examined for their ability to inhibit G. gallus AIR carboxylase using the CAIR decarboxylation assay (Table I). The high degree of sequence identity (>95%) between the G. Gallus enzyme and the human enzyme indicates that these data are also relevant to understanding how these compounds would interact with the human enzyme. All of the nucleotides investigated displayed a significant decrease (1,500–75,000 fold) in binding to the enzyme when compared to NAIR. Like NAIR, all compounds were competitive with CAIR, but unlike NAIR, none of the compounds displayed slow, tight binding kinetics. Because these compounds are competitive with CAIR, it is reasonable to assume that these compounds bind to the active site of the enzyme and that the relative Ki values relate to the differences in free energy.
Table 1.
Inhibition constants against G. gallus AIR carboxylasea.
| Number | Name | Ki (μM) or IC50 (μM) | ΔΔGc |
|---|---|---|---|
| 3 | NAIR | 0.00034 | 0 |
| 4 | NIR | 1.3 | 5.1 |
| 5 | FAIR | 62 | 7.4 |
| 6 | NpyR | >100b | >7.7 |
| 7 | NPzR | 22 | 6.8 |
| 8 | CAPzR | 6.0 | 6.0 |
| 9 | NAPzR | 0.39 | 4.3 |
| 10 | CATR | 2.8 | 5.5 |
Values determined using the CAIR decarboxylation assay.
IC50 value.
calculated by ΔGNAIR−ΔGInhibitor
The changes in affinity as measured by Ki values indicate that binding to the enzyme is remarkably sensitive to changes in the structure of the compounds. For example, the movement of the N3 nitrogen in NAIR to yield NAPzR results in a 1,000-fold decrease in Ki value. The same modification in converting CAIR into CAPzR results in a 3-fold difference in binding affinity and CAPR binds with greater affinity than CAIR. These data outlined in Table I indicate that there is no direct correlation between the presence or absence of certain functional groups and binding affinity.
CAPzR and CATR are not alternative substrates for G. gallus AIR carboxylase
Compounds CAPzR and CATR could also act as alternative substrates for AIR carboxylase. We tested both of these agents as substrates for AIR carboxylase. UV and HPLC analysis indicates that neither compound is an alternative substrate for the enzyme.
Electrostatic calculation of inhibitors of AIR carboxylase
Previous studies of binding of NAIR to AIR carboxylase revealed a 10 nm shift in the UV-vis spectrum with no change in the extinction coefficient for NAIR upon binding8. While the exact mechanism for this change is unknown, the observed shift does indicate that there are environment changes in the chromophore upon NAIR binding to the enzyme but are not consistent with alterations in the bonding framework. These observations suggest that desolvation and/or the electronic structure of NAIR may be critical for tight-binding. To probe this concept, we investigated two questions: a) does NAIR electronically resemble a reaction intermediate in the conversion of AIR to CAIR and b) do the compounds prepared in this report display electrostatic properties that are similar or dissimilar to these reaction intermediates?
Since the pKa value of N3 of NAIR is so low, we consider it unlikely that NAIR would be protonated in the active site12. This assumption is supported by x-ray structural data which indicate that there are no acidic amino acids near the proposed substrate binding site in AIR carboxylase. Thus, we chose to disregard the ylide mechanism for electrostatic calculations.
The molecular electrostatic potential (MEP) surface of the 1-methyl derivatives of CAIR and the proposed reaction intermediate iso-CAIR (figure 2A) were calculated using Gaussian 03 with the BL3yLP basis set. We performed these calculations in both the gas phase and in water. This was done because x-ray structural data on human AIR carboxylase revealed that the active site was remarkably solvent exposed, yet contained a hydrophobic pocket13. Therefore, it is likely that some parts of the molecule would be exposed to an environment with a higher dielectric constant while other would be in a low dielectric constant.
Minimization and calculation of the MEP for methyl-isoCAIR was difficult because of decarboxylation of the intermediate to generate methyl-AIR and CO2. This feature in reactivity has been observed by others6. To address this problem, we allowed the bond length to vary from 1.7 Å̇ and 2.0 Å̇ which resulted in converged, minimized structures. Computational investigations on carboxylases and decarboxylases by other groups have shown that the bond distance between the carbon and CO2 group is between 1.75–2.6 Å̇ in the transition state28–30. We interpret these results to indicate that the 1.7 Å̇ minimized structure provided a reasonable representation of the intermediate methyl-isoCAIR, while the 2.0 Å̇ structure likely resembles the transition state for its synthesis (iso-CAIRTS).
Regardless of the exact bond distance for the C4-CO2 bond, conversion of AIR into iso-CAIR results in the formation of an sp3 center altering the geometry around the C4 carbon. None of our compounds contain this feature of the intermediate and as discussed above, we feel that it is unlikely that the enzyme would catalyze protonation at this position. However, substantial electronic changes also occur during this conversion (Figure 3). The exocyclic amine of AIR becomes increasingly cationic with a developing negative charge on the carboxylic acid of iso-CAIR. Removal of the C4 proton from iso-CAIR to give CAIR results in substantial negative charge on the carboxylic acid with a decrease in the positive charge on the exocyclic amine (data not shown). In addition, there is a change in the conformation of the exocyclic amine during the conversion from AIR to iso-CAIR. The exocyclic amine rotates such that one of the hydrogens on the amine is within hydrogen bonding distance to the carboxylic acid. It is reasonable to anticipate that complementary chemical features would exist in the active site of AIR carboxylase.
