

Abstract
Dopamine (DA)-dependent corticostriatal plasticity is thought to underlie incremental procedural learning. A primary effector of striatal DA signaling is cAMP, yet its role in corticostriatal plasticity and striatum-dependent learning remains unclear. Here, we show that genetic deletion of a striatum-enriched isoform of adenylyl cyclase, AC5 knock-out (AC5KO), impairs two forms of striatum-dependent learning and corticostriatal synaptic plasticity. AC5KO mice were severely impaired in acquisition of a response strategy in the cross maze, a striatum-dependent task requiring a correct body turn to find a goal arm. In addition, AC5KO mice were impaired in acquisition of a motor skill, as assessed by the accelerated rotarod. Slice electrophysiology revealed a deficit in corticostriatal long-term depression (LTD) after high-frequency stimulation of tissue from AC5KO mice. LTD was rescued by activation of either presynaptic cannabinoid type 1 (CB1) receptors or postsynaptic metabotropic glutamate receptors (mGluRs), suggesting a postsynaptic role of AC5–cAMP, upstream of endocannabinoid release. In striatopallidal-projecting medium spiny neurons, DA D2 receptors are negatively coupled to cAMP production, and activation of these receptors is required for endocannabinoid release and corticostriatal LTD. Recordings from striatopallidal neurons indicated that this is mediated by AC5, because coactivation of D2 and mGluRs could induce LTD in wild-type but not in AC5KO neurons. To further examine the role of cAMP in corticostriatal plasticity, we elevated cAMP in striatal neurons of wild-type mice via the recording electrode. Under these conditions, corticostriatal LTD was eliminated. Together, these data suggest an AC5–cAMP–endocannabinoid–CB1 signaling pathway in corticostriatal plasticity and striatum-dependent learning.
Introduction
In humans, the dorsal striatum plays a critical role in procedural learning, the memory for motor skills and stimulus-response habits, that is dissociable from hippocampus-dependent declarative learning, the memory for facts and events (Packard et al., 1989; Packard and White, 1991; Packard and McGaugh, 1996; Packard and Knowlton, 2002; Schroeder et al., 2002; Squire, 2004; Yin et al., 2004, 2005). In rodents, lesion studies have indicated that striatum-dependent learning of stimulus-response habits is dissociable from hippocampus-dependent learning of spatial information (Packard and McGaugh, 1996; Packard and Knowlton, 2002; Yin and Knowlton, 2006). This dissociation has been documented through use of a cross maze, in which inactivation of the dorsal striatum impairs the animal's ability to learn to make the correct body turn at a choice point to find the goal arm (Packard and McGaugh, 1996). The dorsal striatum receives dense glutamatergic innervation from cortical areas, as well a dopaminergic input from midbrain nuclei. The interplay between dopamine (DA) and glutamate input to medium spiny neurons (MSNs) of the dorsal striatum has provided the basis for models of reinforcement learning that suggest DA-dependent plasticity at corticostriatal synapses is necessary for striatum-dependent learning (Calabresi et al., 1992; Schultz et al., 1997; Reynolds et al., 2001; Reynolds and Wickens, 2002). However, few studies have addressed the role of DA-dependent intracellular signaling pathways that underlie this form of striatum-dependent learning and how they contribute to DA-dependent corticostriatal plasticity.
In brain slice recordings, the predominant form of synaptic plasticity seen in the dorsolateral striatum is long-term depression (LTD), which requires DA D2 receptor activation, increases in intracellular calcium, and retrograde release of endocannabinoids that inhibit glutamate release via presynaptic cannabinoid type 1 (CB1) receptors (Calabresi et al., 1994, 2007; Gerdeman et al., 2002; Kreitzer and Malenka, 2007). Although modulation of intracellular cAMP content is a direct downstream effect of DA receptor activation (Neve et al., 2004), the role of cAMP in corticostriatal plasticity has remained essentially uninvestigated. Two downstream targets of cAMP-dependent protein kinase (PKA), cAMP response element binding protein (CREB), and the dopamine and cAMP-regulated phosphoprotein 32 kDa (DARPP-32) have been implicated in corticostriatal LTD (Calabresi et al., 2000; Pittenger et al., 2006), but these proteins are also regulated by non-cAMP-dependent signaling pathways (Kang et al., 2001; Svenningsson et al., 2004), leaving no direct evidence for a contribution of cAMP to corticostriatal LTD.
The highly conserved cAMP second-messenger system has been implicated in synaptic plasticity and learning in many different organisms (Kandel, 2001). The enzyme that catalyzes cAMP production, adenylyl cyclase (AC), has at least nine membrane-bound isoforms that have different expression patterns and regulatory properties (Cooper et al., 1998; Hanoune and Defer, 2001; Wang and Storm, 2003). Calcium/calmodulin-dependent AC isoforms have been extensively studied and are essential for induction of long-term potentiation (LTP) in the hippocampus, because they couple glutamate-mediated increases in intracellular calcium to cAMP accumulation (Xia et al., 1993; Wu et al., 1995; Matsuoka et al., 1997; Wong et al., 1999; Wang and Storm, 2003; Nicol et al., 2005). The dorsal striatum expresses high levels of AC5 (Matsuoka et al., 1997), a primary downstream effector of DA receptor signaling in the striatum. Mice that lack AC5 expression [AC5 knock-outs (AC5KO)] show severely impaired striatal D1 and D2 receptor modulation of cAMP signaling (Iwamoto et al., 2003). In addition, recent studies have indicated that AC5KO mice are unable to predict reward delivery in a DA-dependent Pavlovian conditioning task (Kheirbek et al., 2008). Thus, the AC5KO mouse provides an opportunity to test the contribution of cAMP to corticostriatal plasticity and dorsal striatum-dependent learning. Here, we demonstrate a critical role for AC5 in a striatum-dependent learning task, response learning in the cross maze. This deficit in striatum-dependent learning was confirmed in the accelerated rotarod, a motor skill learning task correlated previously with plasticity in the dorsal striatum. In addition, we reveal a role for AC5 and cAMP signaling in corticostriatal LTD by regulating D2-mediated retrograde release of endocannabinoids (Costa et al., 2004; Dang et al., 2006; Yin et al., 2009).
Materials and Methods
Mice
AC5KO mice were generated as described previously (Iwamoto et al., 2003) and backcrossed to C57BL/6 for nine generations. All mice tested were 8–12 weeks of age, and behavioral experiments were conducted during the light phase. Wild-type (WT) littermates were used as controls, and independent groups were used for each experiment. Drd2–enhanced green fluorescent protein (EGFP) bacterial artificial chromosome transgenic mice were obtained from the Mutant Mouse Regional Resource Center. Drd2–EGFP mice were crossed to AC5KO background to generate AC5KO/Drd2–EGFP and WT/Drd2–EGFP littermate controls for electrophysiological experiments. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Chicago.
In situ hybridization
Mice were decapitated, and brains were fresh frozen on dry ice. Coronal sections (20 μm) were cut and thaw mounted on slides and stored at −80° until hybridization. Sections were fixed in 4% paraformaldehyde for 10 min and digested with proteinase K, followed by acetylation for 10 min and prehybridization for 2 h at 65°C in a buffer containing 50% formamide, 20× SSC, 5× Denhardt's solution, 250 μg/ml tRNA, and 500 μg/ml fish sperm DNA. For radiolabeled probe, 106 cpm [33P]rUTP probe per 50 μl of hybridization buffer was used for overnight incubation at 65°C. Signal was visualized after 48 h development on Kodak BioMax MR film. For digoxigenin (DIG)-labeled probes, sections were incubated with 400 ng/ml probes overnight at 65°C. Slides were washed and incubated in 1:5000 dilution of anti-DIG–alkaline phosphatase (AP) antibody (Roche) for 2 h and then visualized using nitroblue-tetrazolium-chloride (NBT)/5-bromo-4-chlor-indolyl-phosphate (BCIP) staining for alkaline phosphatase. For double labeling in situ, sections were hybridized with DIG and biotin-labeled mRNA probes simultaneously. After washing, the sections were incubated in 1:5000 dilution anti-biotin–AP antibody, followed by visualization with Fast Red (Sigma). Slides were photographed, then washed in 0.1 m glycine, pH 2.2, and incubated in anti-DIG–AP, followed by development in NBT/BCIP solution.
