

Abstract
Within antigen presenting cells (APC), ubiquitination regulates the trafficking of immune modulators such as MHC class II and CD86 (B7.2) molecules. MARCH1 (membrane-associated RING-CH), a newly identified ubiquitin E3 ligase expressed in APC, ubiquitinates MHC class II thereby reducing its surface expression. Following LPS-induced maturation of dendritic cells (DC), MARCH1 mRNA is downregulated and MHC class II is redistributed to the cell surface from endosomal compartments. Here, we show that MARCH1 expression is also regulated at the post-transcriptional level. In primary DC and APC cell lines of murine origin, MARCH1 had a half-life of less than 30 min. MARCH1 degradation appears to occur partly in lysosomes, since inhibiting lysosomal activity stabilized MARCH1. Similar stabilization was observed when MARCH1-expressing cells were treated with cysteine protease inhibitors. Mutational analyses of MARCH1 defined discrete domains required for destabilization, proper localization, and functional interaction with substrates. Together, these data suggest that MARCH1 expression is regulated at a post-transcriptional level by trafficking within the endo-lysosomal pathway where MARCH1 is proteolysed. The short half-life of MARCH1 permits very rapid changes in the levels of the protein in response to changes in the mRNA, resulting in efficient induction of antigen presentation once APC receive maturational signals.
INTRODUCTION
Antigen presentation is strictly regulated to ensure immune priming only under the appropriate circumstances. This is true of both the MHC class I and class II presentation pathways, which share a requirement for costimulation in order to efficiently activate naïve CD8 and CD4 T cells, respectively. In the case of MHC class II-expressing professional antigen presenting cells (APC), such as dendritic cells, macrophages, and B cells, the ability to prime CD4 T cells is coupled to their maturational state. Immature APC are characterized by relatively low levels of MHC class I and class II, and costimulatory molecules including CD80 (B7.1) and CD86 (B7.2). Various stimuli, which include Toll-like Receptor ligands such as LPS, induce rapid changes in APC which result in enhanced priming capacity. Though these changes are manifold, notable among them is a substantial increase in MHC class II and CD80/86 levels (1, 2). Consequently, matured APC are much more potent in their T cell-activating ability (2). In large part, the rapid, maturation-induced changes in MHC class II (and probably CD86) levels are the result of changes in intracellular trafficking pathways (3, 4).
Extensive work has shown that in immature APC, MHC class II molecules are sorted into the endo-lysosomal system, either directly from the trans-Golgi network and/or after transient appearance at the plasma membrane (5, 6). This sorting process requires specific information in the cytosolic tail of the MHC class II “chaperone”, the invariant-chain (3). In immature dendritic cells (DC), MHC class II molecules are primarily found in late endosomes containing internal vesicles (7). When DC are matured with stimuli such as LPS, MHC class II molecules leave endosomes and traffic to the cell surface (6, 8, 9), where they maintain high levels of expression. Interestingly, in immature DC, MHC class II beta-chains are constitutively ubiquitinated on their cytosolic tails, causing MHC class II to be retained within the endo-lysosmal system; ubiquitination is lost once DC mature (10–13). MHC class II beta-chains that cannot be ubiquitinated are expressed at high levels at the cell surface even in immature DC (11, 12). It has recently become clear that Membrane-associated RING-CH protein 1 (MARCH1) is the E3 ligase responsible for ubiquitinating MHC class II in immature APC (13, 14). Maturation of APC results in a decrease in MARCH1 mRNA and redistribution of MHC class II to the cell surface (13, 15). Thus, MARCH1 appears to function as a negative regulator of antigen presentation. In addition to affecting antigen display (MHC class II), MARCH1 also regulates the expression of the costimulatory molecule, CD86 (13, 16).
MARCH1 is a member of a family of RING domain-containing E3 ligases which were identified by virtue of their relatedness to viral immune evasion molecules (16, 17). Like most of its cellular relatives, MARCH1 is membrane-anchored and possesses a RING domain of the RING-CH subtype (18, 19). MARCH1 and its closest homolog, MARCH8 (c-MIR), regulate the surface expression of MHC class II and CD86 through ubiquitin-dependent mechanisms (10, 14). While MARCH8 is broadly expressed (16, 20), MARCH1 expression is highly enriched in lymphoid tissues (16) and appears to be especially prominent in APC (14). Importantly, in the absence of MARCH1, MHC class II and CD86 levels increase at the cell surface on immature APC (13–15). MARCH1 has been reported to localize to LAMP-1-positive late endosomes/lysosomes (16), which is consistent with the finding that MHC class II molecules are ubiquitinated after processing of the invariant-chain (11), which occurs in a similar compartment. MHC class II beta chains appear to be direct substrates of MARCH1, and an interaction between the two proteins has been reported (21). However, it is noteworthy that direct detection of endogenous MARCH1 by immunoblot has proven difficult; this appears to reflect the relatively low expression levels of this protein (13, 14).