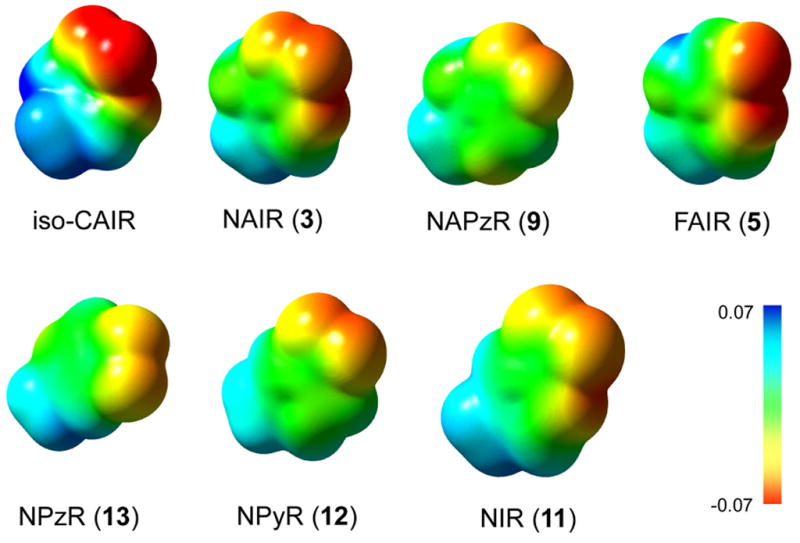
Using the calculations for iso-CAIR as a comparison, we calculated the MEP surfaces for NAIR and each of the charge neutral inhibitors using the same protocol as above. The results of these calculations are shown in figure 4. Visual examination of the MEP surfaces for methyl-iso-CAIR compared to the charge neutral inhibitors reveal that NAIR is the closest match to methyl-isoCAIR. Almost all other compounds display a substantial decrease in negative charge on the C4 substituent when compared to methyl-isoCAIR and methyl-NAIR. Methyl-FAIR is the only other compound besides NAIR that displays a negative charge on the substituents that is close to methyl-isoCAIR; however, methyl-FAIR contains only one electronegative atom at C4 compared with two in methyl-isoCAIR. Furthermore, NMR studies have demonstrated that FAIR undergoes pH-dependent structural changes through at least one, ill-defined, intermediate31. This introduces ambiguity into a direct comparison of the binding affinity for this compound with its electrostatic features since it is likely that FAIR exists in multiple configurations/conformations in solution. Methyl-NAIR contains a large negative charge on the N3 nitrogen of the ring, a feature which mimics that found in methyl-isoCAIR. The same trend holds for calculations done with water as the solvent, but the comparison between methyl-NAIR and methyl-isoCAIR is more striking (see supplemental information). A hypothesis consistent with the results of these calculations and the experimental data is that NAIR likely mimics either isoCAIR or the transition state going from AIR to isoCAIR.
Figure 4.

Comparison of NAIR to the proposed reaction intermediates in the AIR carboxylase reaction. A. The proposed first step of the reaction catalyzed by AIR carboxylase. AIR is converted to the tetrahedral intermediate isoCAIR. B. MEP surface of AIR, isoCAIRTS and iso-CAIR. Surfaces were calculated in the gas phase. The second set of MEP surfaces are the MEP surfaces looking down the C4 bond. C. Comparison of iso-CAIRTS and isoCAIR with NAIR. Notice the similarity between NAIR and iso-CAIRTS.
Discussion
The synthesis and evaluation of a variety of heterocyclic analogs of the AIR, CAIR and the slow, tight-binding inhibitor NAIR are reported here. All of the compounds show a decrease in binding affinity when compared to NAIR, despite the fact that only conservative structural analogs were designed. For example, moving a single nitrogen from N3 to N2 (NAIR vs. NAPzR) results in a 1000-fold decrease in binding affinity as measured by Ki. Larger structural changes such as removal of the exocyclic amine (NIR, NPyR, NPzR) or replacement of the nitro group with either an aldehyde or carboxylic acid group resulted in a greater loss of binding affinity. Overall, the range of ΔΔG values for the inhibitor analog binding to AIR carboxylase with respect to NAIR is 4 to >7.7 kcal/mole.
Why do all of these conservative substitutions in NAIR have large quantitative impacts on binding? We have previously suggested that NAIR is a transition state analog based upon the fact that this compound displays slow, tight binding kinetics8. However, NAIR is not geometrically similar to either the proposed transition state, or the intermediate iso-CAIR. In addition, other compounds, such as NAPzR, contain all of the necessary functional groups to mimic the transition state yet are only micromolar, competitive, steady-state inhibitors. Based upon the results presented here, it is reasonable to surmise that neither geometric similarity nor functional group presence is the sole determinate for tight binding to the G. gallus AIR carboxylase.
We examined the molecular electrostatic potential surface of the methyl derivatives of the compounds prepared in this study. Qualitative comparison of MEP surfaces to the proposed intermediate iso-CAIR and the transition state for its synthesis, revealed that NAIR closely matched both. NAIR, however, possessed less negative charge on the C4 nitro group than the C4 carboxyl of the intermediate. For the putative transition state model, methyl-isoCAIRTS, a strong electronic similarity between the transition state model and NAIR was clear. Other compounds, such as NIR and NAPzR also have some similarity to methyl-isoCAIRTS and these two compounds are the most potent, new nucleotides described in this work. Therefore, the data presented here indicate that AIR carboxylase is sensitive to the electronic character of the inhibitors.
The concept of complementary electrostatic recognition in enzyme inhibitor binding and catalysis has been discussed for several important enzyme systems. For example, work by the Schramm group has highlighted the importance of electrostatic complementarity in the design of transition state analogs for deaminases, nucleosidases and phosphorylases32–35. Electrostatics have been suggested to be important for the design of haptens for the creation of catalytic antibodies36 and an ultra high-resolution crystallographic study of aldose reductase has demonstrated electrostatic complementarity for cofactor binding37. However, a recent study on transition state analogs binding to ketosteroid isomerase showed that changes in the electrostatics of the compounds had small effects on binding to the enzyme38. This work suggests that electrostatics contributed only a small amount to the binding energy of the compound, while geometrical complementarity provides a more significant contribution. For AIR carboxylase, geometrical complementarity alone does not appear a major source of binding energy.
Here, we have proposed that NAIR is an electrostatic mimic of either iso-CAIR or the transition state leading to its synthesis. Previous studies have shown that AIR carboxylase is evolutionarily related to the bacterial enzyme N5-CAIR mutase which also produces CAIR. Structural analysis has shown that the two enzymes display a remarkable degree of identity in the active site and recent research on N5-CAIR mutase has also postulated the involvement of iso-CAIR in its mechanism6. These data indicate that NAIR should be a potent inhibitor of N5-CAIR mutase as well, yet NAIR binds 4.5 kcal/mol weaker to the mutase enzyme than AIR carboxylase6,39.
What accounts for this difference? Although both enzymes are related and likely proceed via the same intermediate, isoCAIR, the enzymes clearly recognize different substrates and are specific for their own reactions. From this perspective, it is not unexpected that each enzyme would have its own reaction coordinate with its own potential energy landscape. Even if both enzymes proceed via the iso-CAIR intermediate, it would not be without precedent that the energy barriers for iso-CAIR synthesis are different for each enzyme40,41. Since we have proposed that NAIR is an electrostatic mimic of either iso-CAIR or iso-CAIRTS, the differences in NAIR binding affinity may be related to the differences in the energy barriers for iso-CAIR synthesis for each enzyme.