AC5 and preproenkephalin (ENK) probes were isolated from a mouse brain cDNA library using the following primer pairs: AC5, 5′CTGGAGGAGCGTCGAGGAAA3′ and 5′GGTCCTGGGAGTTGGTGTGC3′; ENK, 5′GGATGCAGATGAGGGAGACA3′and 5′AAACCCGTGACCCCAACTGC3′. The cDNA fragment was cloned into PCRII-TOPO vector (Invitrogen) and linearized, and antisense probe was transcribed using the appropriate RNA polymerase.
Behavioral experiments
Water cross maze.
Experiments were conducted in a water tank (white walls, 100 cm in diameter), with white Plexiglas walls inserted in the maze to make alleyways. Water was at room temperature, and white nontoxic paint (FunStuff) was added to obscure the hidden platform. A black curtain surrounded the maze to reduce spatial cues. The walls of the maze were rotated every 10 trials to further reduce the contribution of spatial cues. A trial was started by placing the mouse in the start arm facing the wall of the tank. Once the mouse made a right or left choice, a divider closed off the arm, and the mouse was allowed to either stand on the platform for 15 s (correct choice) or explore the arm of the maze without the platform for 15 s (incorrect choice). Mice were then removed from the maze and placed under a heat lamp for 30 s before beginning the next trial. Trials were started randomly from either the north or the south end of the maze with the goal arm always to the right of the start arm during initial acquisition and to the left of the start arm during reversal learning. Correct or incorrect choices were recorded.
Morris water maze.
Experiments were conducted as described previously (Vorhees and Williams, 2006). Experiments were conducted in a water tank (white walls, 100 cm in diameter), with white Plexiglas walls inserted in the maze to make alleyways. Water was at room temperature, and white non-toxic paint (FunStuff) was added to obscure the hidden platform. Around the tank, white paper cutouts of stars and arrows were placed on the black surrounding curtain to serve as spatial cues. Mice were first trained for 2 d, three trials a day with a visible platform (8 cm diameter) for habituation purposes and to determine any swimming deficits. After habituation, training trials began. A 1 cm submerged platform (8 cm diameter) was placed in one quadrant. During training, mice were started from random start positions, and latency to reach the hidden platform was measured. Mice were given three training trials a day for 7 consecutive days. All measurements were made using a live camera and EthoVision (Noldus) software. Twenty-four hours after the last days of training, mice were given a 1 min probe test (Vorhees and Williams, 2006) in which the platform was removed, and time spent in the target quadrant was measured by the EthoVision software.
Accelerating rotarod.
Mice were placed on a rotarod apparatus (Columbus Instruments) accelerating from 4 to 40 rpm in 5 min. Trials began by placing the mouse on the rod and beginning rotation. Each trial ended when the mouse fell off the rod, and latency was recorded. Mice were tested for four trials a day (1 min intertrial interval) for 3 consecutive days.
Locomotor activity.
Each mouse was placed in an acrylic open-field chamber 40 cm long × 40 cm wide × 37 cm high (Med Associates). The floor of the chamber is white, and the sides were transparent. Each chamber was surrounded by a black drop cloth obscuring views beyond the chamber. Illumination of open field was set to 16 lux. No background noise was provided. Infrared beams recorded the animal's location and path (locomotor activity) and recorded distance traveled. Data were collected in 5 min bins during 60 min trials.
Electrophysiological and electrochemical recordings
Slice preparation.
Coronal brain slices (250 μm thick) were obtained from 6- to 10-week-old mice. The mice were anesthetized with isoflurane (Abbott Laboratories) and rapidly decapitated. Their brains were transferred to cold, sucrose–artificial CSF (ACSF) containing the following (in mm): 200 sucrose, 25 NaHCO3, 20 glucose, 10 ascorbic acid, 2.5 KCl, 2.5 CaCl2, 1 MgCl2, and 1 NaH2PO4, pH 7.4 (saturated with 95% O2 and 5% CO2) (Fagen et al., 2007). Striatal brain slices were made on a vibratome (VT100S; Leica). Slices were incubated for at least 1 h in a bath circulated at 20 ml/min with normal, 32°C ACSF containing the following (in mm): 125 NaCl, 25 NaHCO3, 20 glucose, 2.5 KCl, 2.5 CaCl2, 1 MgCl2, 1 NaHCO3, and 1 ascorbic acid, pH 7.4 (saturated with 95% O2 and 5% CO2) (Fagen et al., 2007). During recording, slices were superfused (2 ml/min) with this same ACSF at ∼34°C but without the ascorbic acid and with 10 μm (−)bicuculline methiodide to block GABAA receptor-mediated synaptic responses. DNQX at 10 μm was added to the ACSF for experiments testing intrinsic membrane excitability. Tetrodotoxin at 1 μm was included in the ACSF for the recording of miniature EPSCs (mEPSCs).
Electrophysiology.
Whole-cell voltage-clamp recordings from MSNs located in the dorsal lateral striatum were obtained under visual control on a differential interference contrast, upright microscope with infrared illumination (Axioskop; Carl Zeiss). Recordings were obtained using 2.5–6 MΩ resistance pipettes made from borosilicate glass capillary tubing (G150-4; Warner Instruments) that was pulled on a Flaming/Brown micropipette puller (model P-97; Sutter Instruments). To test membrane excitability and mEPSCs, recording pipettes were filled with an internal solution containing the following (in mm): 154 K-gluconate, 1 KCl, 1 EGTA, 10 HEPES, 10 glucose, 5 ATP, and 0.1 GTP, brought to pH 7.4 with KOH (Mansvelder and McGehee, 2000). To test stimulated synaptic currents, recording pipettes were filled with an internal solution containing the following (in mm): 120 CsMeSO3, 10 tetraethylammonium-Cl, 10 HEPES, 10 sucrose, 5 NaCl, 5 QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine], 4 ATP-Mg2, 1.1 EGTA, and 0.3 GTP-Na2, brought to pH 7.25 with CsOH (Wang et al., 2006). Currents were measured using a Multiclamp 700A amplifier (2 kHz low-pass Bessel filter) with a DigiData 1322A interface (10 kHz digitization) and pClamp 9.2 software (Molecular Devices). MSNs in the dorsal lateral striatum, identified by their morphology and hyperpolarized resting membrane potential, were voltage clamped at −80 mV, unless noted otherwise. A bipolar tungsten electrode with a 500 μm tip separation (FHC) was placed in the white matter 200–300 μm dorsal to the recording electrode. Test stimuli (two pulses 25 ms apart) were delivered through a Master-8 stimulator (A.M.P.I.) every 20 s. Stimulus intensity ranged from 0.1 to 1 mA, and pulse duration ranged from 50 to 400 μs. Series resistance in the recorded cells was monitored online from a 5 mV, 20 ms depolarizing pulse delivered 60 ms after the second of the paired stimuli. The series resistance was 60% compensated, and cells were discarded if this varied >20% or increased to >30 MΩ during the course of the experiment. After evoked EPSC amplitudes remained relatively stable for 10 min, high-frequency stimulation (HFS) was delivered in some experiments, which consisted of four 1 s, 100 Hz trains delivered every 10 s, during which the postsynaptic cell was depolarized to 0 mV. EPSC amplitudes are expressed as a percentage of the baseline values, mean ± SEM. For prevalence data, plasticity was determined by paired t tests comparing the 10 min baseline period with the last 10 min of recording.