Implications from the present work
Previous research on de novo purine biosynthesis has revealed that this pathway is different in prokaryotes and lower eukaryotes than in humans2,3,39. This difference has significant implications for the development of novel antimicrobial agents42. To develop selective antimicrobial compounds, agents targeting N5-CAIR mutase over AIR carboxylase must be created. In this report, we have shown here that AIR carboxylase is sensitive to the electrostatic nature of inhibitors and we surmise that compounds that mimic the electrostatic character of the transition state for the decarboxylation of N5-CAIR should be excellent inhibitors of bacterial and fungal N5-CAIR mutase. Investigations are currently underway to either validate or refute this hypothesis.
Experimental Section
Melting points were determined using either a Melt-Temp or Thomas Hoover apparatus and are uncorrected. Infrared spectra were acquired using a Perkin Elmer 1600 series FTIR. Mass spectra were acquired using a Kratos MS 50 spectrometer. NMR spectra were obtained on a Bruker 300 MHz or Varian 500 MHz spectrometer. All NMR spectra acquired in D2O were referenced to an external sample containing TSP for 1H, 85% H3PO4 for 31P. Elemental analysis were conducted at the Purdue Microanalysis facility and all values are ± 0.40% of the calculated value. All chemical shifts are listed in ppm (δ) with the following abbreviations: s-singlet, bs-broad singlet, d-doublet, t-triplet, q-quartet, dd-doublet of doublets, m-multiplet. Silica thin layer chromatography plates containing 254 nm fluorescent indicator were purchased from Sigma. Whatman cellulose thin layer chromatography plates with 254 nm fluorescent indicator and bulk cellulose (CF-11 or CC-31) were purchased from VWR. TLC plates were visualized by fluorescence, staining with I2, 5% phosphomolibdic acid (PMA) in ethanol or 5-sulfosalicilic acid/FeCl3. Dowex AG 50W-X8 was purchased from Sigma and the Dowex resin was treated with a 5 mM EDTA solution before exchanging to the desired ion. Ion chromatography resins, Q and DEAE Sepharose were purchased from Pharmacia. Affi-Gel boronate gel was purchased from Bio-Rad. Aqueous ion exchange chromatography were conducted using an Isco 2350 series HPLC pump with an Isco 2360 series gradient maker. The eluent was monitored at the indicated wavelengths using an Isco V4 series UV-vis detector. Analytical and preparative PRP-1 columns were purchased from Hamilton. Semi-preparative C18 columns were purchased from Ranin. All solvents, unless otherwise indicated are HPLC grade. 1-O-Acetyl-2,3,5-tribenzoyl-ribofuranoside was purchased from Sigma and used without purification. Triethyl phospate was purchased from Aldrich and distilled from BaO before use. Hexamethyldisilizane was purchased from Aldrich and distilled before use. Carbon tetrachloride, acetonitrile, methylene chloride and 1,2-dichloroethane were distilled from P2O5 before use. Hexamethylphosphoric acid triamide was purchased in technical grade, purified by vacuum distillation and stored at −20 °C. Hexamethylphosphorus triamide and tert-butyl alcohol were dried over CaH2 and distilled before use. Dry MeOH was produced by distillation from sodium. Diisopropylethylamine was purchased from Aldrich and distilled from KMnO4 and acetic anhydride before use. Toluene was dried over sodium/benzophenone and distilled before use. Phosphorous oxychloride, trifluoroacetic acid and phosphorus pentoxide was purchased from Fisher. Ammonium bicarbonate, ammonia, hydrogen chloride, Celite, NaH (60% in oil), trimethylsilyl chloride, t-butyldimethylsilyl chloride and mercuric chloride were purchased from Aldrich. All solvents used for NMR analysis were purchased from Cambridge Isotopes.
Ethyl-5-nitro-1-(5′-O-tert-butyldimethylsilyl-2′,3′-O-isopropylidene-β-D-ribofuranosyl)imidazole-4-carboxylate (18)
In a flame-dried 100 mL flask, 5′-O-tert-butyldimethylsilyl-2′,3′-isopropylidene-β-D-ribofuranose (2.7 g, 8.9 mmol) was dissolved in 20 mL of THF containing 1.3 mL of freshly distilled CCl4 (13.4 mmol). The solution was cooled to −78 °C and 2.1 mL of HMPT (11.6 mmol) was added slowly via an addition funnel. Occasional gel formation was reduced by momentary increases of the temperature to −40 ° C. To another flame-dried 50 ml flask, sodium hydride (0.36 g, 8.9 mmol) was added to a solution of ethyl-4(5)-nitroimidazole-5(4)-caboxylate (1.5 g, 8.1 mmol) in 10 mL of THF and 1 mL of HMPA. After stirring for 30 min, the sodium salt solution was added via an addition funnel to the above solution of the ribosyl chloride at −78 ° C. The reaction was complete after 30 min. The reaction was warmed to room temperature over a 30 min period, quenched with water, extracted with ethyl acetate and dried over MgSO4. The crude product was purified by flash chromatography (silica, 8:1:0.1 Hexane:EtOAc:MeOH Rf 0.2–0.3) to yield the 5-nitroisomer (1.26 g, 34 %) 18 and the 4-nitroisomer (1.32 g, 36 %) as yellow oils. 5-Nitro isomer 18: UV (λmax in MeOH) 306 nm; 1H NMR (300 MHz, CDCl3) δ 7.89 (1H, s, C2), 6.47 (1H, d, J=4.3 Hz, C1′), 5.04 (1H, t, J=5.8 Hz, C2′), 4.80 (1H, m, C3′), 4.59 (1H, m, C4′), 4.45 (2H, q, J=7.14 Hz, CH3CH2O−), 3.92 (1H, dd, J=2.1 and 11.2 Hz, C5′), 3.79 (1H, dd, J=2.1 and 11.2 Hz, C5′), 1.40 (3H, t, J=7.7 Hz, CH3CH2O−), 1.27, 1.06 (6H, 2s, isopropylidene), 0.94 (9H, s, (CH3)3CSi−), 0.11 (6H, s, (CH3)2Si−); 13C NMR (75 MHz, CDCl3) δ160.7, 136.1, 135.6, 132.8, 112.8, 90.8, 83.9, 81.6, 80.6, 65.4, 61.6, 25.4, 24.8, 23.9, 17.7, 13.6, −5.9, −6.1; HRMS (CI) calcd for C20H34N3O8Si 472.2115 [M+H]+, found 472.2124; 4-Nitro isomer: 1H NMR (300 MHz, CDCl3) 7.26 (1H, s, C2), 6.40 (1H, d, J=4.5 Hz, C1′), 5.04 (1H, t, J=4.4 Hz, C2′), 4.85 (1H, m, C3′), 4.59 (1H, bs, C4′), 4.39 (2H, q, j=7.0 Hz, CH3CH2O), 3.90 (1H, dd, J=2.8 and 11.1 Hz, C5′), 3.81 (1H, dd, J=2.8 and 11.1 Hz, C5′), 1.40, 1.27 (6H, 2s, isopropylidene), 1.36 (3H, t, J=7.0 Hz, CH3CH2O), 0.93 (9H, s, CH3)3CSi−), 0.12 (6H, s, (CH3)2Si−), 13 C NMR (75 MHz, CDCl3) δ 157.9, 147.9, 134.5, 117.0, 112.6, 89.9, 83.8, 81.8, 80.9, 64.8, 61.2, 25.4, 24.8, 23.9, 17.7, 13.3, −6.0 and −6.1 (Si(CH3)2); HRMS (CI) calcd for C20H34N3O8Si 472.2115 [M+H]+, found 472.2120.