Electrochemistry.
Carbon fiber microelectrodes were fabricated by threading a 7-μm-diameter carbon fiber (Fortafil Fibers) through borosilicate glass capillary tubing (G150-4; Warner Instruments) that was then pulled on a Flaming/Brown micropipette puller (model P-97; Sutter Instruments). The carbon fiber was subsequently cut to restrict the length of the exposed fiber in the slice to ∼50 μm. Recording electrodes were placed in the dorsal striatum ∼250 μm from a bipolar tungsten electrode with a 500 μm tip separation (FHC). Dopamine release was evoked by electrical stimulation (400 μA, 1 ms) delivered every 2 min with an Iso-Flex stimulus isolator triggered by a Master-8 pulse generator (A.M.P.I.). Currents were measured using a Multiclamp 700A amplifier (10 kHz low-pass Bessel filter) with a DigiData 1322A interface (50 kHz digitization) and pClamp 9.2 software (Molecular Devices). For fast-scan cyclic voltammetry (FSCV), the electrode voltage was ramped from the baseline voltage of −400 to +1000 mV and then back again at 300 V/s versus Ag/AgCl at 100 ms intervals. Background-subtracted FSCV was used to visualize oxidation and reduction currents, allowing for identification of the oxidized substance and for calibration with 5 μm dopamine at the end of each experiment.
Results
Expression of AC5 in the adult striatum
We first confirmed the enrichment of AC5 mRNA in the adult mouse striatum using in situ hybridization with 33P-labeled probes (Fig. 1A,B). AC5 mRNA expression was very high in the dorsal striatum, nucleus accumbens, and olfactory tubercle (Fig. 1A). AC5 message was detected at low levels in hippocampus and cortex (Fig. 1B). AC5 levels were low in other regions of the basal ganglia, including the globus pallidus and substantia nigra (data not shown). To examine AC5 expression in the dorsal striatum in more detail, we conducted double-labeling in situ hybridization for AC5 and ENK to ask whether AC5 was differentially expressed in subsets of MSNs in the striatum (Fig. 1C,D). ENK is expressed in neurons projecting via the striatopallidal indirect pathway to the substantia nigra and is absent in those neurons projecting via the striatonigral direct pathway (Beckstead and Kersey, 1985; Gerfen and Young, 1988). AC5 mRNA expression was seen in both ENK-positive and ENK-negative cells (Fig. 1C,D), suggesting that AC5 was expressed in both subtypes of striatal projection neurons.
Figure 1.
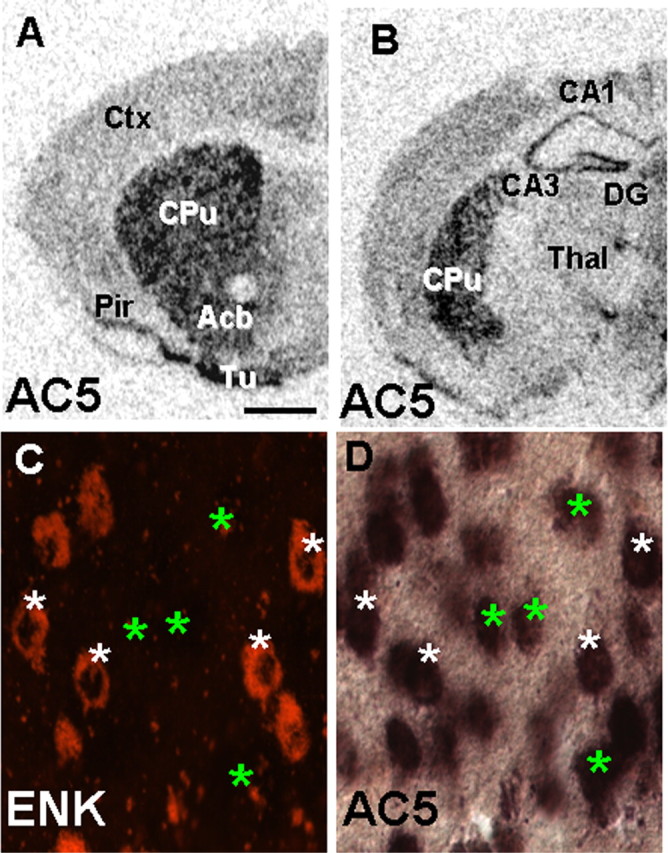
Expression of AC5 in the striatum. A, B, In situ hybridization with 33P-labeled AC5 mRNA probe revealed high AC5 expression in dorsal striatum, nucleus accumbens, and olfactory tubercule. AC5 was also detectable at very low levels in CA1, CA2, CA3, and dentate gyrus regions of the hippocampus. C, D, Double-labeled in situ hybridization for ENK and AC5 in the adult striatum. White asterisks indicates cells positive for both ENK and AC5; green asterisks indicate cells negative for ENK and positive for AC5. Ctx, Cortex; Cpu, caudate–putamen; Acb, nucleus accumbens; Tu, olfactory tubercule; Pir, piriform cortex; Thal, thalamus; DG, dentate gyrus. Scale bar: A, B, 1 mm; C, D, 20 μm.
Response learning in AC5KO mice
AC5-deficient mice (AC5KO) have impaired DA receptor-mediated modulation of cAMP levels in the striatum (Iwamoto et al., 2003). In addition, recent studies have indicated that AC5 plays a role in appetitive Pavlovian learning (Kheirbek et al., 2008), a task attributed to DA in the ventral striatum (Parkinson et al., 2002). To examine whether AC5 also plays a role in dorsal striatum-dependent learning, we tested mice in a well established dorsal striatum-dependent response learning task (Tolman and Gleitman, 1949; Restle, 1957; Packard and McGaugh, 1996; Packard, 1999; Schroeder et al., 2002). In a water-filled cross maze, mice were trained to swim to find a submerged hidden platform using a response (egocentric) strategy, i.e., making the same body turn on each trial to find the goal arm. In the first phase of the experiment, mice were randomly started from either the north or south arm of the maze and trained to find the hidden platform in the arm to the right of the start arm. Mice were given 10 trials a day until they reached a learning criterion of 9 of 10 correct choices for 3 consecutive days. WT mice learned the task in approximately three sessions, whereas AC5KO mice required two to three times as many sessions to reach learning criterion, reflecting severe impairment in response learning (Fig. 2A,C) [n = 8 AC5KO and 8 WT; number of trials to reach criterion (mean ± SEM): WT, 2.9 ± 0.5; AC5KO, 8.6 ± 2.4; t test, genotype effect, t(14) = 2.31; p = 0.035]. After learning the task, mice were trained to find the hidden platform on the opposite side, i.e., the arm to the left of the start arm, to test their ability to learn a new response strategy. AC5KO mice required significantly more trials to reach learning criterion, again demonstrating severely impaired learning (Fig. 2A,D) [number of trials to reach criterion (mean ± SEM): WT, 10.38 ± 2.1; AC5KO, 24.75 ± 2.4; t test, genotype effect, t(14) = 4.49; p = 0.0005]. During reversal learning, AC5KO mice required significantly more trials to extinguish previous choice and reach chance performance (Fig. 2B) [number of trials to reach chance (mean ± SEM): WT, 6 ± 1.0, AC5KO, 14.8 ± 3.8; t test, genotype effect, t(14) = 2.24; p = 0.04] and, once chance performance was reached, significantly more trials to reach learning criterion (Fig. 2B) [number of trials to reach criterion after chance (mean ± SEM): WT, 3.75 ± 1.1; AC5KO, 10.3 ± 2.3; t test, t(14) = 2.59; p = 0.021]. Interestingly, AC5KO mice do eventually learn the task, suggesting that either non-AC5 mechanisms in the striatum or non-striatal-dependent mechanisms can compensate for loss of AC5. Similar phenomena have been well documented in other systems (Wiltgen et al., 2006).
Figure 2.

Response learning in AC5KO mice. A, Impaired response learning in AC5KO mice. Number of trials required to reach learning criterion during initial and reversal acquisition. B, During reversal acquisition, number of trials required to extinguish the previous strategy and reach chance performance, and number of trials required to reach criterion after chance performance was reached. *p < 0.05. C, D, Acquisition of response learning during initial acquisition and reversal acquisition in individual subjects, number of correct turns of 10 trials in a session. E, Spatial learning in the Morris water maze. Both genotypes learned to find the hidden platform during training days as indicated by latency, with each bar representing average of three trials on each day. F, During a probe trial, both genotypes spent significantly more time in the quadrant (Q) of the maze that previously contained the hidden platform during training. Summary data are presented as mean ± SEM.
To test whether the above behavioral deficits were attributable to a global deficit in learning, mice were subjected to a spatial learning task in the Morris water maze. Mice were trained for 7 d, three trials a day to find a hidden platform using spatial cues situated around the room. Both genotypes reduced latency to find the hidden platform across training days (Fig. 2G) (n = 6 AC5KO and 8 WT; ANOVA, genotype × day interaction, F(6,72) = 0.572; p = 0.75). To test spatial memory, 24 h after the last training day, mice were given a 1 min probe trial to measure percentage of total time spent in each quadrant. Both AC5KO and WT mice spent significantly >25% of their time in the target quadrant (Fig. 2H) (single-sample t test: WT, p = 0.012; AC5KO, p = 0.008). In addition, there was no difference between genotypes in time spent in the target quadrant (genotype main effect, F(1,12) = 0.13, p = 0.7). This suggests that spatial memory in the Morris water maze is intact in AC5KO mice. There was an insignificant genotype effect in latency to find the hidden platform during training sessions (Fig. 2G) (ANOVA, genotype main effect, F(1,12) = 4.62, p = 0.053), indicating a performance deficit and not a learning deficit. This could be attributed to a slight motor skill deficit associated with swim speed, because a similar deficit was seen during habituation sessions when mice were tested for latency to find a visible platform (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) (ANOVA, genotype main effect, F(1,12) = 5.08, p = 0.04; genotype × trial interaction, F(1,12) = 0.103, p = 0.75).
Motor skill learning in AC5KO mice
Plasticity in corticostriatal circuits has also been implicated in motor learning in the accelerating rotarod task (Costa et al., 2004; Dang et al., 2006; Yin et al., 2009). In this task, mice were placed on an accelerating rotarod (4–40 rpm in 5 min), and staying on the rod required learning a sequence of coordinated movements. To confirm and extend previous results reporting rotarod deficits in AC5KO mice (Iwamoto et al., 2003), mice were trained for four trials a day for 3 consecutive days, and learning was assessed by an increase in latency to fall off the rod across trials. AC5KO mice exhibited a severe deficit in performing this task (Fig. 3A) (n = 8 AC5KO and 8 WT; ANOVA, genotype main effect, F(1,14) = 17.31, p = 0.001; genotype × day interaction, F(2,28) = 0.07; genotype × trial interaction, F(3,42) = 2.49, p = 0.07; genotype × day × trial interaction, F(6,84) = 1.4, p = 0.22). Although both genotypes performed similarly on the first trial, WT mice greatly improved their performance across the four trials on the first day, whereas AC5KO mice did not (first day ANOVA, genotype main effect, F(1,14) = 5.12, p = 0.04; genotype × trial interaction effect, F(3,42) = 3.65, p = 0.02). In subsequent days, performance in AC5KO was significantly impaired compared with WT mice as seen by comparison of the average time on the rod across days (Fig. 3B) (ANOVA, genotype main effect, F(1,14) = 17.31, p = 0.001; genotype × day interaction, F(2,28) = 2.9, p = 0.07) as well as a failure to increase their performance on the first trial each day (Fig. 3C) (ANOVA, genotype main effect, F(1,14) = 8.81, p = 0.01; day × genotype interaction, F(2,28) = 5.27, p = 0.01). This deficit was not attributable to a general locomotor deficit, because activity in AC5KO mice did not differ significantly from their WT littermates when tested in 1 h sessions for 3 consecutive days for distance traveled in the open field (Fig. 3D) (ANOVA, genotype main effect, F(1,14) = 1.9, p = 0.2). In addition, AC5KO and WT mice habituated similarly to the novel environment of the locomotor chamber during the 1 h session, on the first day of testing (Fig. 3E) (ANOVA, genotype × blocks interaction effect, F(1,14) = 3.14, p = 0.1).
Figure 3.

Motor skill learning on the accelerated rotarod in AC5KO mice. A, Motor skill learning on the accelerated rotarod is impaired in AC5KO mice. Mice were tested for 3 d, four trials a day (n = 8 AC5KO and 8 WT). T, Trials. B, Average latency to fall from the rotarod across days. C, Latency to fall off the rotarod in the first trial of each of the 3 d of testing. D, AC5KO mice were not significantly different in total distance traveled in a 1 h open field locomotor activity session when tested for 3 consecutive days. E, Five minute blocks of distance traveled in the first session of locomotor testing reveals no significant difference between genotypes in habituation to a novel environment. Summary data are presented as mean ± SEM.
Striatal DA release in AC5KO mice
To assess potential differences in striatal dopaminergic transmission that may account for the observed behavioral deficits in AC5KO mice, we used fast-scan cyclic voltammetry to compare electrical stimulus-evoked DA overflow in tissue slices from each genotype (Fig. 4A). The amount of DA overflow observed with one, two, and four local striatal stimulations given at 25 Hz did not significantly differ between WT and AC5KO mice (Fig. 4B) (ANOVA, genotype effect, F(1,38) = 0.02, p = 0.90; stimulus number effect, F(1,38) = 4.31, p = 0.04; interaction effect, F(1,38) = 0.04, p = 0.85). We also examined the DA overflow elicited by paired pulses with interstimulus intervals ranging between 2 and 60 s and found no differences between the genotypes (Fig. 4C,D) (ANOVA, genotype effect, F(1,36) = 0.07, p = 0.79; interstimulus interval effect, F(1,36) = 77.90, p < 0.001; interaction effect, F(1,36) = 0.01, p = 0.94). The lack of effect of AC5 deletion on DA release suggests that the D2 autoreceptor function is intact in these mice, signaling through an AC5-independent mechanism, likely via other isoforms of AC. This is consistent with our previous report indicating that injection of a low dose of quinpirole to preferentially activate presynaptic D2 autoreceptors significantly reduces locomotor activity in AC5KO mice (Kheirbek et al., 2008).
Figure 4.

Dorsal striatal DA overflow in AC5KO mice. A, Sample traces of dopamine oxidation currents elicited with one, two, and four local striatal stimulations given at 25 Hz in WT and AC5KO mice (top). Subtracted voltammograms obtained from the sweeps that elicited the peak dopamine oxidation currents (bottom). B, The amount of dopamine overflow elicited with one, two, or four local electrical stimulations does not significantly differ between genotypes (n = 8 WT and 6 AC5KO). C, Sample traces of dopamine oxidation currents elicited with paired pulses using interstimulus intervals ranging between 2 and 60 s. Paired pulses were delivered in 2 min intervals but concatenated here for display purposes. D, The relative amount of dopamine overflow obtained from a second stimulus given at varying interstimulus intervals does not differ between the genotypes (n = 4 WT and 4 AC5KO).