5-Nitro-1-(5′-O-tert-butyldimethylsilyl-2′,3′-O-isopropylidene-β-D-ribofuranosyl) imidazole-4-carboxaldehyde (19)
Compound 18 (1 g, 2.1 mmol) was dissolved in 20 mL of diethyl ether and chilled to −78°C before the dropwise addition of 2.3 mL of diisobutylaluminum hydride (1M in hexane). The reaction, which was completed within 10 min, was quenched with 20 mL of tartaric acid solution (0.1 M, pH 6.8) and the mixture was stirred for 5 h at room temperature after which time the phases became separated. The phases were separated by a separatory funnel and the aqueous layer was extracted with diethyl ether. The combined organic layers was dried over MgSO4, concentrated and purified by flash chromatography to give the pure product (635 mg, 70 %) which required storage at −20 °C in the dark to prevent decomposition. UV (λmax in MeOH): 221, 304 nm; 1H NMR (300 MHz, CDCl3)δ 10.47 (1H, s, CHO), 8.04 (1H, s, C2), 6.58 (1H, d, J=4.8 Hz, C1′), 5.11 (1H, ψt, J=5.26 Hz, C2′), 4.82 (1H, m, C3′), 4.64 (1H, bs, C4′), 3.84 (1H, dd, J=2.1 and 11.1 Hz, C5′), 3.69 (1H, dd, J=1.2 and 11.1 Hz, C5′), 1.27, 1.00 (6H, 2s, isopropylidene), 0.96 (9H, s, (CH3)3CSi−), 0.13 (6H, s, (CH3)2Si−); 13C NMR (75 MHz, CDCl3) δ 183.5, 137.5, 113.3, 91.7, 84.5, 81.9, 81.0, 65.0, 25.8, 25.7, 24.2, −0.1 HRMS (ESI) calcd for C18H30N3O7Si 428.1853 [M+H]+, found 428.1857.
5-Amino-1-(5′-O-tert-butyldimethylsilyl-2′,3′-O-isopropylidene-β-D-ribofuranosyl) imidazole-4-carboxaldehyde (20)
To a 50 mL flask, 19 (600 mg, 1.4 mmol) was dissolved in 20 mL of THF and 10 % Pd/C (600 mg, 100 %, w/w) was added in one portion. The reaction flask was flushed with hydrogen gas and placed under a positive hydrogen atmosphere. After 2.5 h at room temperature, the starting material was consumed. The reaction mixture was filtered through a sintered glass filter funnel to remove the catalyst and concentrated in vacuo to give a pale yellow oil (464 mg, 83 %) which required no furthur purification. UV(λmax in MeOH) 215, 308 nm; 1H NMR (300 MHz, CDCl3) δ 9.72 (1H, s, CHO), 7.16 (1H, s, C2), 5.92 (1H, d, J=4.2 Hz, C1′), 5.72 (1H, bs, NH2), 4.87 (2H, m, C2′ and C3′), 4.44 (1H, bs, C4′), 3.91 (1H, dd, J=2.2 and 11.3 Hz, C5′), 3.73 (1H, dd, J=2.0 and 11.1 Hz, C5′), 1.39, 1.34 (6H, 2s, isopropylidene), 0.93 (9H, s, (CH3)3CSi−), 0.11(6H, s, (CH3)2Si−);13C NMR (75 MHz, CDCl3) δ 185.2, 145.9, 131.0, 122.9, 87.9, 83.4, 82.0, 80.5, 65.2, 25.7, 25.3, 24.0, 17.9, −5.6, −5.7; HRMS (CI) calcd for C18H32N3O5Si 428.1853 [M+H]+, found 428.1857.
5-Amino-1-β-D-ribofuranosylimidazole-4-carboxaldehyde (15)
To 20 (153 mg, 0.385 mmol), a precooled solution of 90 % trifluoroacetic acid (1 mL) was added and the reaction was stirred for 5 h at 0–4 °C. The reaction was poured into 50 mL of diethyl ether and the resulting yellow precipitate was collected by centrifugation and washed with 10 mL of diethyl ether (2X). The solid was dried in vacuo to remove trace amounts of trifluoroacetic acid, dissolved in 1% ammonia in MeOH and concentrated again to a yellow solid. The resulting solid (97.5 mg) was purified by flash chromatography (silica, 65:35:10 CHCl3:MeOH:H2O, lower phase, Rf 0.4) and recrystallized from methanol to yield yellow plate crystals (40 mg, 43 %). The material spontaneously decomposed to yield a dark red liquid after prolonged exposure to air and light at room temperature. UV (λmax in H2O) 212, 306 nm; 1H NMR (300 MHz, D2O) δ 9.41 (1H, s, CHO), 7.58 (1H, s, C2), 6.01 (1H, d, J=4.5 Hz, C1′), 4.82 (1H, ψt, J=4.2 Hz, C2′), 4.60 (2H, bs, C3′ and C4′), 3.92 (1H, dd, J=2.4 and 12.8 Hz, C5′), 3.76 (1H, dd, J=2.0 and 12.7 Hz, C5′); 13C NMR (75 MHz, D2O) δ 168.5, 118.3, 105.1, 69.2, 67.9, 55.3, 54.1, 44.8 HRFABMS (−Ve, DTT/DTE) calcd for C9H14N3O5 244.0933, found 244.0944
5-Amino-1-(β-D-ribofuranosyl)-4-nitropyrazole (16)
In a 250 mL flask, 3(5)-amino-4-nitropyrazole (4.10 g, 0.032 mol) was dissolved in a solution of 75 mL acetonitrile containing 25 mL of HMPA. To the clear solution, NaH (1.41 g, 60% in oil) was added in large portions and after 15 min a clear orange solution of the sodium salt formed, which was used immediately in the next reaction.