Striatal MSN physiology in AC5KO mice
Using striatal tissue slices, we investigated possible differences in the physiological properties of striatal MSNs between WT and AC5KO mice. Whole-cell voltage-clamp recordings from MSNs in the dorsolateral striatum revealed that genetic deletion of AC5 did not significantly alter MSN resting membrane potential (WT, −76 ± 1 mV; KO, −77 ± 1 mV; t(62) = 0.51, p = 0.61), cell capacitance (WT, 35 ± 2 pF; KO, 32 ± 2 pF; t(60) = 1.00, p = 0.32), or cell input resistance (WT, 66 ± 7 MΩ; KO, 77 ± 6 MΩ; t(62) = 1.11, p = 0.27). To test the intrinsic membrane excitability of striatal MSNs, we measured action potential frequency during depolarizing current injections (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). The average firing rate during 500 ms current injections did not differ between the genotypes (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material) (ANOVA, genotype effect, F(1,237) = 0.11, p = 0.74; input current effect, F(1,237) = 53063, p < 0.001; interaction effect, F(1,237) = 4.31, p = 0.65). MSNs from AC5KO mice did show an exaggerated adaptation in spike frequency (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material) (ANOVA, genotype effect, F(1,158) = 0.64, p = 0.43; spike number effect, F(1,158) = 85.23, p < 0.001; interaction effect, F(1,158) = 24.02, p < 0.001), which suggests possible differences in expression of small conductance calcium-activated potassium current (Stocker et al., 1999). Although the AC5KO mice showed larger mEPSC amplitudes and less spike-frequency adaptation, it is unlikely that these differences in baseline excitability contributed to the observed differences in synaptic plasticity.
Corticostriatal plasticity in AC5KO mice
To examine the synaptic properties of the glutamatergic afferents to MSNs, we recorded mEPSCs in MSNs from both WT and AC5KO mice (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). There was no significant difference in mEPSC frequency between the genotypes (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material) (t(30) = 1.08, p = 0.29), but AC5KO mice did have larger-amplitude mEPSCs (Fig. 5C) (t(30) = 2.96, p = 0.006). This suggests that the AC5KO mice may have more synaptic AMPA receptors (AMPARs) on MSNs than the WT mice. To assess possible differences in the status of synaptic plasticity and presynaptic function, we measured the AMPAR/NMDAR ratio and paired-pulse ratio (PPR) between the two genotypes. For these studies, we electrically stimulated the white matter adjacent to the dorsolateral striatum to preferentially activate cortical afferents to MSNs (Fig. 5A). The AMPAR/NMDAR ratio was not different between the WT and AC5KO mice (Fig. 5B) (t(19) = 0.44, p = 0.66). The fact that AC5KO mice exhibited larger mEPSC amplitudes without any difference in AMPAR/NMDAR ratio suggests that the MSNs in the AC5KO mice had an increased density of both AMPA and NMDA receptors. This may be a consequence of homeostatic plasticity attributable to differences in network activity in the absence of AC5 (Turrigiano, 2007). Consistent with the similarity in mEPSC frequency, the paired-pulse ratio did not differ between the genotypes (Fig. 5C) (ANOVA, genotype effect, F(1,110) = 0.45, p = 0.50; interstimulus interval effect, F(3,110) = 0.92, p = 0.43; interaction effect, F(3,110) = 0.19, p = 0.91), suggesting that presynaptic release probability is similar between the genotypes.
Figure 5.

Basal and stimulated synaptic plasticity in AC5KO mice. A, Typical evoked EPSC traces recorded from MSNs held at −70 and +40 mV. B, Ratio of AMPAR EPSC (measured at peak, −70 mV holding potential) to NMDAR EPSC (measured 40 ms after AMPAR EPSC peak, +40 mV holding potential) for each genotype (n = 11 neurons for each genotype). C, Summary graph of PPRs (EPSC2/EPSC1) plotted against interstimulus interval (n = 9–30). D, Averages of 15 traces from representative recordings showing paired EPSCs before and after LTD induction protocol. E, The high prevalence of LTD exhibited in WT mice is reflected in the persistent suppression of the average EPSC amplitudes (n = 11 recordings). In contrast, the average EPSC amplitudes from AC5KO mice showed no long-term changes after high-frequency stimulation paired with postsynaptic depolarization (n = 9 recordings). Summary data are presented as mean ± SEM.
The best characterized form of plasticity in the dorsal striatum is LTD of corticostriatal synapses (Calabresi et al., 2007). This form of plasticity is induced by pairing high-frequency stimulation of glutamatergic afferent fibers with postsynaptic depolarization (Choi and Lovinger, 1997). To assess possible differences in corticostriatal plasticity in AC5KO mice, we compared LTD induction in brain slices from AC5KO and WT mice. Cortical glutamatergic inputs to MSNs in the dorsolateral striatum were stimulated by an extracellular electrode placed in the adjacent white matter (Fig. 5D). High-frequency stimulation (four 1 s, 100 Hz trains delivered every 10 s) paired with postsynaptic depolarization (to 0 mV) produced LTD on average in recordings from WT mice (Fig. 5E) (73 ± 8% of baseline at 30–40 min; n = 11 cells/slices from 8 animals; t(10) = 2.83, p = 0.02). This LTD accompanied an increase in the paired-pulse ratio (109 ± 2% of baseline at 30–40 min; t(10) = 2.53, p = 0.03), which is consistent with previous observations that corticostriatal LTD is mediated presynaptically by a decreased release probability (Choi and Lovinger, 1997). In tissue slices from AC5KO mice, the same HFS protocol did not induce long-term plasticity on average (Fig. 5E) (105 ± 8% of baseline at 30–40 min; n = 9 cells/slices from 7 animals; t(8) = 1.79, p = 0.86). Individual recordings showed some heterogeneity in the type and magnitude of plasticity observed after the LTD induction protocol. In WT recordings, 7 of 11 cells showed significant LTD (t test, p < 0.05) that averaged 59 ± 7% of baseline at 30–40 min, and 4 of 11 cells showed no significant change in synaptic strength (t test, p > 0.05). Such heterogeneity is in agreement with previous studies (Kreitzer and Malenka, 2007). In AC5KO slices, only two cells exhibited a low-magnitude LTD (85% for both), four cells showed no significant long-term changes, and three cells exhibited low-magnitude LTP (131 ± 14%). Thus, the magnitude and prevalence of striatal LTD were both significantly lower in AC5KO mice relative to WT. Although the AC5KO mice showed larger mEPSC amplitudes and less spike-frequency adaptation (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material), it is unlikely that these differences in baseline excitability contributed to the observed differences in synaptic plasticity. The firing properties to prolonged depolarization would not affect the presynaptic stimulation, because each stimulus was ≤0.4 ms, even during HFS. Whereas stronger quantal responses could affect plasticity, stronger postsynaptic excitation and calcium entry should have enhanced plasticity, but we saw the opposite effect in the slices from AC5KO mice.
To determine whether the impairment in corticostriatal plasticity was a result of presynaptic deficits, we bath applied the CB1 agonist Win 55,212-2 [R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl) methanone monomethanesulfonate] (1 μm) while monitoring evoked EPSCs. Both WT and AC5KO mice exhibited robust, persistent suppression of EPSC amplitude after Win 55,212-2 application, demonstrating that the presynaptic glutamatergic terminals were capable of depression in response to CB1 stimulation in AC5KO mice (Fig. 6A) (WT, 52 ± 1% of baseline at 35–45 min, n = 5 cells/slices from 3 animals, t(4) = 4.61, p = 0.01; KO, 59 ± 1% of baseline at 35–45 min, n = 8 cells/slices from 3 animals, t(7) = 7.73, p < 0.001). The lack of HFS-induced LTD in AC5KO mice could either be attributable to a lack of AC5-mediated modulation in endocannabinoid synthesis or a deficit in endocannabinoid release. To address this issue, we tested DA-independent endocannabinoid-mediated LTD in MSNs using the metabotropic glutamate receptors (mGluR) agonist (RS)-3,5-dihydroxyphenylglycine (DHPG). Bath application of DHPG (100 μm) elicited LTD in both genotypes while the cells were voltage clamped at −50 mV (Fig. 6B) (WT, 78 ± 1% of baseline at 30–40 min, n = 8 cells/slices from 4 animals, t(7) = 2.45, p = 0.04; KO, 80 ± 1% of baseline at 30–40 min, n = 11 cells/slices from 5 animals, t(10) = 2.46, p = 0.03). This finding demonstrates that endocannabinoid-mediated synaptic plasticity can be induced in MSNs from AC5KO mice.
Figure 6.

Endocannabinoid and cAMP modulation of corticostriatal plasticity in WT and AC5KO mice. A, Both genotypes show robust LTD in response to a 15 min bath application of the CB1 receptor agonist Win 55,212-2 (1 μm; n = 5 WT and 8 AC5KO), which did not reverse during a 20 min bath application of the CB1 receptor antagonist AM251 [N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide] (10 μm). B, Both genotypes show significant LTD in response to a 10 min bath application of the mGluR agonist DHPG (100 μm; n = 8 WT and 11 AC5KO). Cells were voltage clamped at −50 mV throughout the recordings. C, A 1 min application of DHPG (100 μm) in the presence of quinpirole (10 μm) induced LTD in GFP-labeled D2 receptor-containing MSNs in WT but not AC5KO mice (n = 7 WT and 6 AC5KO). Cells were voltage clamped at −50 mV throughout the recordings. D, Including cAMP (1 mm) in the internal solution changes the type of plasticity induced by HFS in recordings from WT mice (n = 8 WT). The average plasticity observed in recordings from KO mice was not altered by including cAMP (1 mm) in the internal solution (n = 7 AC5KO). Summary data are presented as mean ± SEM.
To address whether D2 receptors functionally couple to AC5 to mediated endocannabinoid release and LTD, we generated AC5KO/D2–EGFP and WT/D2–EGFP littermate control mice (see Materials and Methods) to selectively record from D2-expressing indirect pathway neurons in both genotypes. In WT, D2-positive MSNs, a brief, 1 min application of DHPG (100 μm) has been demonstrated to induce a lasting depression of EPSCs if applied in the presence of the D2 receptor agonist quinpirole (10 μm) (Kreitzer and Malenka, 2007). This manipulation effectively depressed EPSCs in GFP-labeled D2 receptor-containing neurons in WT but not AC5KO mice (Fig. 6C) (WT, 68 ± 4% of baseline at 30–40 min, n = 7 cells/slices from 3 animals, t(6) = 4.24, p = 0.01; KO, 97 ± 5% of baseline at 30–40 min, n = 6 cells/slices from 3 animals, t(5) = 0.92, p = 0.40), suggesting that D2 receptors couple to AC5 to induce endocannabinoid release and LTD.
Considerable evidence exists that corticostriatal LTD and retrograde signaling via endocannabinoid release is dependent on D2 receptor activation, yet the signaling cascades downstream of D2 receptor activation that are necessary for LTD induction are unknown (Piomelli, 2003; Yin and Lovinger, 2006; Calabresi et al., 2007). Because D2 receptors are negatively coupled to cAMP production by Gi/o inhibition of AC5 (Iwamoto et al., 2003), we hypothesized that D2 receptors reduce AC5 activity and cAMP levels during LTD induction. To test this, we attempted to induce LTD in WT neurons in which cAMP levels were clamped to 1 mm by including cAMP in the internal solution. High-frequency stimulation paired with postsynaptic depolarization in WT cells that were dialyzed with 1 mm cAMP in the internal solution induced LTP (Fig. 6D) (123 ± 1% of baseline at 35–45 min, n = 8 cells/slice from 5 animals, t(7) = 2.45, p = 0.04). Tabulation of individual responses revealed that half the cells exhibited LTP and averaged 140 ± 6% of baseline, and half the cells showed no significant changes in synaptic strength and averaged 100 ± 2% of baseline at 30–40 min. In slices from AC5KO mice, the same induction protocol in combination with 1 mm cAMP in the internal solution did not, on average, induce any long-term changes in synaptic strength (Fig. 6D) (97 ± 1% of baseline at 35–45 min, n = 7 cells/slices from 3 animals, t(7) = 0.24, p = 0.82). Of this population, two cells exhibited significant LTP (131 ± 9%), two cells exhibited significant LTD (82 ± 7%), and three cells did not show any significant long-term changes (102 ± 2%). Thus, cells from AC5KO mice after cAMP loading behaved similarly to cells without cAMP loading.
Discussion
Here we demonstrate that striatum-dependent response and skill learning, as well as LTD of the cortical inputs to dorsolateral striatum, are dependent on AC5 and cAMP signaling. Our data show that loss of AC5, an isoform of AC highly expressed in the striatum, severely impairs response and motor skill learning but not spatial learning. These behavioral deficits are correlated with a loss of LTD in the dorsal striatum of AC5KO mice. This deficit in LTD could be rescued by agonist activation of presynaptic cannabinoid receptors or by DA-independent stimulation of postsynaptic endocannabinoid release. Based on the results presented here, we hypothesize that AC5 regulation of cAMP production during LTD induction facilitates endocannabinoid release and activation of presynaptic CB1 signaling to suppress glutamatergic transmission. In addition, we hypothesize that impairment in these plasticity mechanisms underlie the deficits in striatum-dependent memory exhibited by AC5KO mice.
Deficits in procedural learning in human patients or experimental animals with either damage or lesions to the dorsal striatum supports the critical role of this brain area (Packard and Knowlton, 2002). In double-dissociation studies, inhibition of the dorsal striatum, but not hippocampus, resulted in impaired motor response learning, whereas hippocampal inhibition interfered with spatial learning (Packard and White, 1991; Packard and McGaugh, 1996). In addition, recent studies have implicated modulation of dorsal striatal circuits during learning a motor skill on the accelerated rotarod (Costa et al., 2004; Yin et al., 2009). Our data indicates that AC5 plays a critical role in learning both these tasks by modulating corticostriatal plasticity. It is important to note that, as with many studies on the molecular basis of behavior, the data presented here provides a correlation between the physiology and behavior. To further strengthen the link between physiology and behavior, future studies using more elegant genetic designs to manipulate other components of this signaling pathway will be required. One possibility would be to conditionally delete AC5 in D2-expressing MSNs to observe whether this manipulation would replicate the behavioral deficits seen here. An alternative strategy could be conditional deletion of CB1 receptors on presynaptic corticostriatal terminals. In addition, care must be taken in interpretation of results with constitutive knock-outs, such as AC5KO, because compensatory developmental changes in striatal signaling may contribute to the deficits seen here. Because AC5 is a direct downstream target of DA signaling (Iwamoto et al., 2003) and the role of DA in regulating corticostriatal plasticity and striatum-dependent behaviors is well established (Packard and Knowlton, 2002; Reynolds and Wickens, 2002), it is likely that the behavioral deficits seen in AC5KO mice is directly attributable to loss of AC5, yet future studies using specific pharmacological inhibition of dorsal striatal AC5 or temporal control of AC5 gene expression in specific cell types will be required to rule out developmental confounds.