In a 500 mL flask, 5-O-tert-butyldimethylsilyl-2,3-O-isopropylidene-β-D-ribofuranose (5.01 g, 0.016 mol) was dissolved in THF (80 mL) containing CCl4 (2.4 mL, 0.24 mol). The reaction was cooled to −78 °C, HMPT (3.78 mL, 0.0208 mol) was added dropwise and the reaction was stirred for 1.5 h at −78 °C with occasional warming to −50 °C to liquefy. The reaction was removed from the bath and a solution of the above nitropyrazole sodium solution was cannulated into the reaction. The reaction is stirred at room temperature for 18 h before concentration in vacuo to a black oil. The oil was dissolved in 300 mL of EtOAc and the solution washed with 400 mL of water. The aqueous layer was extracted with EtOAc (2 × 300 mL), the combined organic layers washed with sat. NaHCO3 (2 × 300 mL) and treated with MgSO4 and activated carbon. The solution was filtered and concentrated in vacuo to a dark orange oil. The mixture was purified by flash silica chromatography using a gradient of 10:1 hexane/ethyl acetate to 2:1 hexane/ethyl acetate in 600 mL fractions. Concentration of the appropriate fractions gave a regioisomeric mixture of both the β and α nucleosides. β nucleosides: 1.615 g, 24%. TLC (silica, 2:1 hexane/ethyl acetate) Rf 0.65 α nucleosides: 3.316 g, 50%. TLC (silica, 2:1 hexane/ethyl acetate) Rf 0.49.
In a 250 mL flask, β-nucleosides (1.468 g, 3.54 mmol) were added along with 50 mL of 10% TFA in water and 10 mL of acetonitrile. The reaction was stirred at room temperature for 2 h, concentrated in vacuo and the residue was evaporated in vacuo from MeOH (3X) to remove TFA. The resulting solid was purified by flash silica chromatography using 9:1 CH2Cl2/MeOH. The appropriate fractions were concentrated to a yellow foam which was triturated with 1% MeOH in water. The remaining solid, which is enriched in the 5-amino isomer 16, was crystallized from water to give yellow needles (132 mg, 29%). HPLC (C18) 1% MeOH in water, flow rate 1.0 mL min−1 gave a retention time of 31 min; m.p. 172–174 °C; 1HNMR (300 MHz, DMSO-d6) δ 7.97 (1H, s, C3), 7.74 (2H, bs, NH2), 5.76 (1H, d, J=4.6 Hz, C1′), 4.44 (1H, t, J=4.6 Hz, C2′), 4.08 (1H, t, J=4.6 Hz, C3′), 3.86 (1H, q, J=4.6 Hz, C4′), 3.50 (1H, dd, J=4.6 and 11.8 Hz, C5′), 3.40 (1H, dd, J=4.65 and 11.75 Hz C5′); 13CNMR (75 MHz, DMSO-d6) δ 146.7, 135.5, 117.6, 89.4, 85.0, 72.5, 70.7, 62.1; HR FABMS (+ Ve, DTT/DTE) calcd 261.0835 found: 261.0830.
tert-Butyl 5-amino-1-(5-O-tert-butyldimethylsilyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-1,2,3-triazole-4-carboxylate (23)
In a 250 mL flask, tert-butyl cyanoacetate (1.86 g, 0.0132 mol) was dissolved in 30 mL of THF. NaH (0.581 g, 0.0145 mol, 60% in oil) was added in several large portions and the resulting solution was allowed to stir for 15 min. To the solution of 22 (1.0 g, 3.04 mmol) in 30 mL of dry tert-butyl alcohol was added the above mixture in one portion and the reaction was heated to reflux for 1.5 h. The reaction was poured into 200 mL of brine and extracted with EtOAc (300 mL, 200 mL). The organic layer was washed with sat. NaHCO3 (2 × 300 mL), dried with MgSO4 and concentrated in vacuo to a white solid. The compound was purified by silica flash chromatography using 5:1 hexane/ethyl acetate to give a white solid (0.696 g, 49%). TLC (9:1 hexane/ethyl acetate) Rf 0.25; m.p. 144–146 °C; 1HNMR (300 MHz, CDCl3) δ 6.0 (1H, d, J=2.2 Hz, C1′), 5.56 (2H, bs, NH2), 5.45 (1H, dd, J=2.2 Hz and 6.3 Hz, C2′), 4.81 (1H, dd, J=2.5 and 6.3 Hz, C3′), 4.27 (1H, m, C4′), 3.63 (1H, dd, J=4.4 and 11.2 Hz, C5′), 3.50 (1H, dd, J=4.5 and 11.3 Hz, C5′), 1.56 (9H, s, t-butyl ester), 1.54 (3H, s, C(CH3)2), 1.34 (3H, s, C(CH3)2), 0.83 (9H, s, (CH3)3CSi−), −0.01 (6H, s, (CH3)2Si−); 13CNMR (75 MHz, CDCl3) δ 162.1, 146.3, 122.6, 114.1, 92.5, 87.3, 82.7, 81.7, 80.6, 62.7, 28.4, 26.9, 25.9, 25.2, 18.4, −5.5; FABMS (+Ve,) 471.8 [M+H]+; Anal. (C21H34N4O6Si) C,H,N.
5-Amino-1-(β-D-ribofuranosyl)-1,2,3-triazole-4-carboxylate (17)
In a 50 mL flask, 23 (100 mg, 0.213 mmol) was dissolved in 10 mL of 90% TFA/H2O. The reaction was stirred at room temperature for 1 h, 10 mL of water was added and the reaction was evaporated in vacuo to dryness. The residue was dissolved in 100 mL of water and adjusted to pH 8.0 with NH4OH and applied to a Q-Sepharose column (2.5 × 5.0 cm, HCO3−). The column was washed with water and then with a gradient of 0–500 mM NH4HCO3 over 120 min at a flow rate of 4.0 mL min−1. The eluent was monitored at 260 nm, the appropriate fractions were collected and dried by lyophilization (3X) to a hydroscopic solid which was dried over P2O5 (65.1 mg, 98%). TLC (cellulose, 2:1 CH3CN/100 mM NH4HCO3) Rf 0.31; 1HNMR (300 MHz, D2O)δ 5.78 (1H, d, C1′), 4.75 (1H, dd, C2′), 4.31 (1H, dd, C3′), 4.07 (1H, m, C4′), 3.7 (1H, dd, C5′), 3.55 (1H, dd, C5′). 13C NMR (75 mHz, D2O) δ 172.8, 149.7, 124.6, 92.1, 88.3, 75.8, 73.4, 64.3. HRMS(FAB) calcd. for C8H16N5O6 278.1101 [M+NH4]+, found 278.109.