In brain slices, LTD is a predominant form of synaptic plasticity at corticostriatal synapses (Calabresi et al., 1992, 1994; Lovinger et al., 1993; Kreitzer and Malenka, 2007). There is general agreement that corticostriatal LTD in the dorsal striatum requires D2 receptor activation, increases in intracellular calcium postsynaptically, and retrograde signaling via endocannabinoids that depress glutamate release via presynaptic CB1 receptors (Lovinger et al., 1993; Calabresi et al., 1994, 1997; Gerdeman et al., 2002; Wang et al., 2006; Kreitzer and Malenka, 2007). A direct downstream target of DA receptor stimulation is AC, and signaling mechanisms downstream of cAMP, such as DARPP-32 and CREB, have been implicated in corticostriatal LTD (Calabresi et al., 2000; Pittenger et al., 2006). However, because these proteins are modulated by calcium-dependent signaling pathways (Kang et al., 2001; Nishi et al., 2005), the contribution of cAMP upstream of these signals is unknown. The data presented here demonstrates that AC5 plays a critical role in this pathway.
A provocative finding in the current study is that LTD could be rescued in AC5KO mice by bath application of the CB1 agonist WIN 55,212-2. CB1 receptors are localized on glutamatergic corticostriatal presynaptic terminals and mediate the retrograde endocannabinoid signal from striatal neurons (Gerdeman et al., 2002). In addition, it has been documented that D2 receptor stimulation produces an increase in anandamide overflow in the striatum (Giuffrida et al., 1999), yet the underlying signaling mechanisms remain unclear (Piomelli, 2003). Because CB1 receptor activation could rescue LTD, our data suggest that AC5–cAMP in corticostriatal LTD is in postsynaptic striatal neurons, upstream of endocannabinoids and CB1 activation. In addition, this result indicates that, although very low levels of AC5 could be detected in cortical areas, CB1 receptors on cortical terminals do not require AC5 for regulating LTD induction. This ability to rescue LTD with CB1 receptor agonist suggested that AC5 may contribute to either the release or biosynthesis of endocannabinoids in the MSN. We examined these possibilities by testing whether the mGluR agonist DHPG could induce LTD in AC5KO mice. In the dorsal striatum, DHPG can trigger LTD in a D2-independent, calcium-dependent manner by promoting release of postsynaptic endocannabinoids from MSNs (Kreitzer and Malenka, 2005). AC5KO mice exhibited a DHPG–LTD comparable with WT mice. This suggests that the impaired HFS-induced LTD in AC5KO mice is not attributable to a deficit in endocannabinoid signaling but rather to impaired DA modulation of endocannabinoid release during the induction protocol. Finally, because D2 receptor stimulation promotes endocannabinoid release in vivo (Giuffrida et al., 1999) and D2 receptor activation is required for LTD induction (Calabresi et al., 1994, 1997), we directly tested whether D2 receptor coupling to AC5 is required for endocannabinoid production and LTD. Using mice with genetically labeled D2-expressing striatopallidal neurons, we showed that, although a short application of DHPG in the presence of quinpirole could induce LTD in WT cells, it could not in AC5KO cells. This suggests that the D2 receptor directly couples to AC5 to regulate endocannabinoid-mediated LTD.
Our data further suggest that regulation of cAMP levels by AC5 may be required for corticostriatal LTD, because corticostriatal LTD can be effectively eliminated by elevated postsynaptic cAMP levels in WT striatal neurons. Such a manipulation would block D2-mediated inhibition of cAMP levels, which is similar to that seen after genetic deletion of D2 receptors or chronic haloperidol treatment that also eliminate corticostriatal LTD (Centonze et al., 2004). The data suggest that D2 activation inhibits tonic AC5 activity during LTD induction. The mechanism of tonic AC5 activation is unknown, but one possibility is the adenosine A2a receptors that colocalize with D2 receptors in the striatum and couple to AC5 to increase cAMP production (Fink et al., 1992; Lee et al., 2002). Future studies examining the consequence of brief application of A2a antagonist on this form of LTD may provide insight into the mechanism of tonic AC5 activation in D2-expressing neurons.
In addition, elevated postsynaptic cAMP levels in WT cells resulted in a net LTP of corticostriatal synapses, which was also seen with D2 receptor knock-out or chronic haloperidol treatment (Centonze et al., 2004). In our experiments, the elevation of cAMP in the MSN may also contribute to LTP induction, because two receptors that are positively coupled to cAMP signaling, A2a and D1 receptors, contribute to LTP induction at corticostriatal synapses (Reynolds et al., 2001; Reynolds and Wickens, 2002; Shen et al., 2008). Elevation of cAMP levels in AC5KO mice did not, on average, alter synaptic strength, suggesting impairment in the ability to strengthen synapses in AC5KO. This is not surprising, because cAMP signaling contributes to LTP induction at these inputs, as mentioned.
Independent of the LTP effects, our data shows that D2-mediated changes in cAMP are necessary for LTD at the glutamatergic inputs to striatal MSNs. A major goal for future investigations will be the identification of downstream cAMP-responsive proteins that couple DA receptors and AC5 activity to the induction of plasticity at corticostriatal synapses. The control of endocannabinoid release is poorly understood, which is a major challenge in the field, as recent studies illustrate (Adermark and Lovinger, 2007). Focusing on cAMP-dependent pathways may help identify novel molecular mechanisms that contribute to this important mediator of striatal plasticity.
In conclusion, the current study elucidates the critical role of AC5, an isoform of AC highly expressed in the striatum, in striatum-dependent response learning. The data provide insight into the molecular mechanisms underlying LTD induction in the dorsal striatum and how DA regulates this form of plasticity via an AC5–cAMP–endocannabinoid–CB1 signaling pathway. It establishes a framework for analysis of the contribution of corticostriatal plasticity to dorsal striatum-dependent behaviors.
Footnotes
This work was supported in part by National Institutes of Health Grants F31MH076422 (M.A.K.), F31DA023340 (J.P.B.), F32DA020427 and RO1DA25875-2 (J.A.B.), HL059139 and GM067773 (Y.I.), DA015918 (D.S.M.), and DA022269 and MH66216 (X.Z.) and the Edward Mallinckrodt Jr. Foundation (X.Z.). We thank Wei-Jen Tang for helpful discussions in preparing this manuscript.
References
- Adermark L, Lovinger DM. Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proc Natl Acad Sci U S A. 2007;104:20564–20569. doi: 10.1073/pnas.0706873104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead RM, Kersey KS. Immunohistochemical demonstration of differential substance P-, met-enkephalin-, and glutamic-acid-decarboxylase-containing cell body and axon distributions in the corpus striatum of the cat. J Comp Neurol. 1985;232:481–498. doi: 10.1002/cne.902320406. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G. Post-receptor mechanisms underlying striatal long-term depression. J Neurosci. 1994;14:4871–4881. doi: 10.1523/JNEUROSCI.14-08-04871.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Saiardi A, Pisani A, Baik JH, Centonze D, Mercuri NB, Bernardi G, Borrelli E. Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J Neurosci. 1997;17:4536–4544. doi: 10.1523/JNEUROSCI.17-12-04536.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Centonze D, Picconi B, Bernardi G, Chergui K, Svenningsson P, Fienberg AA, Greengard P. Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J Neurosci. 2000;20:8443–8451. doi: 10.1523/JNEUROSCI.20-22-08443.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Centonze D, Usiello A, Costa C, Picconi B, Erbs E, Bernardi G, Borrelli E, Calabresi P. Chronic haloperidol promotes corticostriatal long-term potentiation by targeting dopamine D2L receptors. J Neurosci. 2004;24:8214–8222. doi: 10.1523/JNEUROSCI.1274-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Lovinger DM. Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc Natl Acad Sci U S A. 1997;94:2665–2670. doi: 10.1073/pnas.94.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DM, Karpen JW, Fagan KA, Mons NE. Ca2+-sensitive adenylyl cyclases. Adv Second Messenger Phosphoprotein Res. 1998;32:23–51. [PubMed] [Google Scholar]
- Costa RM, Cohen D, Nicolelis MA. Differential corticostriatal plasticity during fast and slow motor skill learning in mice. Curr Biol. 2004;14:1124–1134. doi: 10.1016/j.cub.2004.06.053. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci U S A. 2006;103:15254–15259. doi: 10.1073/pnas.0601758103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagen ZM, Mitchum R, Vezina P, McGehee DS. Enhanced nicotinic receptor function and drug abuse vulnerability. J Neurosci. 2007;27:8771–8778. doi: 10.1523/JNEUROSCI.2017-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, Reppert SM. Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Brain Res Mol Brain Res. 1992;14:186–195. doi: 10.1016/0169-328x(92)90173-9. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Young WS., 3rd Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 1988;460:161–167. doi: 10.1016/0006-8993(88)91217-6. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodríguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Okumura S, Iwatsubo K, Kawabe J, Ohtsu K, Sakai I, Hashimoto Y, Izumitani A, Sango K, Ajiki K, Toya Y, Umemura S, Goshima Y, Arai N, Vatner SF, Ishikawa Y. Motor dysfunction in type 5 adenylyl cyclase-null mice. J Biol Chem. 2003;278:16936–16940. doi: 10.1074/jbc.C300075200. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kang H, Sun LD, Atkins CM, Soderling TR, Wilson MA, Tonegawa S. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell. 2001;106:771–783. doi: 10.1016/s0092-8674(01)00497-4. [DOI] [PubMed] [Google Scholar]
- Kheirbek MA, Beeler JA, Ishikawa Y, Zhuang X. A cAMP pathway underlying reward prediction in associative learning. J Neurosci. 2008;28:11401–11408. doi: 10.1523/JNEUROSCI.4115-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lee KW, Hong JH, Choi IY, Che Y, Lee JK, Yang SD, Song CW, Kang HS, Lee JH, Noh JS, Shin HS, Han PL. Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J Neurosci. 2002;22:7931–7940. doi: 10.1523/JNEUROSCI.22-18-07931.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Tyler EC, Merritt A. Short- and long-term synaptic depression in rat neostriatum. J Neurophysiol. 1993;70:1937–1949. doi: 10.1152/jn.1993.70.5.1937. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Matsuoka I, Suzuki Y, Defer N, Nakanishi H, Hanoune J. Differential expression of type I, II, and V adenylyl cyclase gene in the postnatal developing rat brain. J Neurochem. 1997;68:498–506. doi: 10.1046/j.1471-4159.1997.68020498.x. [DOI] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Nicol X, Muzerelle A, Bachy I, Ravary A, Gaspar P. Spatiotemporal localization of the calcium-stimulated adenylate cyclases, AC1 and AC8, during mouse brain development. J Comp Neurol. 2005;486:281–294. doi: 10.1002/cne.20528. [DOI] [PubMed] [Google Scholar]
- Nishi A, Watanabe Y, Higashi H, Tanaka M, Nairn AC, Greengard P. Glutamate regulation of DARPP-32 phosphorylation in neostriatal neurons involves activation of multiple signaling cascades. Proc Natl Acad Sci U S A. 2005;102:1199–1204. doi: 10.1073/pnas.0409138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard MG. Glutamate infused posttraining into the hippocampus or caudate-putamen differentially strengthens place and response learning. Proc Natl Acad Sci U S A. 1999;96:12881–12886. doi: 10.1073/pnas.96.22.12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard MG, Knowlton BJ. Learning and memory functions of the basal ganglia. Annu Rev Neurosci. 2002;25:563–593. doi: 10.1146/annurev.neuro.25.112701.142937. [DOI] [PubMed] [Google Scholar]
- Packard MG, McGaugh JL. Inactivation of hippocampus or caudate nucleus with lidocaine differentially affects expression of place and response learning. Neurobiol Learn Mem. 1996;65:65–72. doi: 10.1006/nlme.1996.0007. [DOI] [PubMed] [Google Scholar]
- Packard MG, White NM. Dissociation of hippocampus and caudate nucleus memory systems by posttraining intracerebral injection of dopamine agonists. Behav Neurosci. 1991;105:295–306. doi: 10.1037//0735-7044.105.2.295. [DOI] [PubMed] [Google Scholar]
- Packard MG, Hirsh R, White NM. Differential effects of fornix and caudate nucleus lesions on two radial maze tasks: evidence for multiple memory systems. J Neurosci. 1989;9:1465–1472. doi: 10.1523/JNEUROSCI.09-05-01465.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson JA, Dalley JW, Cardinal RN, Bamford A, Fehnert B, Lachenal G, Rudarakanchana N, Halkerston KM, Robbins TW, Everitt BJ. Nucleus accumbens dopamine depletion impairs both acquisition and performance of appetitive Pavlovian approach behaviour: implications for mesoaccumbens dopamine function. Behav Brain Res. 2002;137:149–163. doi: 10.1016/s0166-4328(02)00291-7. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Fasano S, Mazzocchi-Jones D, Dunnett SB, Kandel ER, Brambilla R. Impaired bidirectional synaptic plasticity and procedural memory formation in striatum-specific cAMP response element-binding protein-deficient mice. J Neurosci. 2006;26:2808–2813. doi: 10.1523/JNEUROSCI.5406-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restle F. Discrimination of cues in mazes: a resolution of the place-vs.-response question. Psychol Rev. 1957;64:217–228. doi: 10.1037/h0040678. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Wickens JR. Dopamine-dependent plasticity of corticostriatal synapses. Neural Netw. 2002;15:507–521. doi: 10.1016/s0893-6080(02)00045-x. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Hyland BI, Wickens JR. A cellular mechanism of reward-related learning. Nature. 2001;413:67–70. doi: 10.1038/35092560. [DOI] [PubMed] [Google Scholar]
- Schroeder JP, Wingard JC, Packard MG. Post-training reversible inactivation of hippocampus reveals interference between memory systems. Hippocampus. 2002;12:280–284. doi: 10.1002/hipo.10024. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR. Memory systems of the brain: a brief history and current perspective. Neurobiol Learn Mem. 2004;82:171–177. doi: 10.1016/j.nlm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- Tolman EC, Gleitman H. Studies in spatial learning; place and response learning under different degrees of motivation. J Exp Psychol. 1949;39:653–659. doi: 10.1037/h0059317. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol. 2007;17:318–324. doi: 10.1016/j.conb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–468. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ, Sanders MJ, Anagnostaras SG, Sage JR, Fanselow MS. Context fear learning in the absence of the hippocampus. J Neurosci. 2006;26:5484–5491. doi: 10.1523/JNEUROSCI.2685-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci U S A. 1995;92:220–224. doi: 10.1073/pnas.92.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Choi EJ, Wang F, Blazynski C, Storm DR. Type I calmodulin-sensitive adenylyl cyclase is neural specific. J Neurochem. 1993;60:305–311. doi: 10.1111/j.1471-4159.1993.tb05852.x. [DOI] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci U S A. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ, Balleine BW. Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur J Neurosci. 2004;19:181–189. doi: 10.1111/j.1460-9568.2004.03095.x. [DOI] [PubMed] [Google Scholar]
- Yin HH, Ostlund SB, Knowlton BJ, Balleine BW. The role of the dorsomedial striatum in instrumental conditioning. Eur J Neurosci. 2005;22:513–523. doi: 10.1111/j.1460-9568.2005.04218.x. [DOI] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilário MR, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM, Costa RM. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]