General phosphorylation procedure
The nucleoside was dissolved triethyl phosphate and freshly distilled POCl3 was added at 0 °C. The reaction mixture was stirred for 2 to 5 h and the mixture was poured into 100 mL of 100 mM NH4HCO3 and extracted with diethyl ether (3 × 200 mL). The aqueous solution was lyophilized to dryness. The resulting solid was purified either by anion exchange chromatography on DEAE and Q sepharose column followed by preparative HPLC (PRP-1) using 50 mM DIPEAA pH 7.0/10% CH3CN at a flow rate of 8.0 mL min−1 while monitoring at 280 nm, or by passing through boronate affinity gel column.
3-Nitro-1-(β-D-ribofuranosyl)imidazole 5′-Phosphate (4)
11 (50 mg, 0.204 mmol) was phosphorylated in 2.0 mL of OP(OEt) with POCl3 (190 μL, 2.04 mmol) for 4 h, and purified on a DEAE Sepharose column (HCO3−, 2.5 × 12 cm) to yield a white solid (38 mg, 58%). TLC(cellulose, 3:1 CH3CN/100 mM NH4HCO3) Rf 0.35; 1H NMR (300 MHz, D2O) δ 8.88 (1H, s, C2), 7.91 (1H, s, C5), 5.65 (1H, d, J=7.5 Hz, C1′), 4.35 (1H, m, C3′), 4.15 (1H, m, C4′), 3.85 (2H, m, C5′); 13C NMR (75 MHz, D2O) δ147.5, 136.5, 119.5, 90.9, 85.1, 76.1, 70.9, 61.7; 31P NMR (121 MHz, D2O) δ 0.56; HRMS (ESI) calcd for C8H13N3O9P 326.0389 [M+H]+, found 326.0393.
5-Amino-1-(β-D-ribofuranosyl)imidazole-4-carboxaldehyde 5′-phosphate (5)
15 (45 mg, 0.19 mmol) was phosphorylated in 1.0 mL of triethyl phosphate with POCl3 (86.2 μL, 0.92 mmol) for 4 h, and purified by Q Sepharose anion exchange chromatography to give yellow solid (47.8 mg, 75 %). UV (λmax in H2O) 215, 310 nm; ε 310 nm=5500 M−1 cm−1 at 25 ° C in 50 mM Tris Buffer, pH 8.0; 1H NMR (300 MHz, D2O) δ 9.37 (1H, s, CHO), 7.56 (1H, s, C2), 6.04 (1H, d, J=4.4 Hz, C1′), 4.69 (1H, ψt, J=3.1 Hz, C2′), 4.47 (2H, bs, C3′ and C4′), 3.94 (2H, m, C5′); 13C NMR (75 MHz, D2O) δ 186.5, 142.5, 137.5, 124.0, 88.2, 86.6 (d, Jc-p=7.5 Hz), 74.5, 73.7, 67.2 (d, Jc-p=4.3 Hz); 31P NMR (121 MHz, D2O) δ0.88; HRFABMS (−Ve, DTT/DTE) calcd for C9H15O8N3P 324.0597, found 324.0600
3-Nitro-1-(β-D-ribofuranosyl)pyrrole 5′-phosphate (6)
12 (50 mg, 0.2 mmol) was phosphorylated in 2.0 mL of triethyl phosphate with POCl3 (0.18 mL, 2 mmol) for 3 h and was purified by Q Sephrose anion exchange chromatography to give the desired product (47 mg, 64 %). UV(λmax in 0.1 N HCl) 287 nm; 1H NMR (300 MHz, CD3OD) δ 7.76 (1H, m, C2), 6.74 (1H, ψt, J=4.8 Hz, C4), 6.54 (1H, m, C5), 5.77 (d, J=4.7 Hz, C1′), 4.28 (1H, ψt, J=4.8 Hz, C2′), 4.22 (1H, m, C3′), 4.14 (1H, ψt, J=5.2 Hz, C4′), 3.69 (2H, m, C5′); 13C NMR (75 MHz, CD3OD) δ138.8, 125.5, 124.3, 107.7, 92.2, 86.1 (d, Jc-p=7.5 Hz), 74.3, 73.2, 66.5 (d, Jc-p=3.8 Hz);31P NMR(121 MHz, D2O) δ0.18; HRMS (FAB) calcd for C9H12N2O9P 323.0280 [M−H]−, found 323.0210.
4-Nitro-1-(-β-D-ribofuranosyl)pyrazole 5′-phosphate (7)
13 (35 mg, 0.14 mmol) was phosphorylated in 1.0 mL of triethyl phosphate with POCl3 (0.13 mL, 1.4 mmol) for 3 h and purified by Q-sepharose anion exchange chromatography to give the desired product (42 mg, 85 %). UV(λmax in 0.1 N HCl) 202, 273 nm; 1H NMR (300 MHz, D2O) δ 8.84 (1H, s, C3), 8.23 (1H, s, C5), 5.81 (1H, d, J=4.2 Hz, C1′), 4.56 (1H, ψt, J=5.1 Hz, C2′), 4.34 (1H, ψt, J=5.2 Hz, C3′), 4.24 (1H, dd, J=4.2 and 4.6 Hz, C4′), 3.96 (2H, m, C5′); 13C NMR (75 MHz, D2O) δ140.0, 132.8, 96.6, 87.87.0 (d, Jc-p=7.6 Hz),77.3, 73.0, 66.8 (d, Jc-p=3.4 Hz); 31P NMR (121 MHz, D2O) 0.61; HRMS (FAB): calcd for C8H11N4O9P 324.0233 [M−H]−, found 324.0263.
5-Amino-1-(β-D-ribofuranosyl)pyrazole-4-carboxylate 5′-phosphate (8)
14 (248 mg, 0.86 mmol) was phosphorylated in 15 mL of triethyl phosphate with POCl3 (403 μL, 4.33 mmol) for 1.5 h and was purified by boronate affinity gel column to give a white solid (249 mg, 72%). 65.6 mg of the ethyl ester (0.163 mmol) was dissolved in 10 mL of water containing NaOH (110 mg, 2.75 mmol). The reaction was heated to reflux for 4 h, cooled to room temperature and then diluted to 50 mL with water. The solution was applied to a Q Sepharose column (1.5 × 13 cm, HCO3−), the column washed with water and then eluted with a gradient of 0–500 mM NH4HCO3 over 30 min at a flow rate of 4.0 mL min−1. The eluent, monitored at 260 nm, was collected in fractions and the appropriate fractions were combined and dried by lyophilization (3X). The solid was dissolved in water (10 mL) and the solution passed through Chelex (NH4+) to remove metal contaminants (59 mg, 93%). TLC; 1HNMR (300 MHz, D2O) δ 7.51 (1H, s, C3), 5.56 (1H, d, J=6.7 Hz, C1′), 4.60 (1H, dd, J=5.5 and 6.7 Hz, C2′), 4.25 (1H, dd, J=3.3 and 5.5 Hz, C3′), 4.08 (1H, m, C4′), 3.76 (2H, m, C5′); 13CNMR (75 MHz, D2O) δ169.3, 152.9, 143.5, 98.1, 66.3, 71.6 (C5′), 73.2 (C2′ or C3′), 84.7 (C2′ or C3′), 90.1 (JC,P=8 Hz, C4′), 98.1 (C4), 143.5 (C3), 152.9 (C5), 169.3 (C=O); 31PNMR (121 MHz, D2O) δ 0.38; HRMS (ESI) calcd for C9H15N3O9P 340.0546 [M+H]+, found 340.0545.
5-Amino-1-(β-D-ribofuranosyl)-4-nitropyrazole 5′-phosphate (9)
16 was phosphorylated in 2.0 mL OP(OEt)3 with POCl3 (154 μL, 1.65 mmol) for 4 h and was purified on a Q Sepharose column to give a white solid (53 mg, 85%). TLC (cellulose, 7:3 CH3CN/100 mM NH4HCO3) Rf 0.32; 1HNMR (300 MHz, D2O) δ 8.36 (1H, s, C3), 6.10 (1H, d, J=5.8 Hz, C1′), 5.07 (1H, dd, J=5.8 Hz, J=5.5 Hz, C2′), 4.74 (1H, dd, J=3.9 Hz, J=5.5, C3′), 4.60 (1H, m, C4′), 4.32 (2H, m, C5′); 13CNMR (75 MHz, D2O) δ 64.2 (JC,P=4.3 Hz, C5′), 69.9 (C2′ or C3′), 71.3 (C2′ or C3′), 83.4 (JC,P=8.6 Hz, C4′), 118.3 (C4), 136.5 (C3), 147.0 (C5); 31PNMR (121 MHz, D2O) δ 1.23; HRMS (ESI): calcd for C8H14N4O9P 341.0498 [M+H]+, found 341.0500.
5-Amino-1-(β-D-ribofuranosyl)-1,2,3-triazole-4-carboxylate 5′-phosphate (10)
17 (19.7 mg, 0.071 mmol) was phosphaorylated in 2.0 mL of OP(OEt)3 with POCl3 (14 μL, 0.15 mmol) for 6 h and was purified first on a Q Sepharose column then on a semi-preparative PRP-1 column to give the desired product (2.7 mg, 20%). 1 HNMR (300 MHz, D2O) δ 5.81 (1H, d, J=5.9 Hz, C1′), 4.73 (1H, dd, J=5.3 and 5.9 Hz, C2′), 4.36 (1H, dd, J=3.8 and 5.3 Hz, C3′), 4.21 (1H, m, C4′), 3.91 (2H, m, C5′); 31PNMR (121 MHz, D2O)δ 0.94; HRMS (FABM+NH4+) calcd for C8H17N5O9P 358.0764, found 368.0760.
Testing for inhibition against AIR carboxylase
All compounds were analyzed as inhibitors as previously described. Briefly, the inhibitor (10 nM to 100 μM) was added to a cuvette containing 50 mM Tris-HCl, 0.5 mM EDTA, pH 8.0 and CAIR (10–100 μM). The reaction was initiated by the addition of 23–46 ng of enzyme and the change in absorbance at 260 nm was monitored. The Ki value was determined from a series of 1/v vs.1/S plots using the program Enzyme Kinetics (Trinity Software). For each compound, three different inhibitor concentrations were used based upon preliminary experiments to determine the IC50 value.
Molecular electrostatic potential calculations
The geometries of each compound were determined by minimization using Gaussian 03 with the B3LyP 6-31G* basis set. The electrostatic potential was calculated from the wave functions generated for the minimized structure using the CUBE option from Gaussian. The color map for each compound was set to the same range of charges (−0.07 to 0.07 in gas phase; −0.1 to 0.1 for water). The range was established to maximize contrast between the compounds.
Supplementary Material
Figure 5.
MEP surfaces for charge neutral compounds calculated in the gas phase.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Meyer E, Leonard NJ, Bhat B, Stubbe J, Smith JM. Biochemistry. 1992;31:5022–32. doi: 10.1021/bi00136a016. [DOI] [PubMed] [Google Scholar]
- 2.Firestine SM, Davisson VJ. Biochemistry. 1994;33:11917–26. doi: 10.1021/bi00205a030. [DOI] [PubMed] [Google Scholar]
- 3.Firestine SM, Misialek S, Toffaletti DL, Klem TJ, Perfect JR, Davisson VJ. Arch Biochem Biophys. 1998;351:123–34. doi: 10.1006/abbi.1997.0512. [DOI] [PubMed] [Google Scholar]
- 4.Firestine SM, Poon SW, Mueller EJ, Stubbe J, Davisson VJ. Biochemistry. 1994;33:11927–34. doi: 10.1021/bi00205a031. [DOI] [PubMed] [Google Scholar]
- 5.Mueller EJ, Meyer E, Rudolph J, Davisson VJ, Stubbe J. Biochemistry. 1994;33:2269–78. doi: 10.1021/bi00174a038. [DOI] [PubMed] [Google Scholar]
- 6.Hoskins AA, Morar M, Kappock TJ, Mathews II, Zaugg JB, Barder TE, Peng P, Okamoto A, Ealick SE, Stubbe J. Biochemistry. 2007;46:2842–2855. doi: 10.1021/bi602436g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Constantine CZ, Starks CM, Mill CP, Ransome AE, Karpowicz SJ, Francois JA, Goodman RA, Kappock TJ. Biochemistry. 2006;45:8193–208. doi: 10.1021/bi060465n. [DOI] [PubMed] [Google Scholar]
- 8.Firestine SM, Davisson VJ. J Med Chem. 1993;36:3484–6. doi: 10.1021/jm00074a033. [DOI] [PubMed] [Google Scholar]
- 9.Bhat B, Groziak MP, Leonard NJ. J Am Chem Soc. 1990;112:4891–4897. [Google Scholar]
- 10.Jordan F. Nat Prod Rep. 2003;20:184–201. doi: 10.1039/b111348h. [DOI] [PubMed] [Google Scholar]
- 11.Miller BG, Wolfenden R. Annu Rev Biochem. 2002;71:847–85. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 12.Blazevic N, Kajfez F, Sunjic V. J Heterocyclic Chem. 1970;7:227–229. [Google Scholar]
- 13.Li SX, Tong YP, Xie XC, Wang QH, Zhou HN, Han Y, Zhang ZY, Gao W, Li SG, Zhang XC, Bi RC. J Mol Biol. 2007;366:1603–14. doi: 10.1016/j.jmb.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 14.Mathews II, Kappock TJ, Stubbe J, Ealick SE. Structure. 1999;7:1395–406. doi: 10.1016/s0969-2126(00)80029-5. [DOI] [PubMed] [Google Scholar]
- 15.Chavis C, Grodenic F, Imbach JL. Eur J Med Chem. 1979;14:123–131. [Google Scholar]
- 16.Harki DA, Graci JD, Korneeva VS, Ghosh SKB, Hong Z, Cameron CE, Peterson BR. Biochemistry. 2002;41:9026–9033. doi: 10.1021/bi026120w. [DOI] [PubMed] [Google Scholar]
- 17.Barascut JL, Tamby C, Imbach JL. Journal of Carbohydrates, Nucleosides, Nucleotides. 1974;1:77–84. [Google Scholar]
- 18.Sanghvi YS, Larson SB, Robins RK, Revankar GR. J Chem Soc, Perkin Trans 1. 1990;11:2943–2950. [Google Scholar]
- 19.Rosemeyer H, Seela F. Helv Chim Acta. 1988;71:1573–1785. [Google Scholar]
- 20.Kissman HM, Child RG, Weiss MJ. J Am Chem Soc. 1957;79:1185–1188. [Google Scholar]
- 21.Acevedo OL, Krawczyk SH, Townsend LB. J Org Chem. 1986;51:1050–1058. [Google Scholar]
- 22.Wamhoff H. In: Comprehensive Heterocyclic Chemistry. Katritzky AR, Rees CW, editors. Pergamon Press; Oxford: 1984. pp. 669–732. [Google Scholar]
- 23.Camarasa MJ, Alonso R, De Las Heras FG. Carbohydr Res. 1980;83:152–156. [Google Scholar]
- 24.Yoshikawa M, Kato T, Takenishi T. Tetrahedron letters. 1967;50:5065–5068. doi: 10.1016/s0040-4039(01)89915-9. [DOI] [PubMed] [Google Scholar]
- 25.Yoshikawa M, Kato T. Bulletin of the Chemical Society of Japan. 1967;40:2849–2853. doi: 10.1246/bcsj.40.2849. [DOI] [PubMed] [Google Scholar]
- 26.Yoshikawa M, Kato T, Takenishi T. Bull Chem Soc Jpn. 1969;42:3505–3508. [Google Scholar]
- 27.Wu W, Bergstrom DE, Davisson VJ. J Org Chem. 2003;68:3860–5. doi: 10.1021/jo020745i. [DOI] [PubMed] [Google Scholar]
- 28.Acevedo O, Jorgensen WL. J Org Chem. 2006;71:4896–902. doi: 10.1021/jo060533b. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Dong H, Li S, He H. J Phys Chem B. 2005;109:18664–18672. doi: 10.1021/jp052802s. [DOI] [PubMed] [Google Scholar]
- 30.Mauser H, King WA, Gready JE, Andrews TJ. J Am Chem Soc. 2001;123:10821–9. doi: 10.1021/ja011362p. [DOI] [PubMed] [Google Scholar]
- 31.Youn H. PhD Dissertation. Purdue University; 1997. [Google Scholar]
- 32.Singh V, Lee JE, Nunez S, Howell PL, Schramm VL. Biochemistry. 2005;44:11647–59. doi: 10.1021/bi050863a. [DOI] [PubMed] [Google Scholar]
- 33.Evans GB, Furneaux RH, Lenz DH, Painter GF, Schramm VL, Singh V, Tyler PC. J Med Chem. 2005;48:4679–89. doi: 10.1021/jm050269z. [DOI] [PubMed] [Google Scholar]
- 34.Ehrlich JI, Schramm VL. Biochemistry. 1994;33:8890–6. doi: 10.1021/bi00196a005. [DOI] [PubMed] [Google Scholar]
- 35.Kline PC, Schramm VL. J Biol Chem. 1994;269:22385–90. [PubMed] [Google Scholar]
- 36.Barbany M, Gutierrez-de-Teran H, Sanz F, Villa-Freixa J, Warshel A. Chembiochem. 2003;4:277–85. doi: 10.1002/cbic.200390048. [DOI] [PubMed] [Google Scholar]
- 37.Muzet N, Guillot B, Jelsch C, Howard E, Lecomte C. Proc Natl Acad Sci U S A. 2003;100:8742–7. doi: 10.1073/pnas.1432955100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. PLoS Biol. 2006;4:e99. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Firestine S. Purdue. 1995 [Google Scholar]
- 40.Lewandowicz A, Schramm VL. Biochemistry. 2004;43:1458–68. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 41.Lewandowicz A, Ringia EA, Ting LM, Kim K, Tyler PC, Evans GB, Zubkova OV, Mee S, Painter GF, Lenz DH, Furneaux RH, Schramm VL. J Biol Chem. 2005;280:30320–8. doi: 10.1074/jbc.M505033200. [DOI] [PubMed] [Google Scholar]
- 42.Donovan M, Schumuke JJ, Fonzi WA, Bonar SL, Gheesling-Mullis K, Jacob GS, Davisson VJ, Dotson SB. Infect Immun. 2001;69:2542–8. doi: 10.1128/IAI.69.4.2542-2548.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.