The fact that MARCH1 protein levels are low in APC may be directly relevant to the regulation of MARCH1 function. MARCH1 must be regulated to permit antigen presentation upon APC maturation. This is accomplished, at least in part, through a decrease in MARCH1 transcription subsequent to LPS stimulation (13). In this report (13), it was observed that MARCH1 mRNA levels dropped substantially 16 hours after LPS treatment of human DC. However, a decrease was also evident as early as 4 hours post-treatment. Though relatively slight at 4 hours (≈2–3-fold decrease), this could well be meaningful to the biology of MARCH1. Indeed, MARCH1 hemizygous mice (MARCH1+/−), which are presumed to have a two-fold reduction in MARCH1 protein levels, have significantly increased surface MHC class II levels on immature APC (14). Thus, the amount of MARCH1 protein that is normally present in immature APC is just sufficient to affect MHC class II expression. Even modest decreases in MARCH1 protein levels could shift the balance to higher MHC class II and CD86 expression. By extension, modest changes in MARCH1 mRNA levels could produce biologically relevant changes in MARCH1 protein levels. In order for this mechanism to account for the rapid induction kinetics of MHC class II and CD86 on DC following maturation, additional requirements must be met. Foremost among these is that MARCH1 protein levels must track closely with its mRNA levels. This would be the case if MARCH1 protein was unstable. Further, this could help explain the fact that it has been difficult to detect endogenous MARCH1. Here, we have explored the post-transcriptional expression of MARCH1. Our results show that MARCH1 turns over in APC with rapid kinetics, and this process is partly dependent on lysosomal acidity and cysteine proteases. Further, we identify regions of MARCH1 which regulate its turnover, function, and localization. Overall, the data support a model wherein the levels of MARCH1 are tuned, through its inherent instability, to provide the minimal levels of E3 ligase function to suppress antigen presentation in immature APC through downregulation of MHC class II and CD86, and also permit rapid induction of antigen presentation following maturation.
MATERIALS AND METHODS
Mice
Three- to five-week-old C57BL/6 mice were obtained from Charles River Laboratories or the Jackson Laboratory and housed in the animal facility at the University of Arizona. All procedures involving mice were done according to protocols approved by the University of Arizona Institutional Animal Care and Use Committee (IACUC).
Antibodies and other reagents
Mouse anti-HA (influenza hemagglutinin epitope) antibody (clone 6E2) and mouse anti-myc antibody (clone 9B11) were purchased from Cell Signaling. Rat anti-CD86 (clone GL1) and goat anti-mouse cathepsin L were obtained from R&D Systems. Rat anti-mouse MHC class II (clone M5/114.15.2), rat anti-mouse invariant chain (In-1), hamster anti-CD80 (16–10A1), and hamster anti-CD11c (clone N418) were obtained from BD Biosciences. Goat anti-mouse cathepsin S (M19) was purchased from Santa Cruz Biotech. Mouse anti-chicken actin antibody (ACTN05), rabbit anti-human EEA-1, and rabbit anti-LAMP-1 were purchased from Abcam. DEC205 was purchased from Cedarlane Laboratories. Rabbit anti-human Derlin-1 was obtained from Medical and Biological Laboratories. Rabbit anti-human furin convertase was obtained from Thermo Scientific. Mouse anti-GFP mAb was obtained from Covance. Cycloheximide (used at 12.5 μg/ml) was purchased from Sigma. Bafilomycin A (used at 0.32 μM) was obtained from Biomol. Inhibitors for cathepsin L (Z-FF-FMK) and S (Z-FL-COCHO) were purchased from Calbiochem and each used at 40 μM.
Cell lines and cell culture
DC2.4 is a DC-like cell line (22) derived from C57BL/6 mice and was provided by Dr. Kenneth Rock (University of Massachusetts Medical School). WT3 is a C57BL/6-derived mouse embryo fibroblast cell line (23) and was obtained from Dr. Ted Hansen (Washington University School of Medicine). The RAW264.7 macrophage cell line (24) and the A20 B cell lymphoma line (25) were obtained from the American Type Culture Collection. Bone marrow-derived DC (BMDC) were generated in a manner similar to that described (26, 27) by culture of C57BL/6 bone marrow cells for 6 days in the presence of 10 ng/ml IL-4 and 10 ng/ml GM-CSF (Peprotech). Non-adherent cells were collected post-culture and phenotypic analysis revealed purity that was consistently ≥80% (CD11c-positive; see Figure 1). The MJDC cell line was generated using C57BL/6 bone-marrow-derived dendritic cell cultures (as above) infected with the J2 retrovirus encoding the v-myc and v-raf oncogenes (28, 29), in a manner similar to that described for DC2.4 cells (22). Replication-defective virus produced by ψCREJ2 cells (obtained from Dr. Howard Young, National Cancer Institute) was used to infect day 2 BMDC cultures. Cultures were maintained for 30 days in the presence of GM-CSF and IL-4. During this time, both adherent and non-adherent cells expanded. However, only the non-adherent fraction was collected at each passage for further expansion. Transformed cells were then cloned by limiting dilution, and clones were screened based on low adherence and expression of markers characteristic of DC. During the cloning steps and all subsequent propagation, only GM-CSF (5 ng/ml) was included in the medium. Clone 2B1 expresses CD11c, DEC205, MHC class II, and CD86, and was selected for these studies (see Supplementary Figure 1), though the expression of MHC class II was relatively low and could be substantially increased following treatment with IFN-γ. All cells were cultured in complete RPMI 1640 (Mediatech) supplemented with 10% fetal calf serum (HyClone), 1 mM HEPES (Invitrogen), 2 mM L-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 100 U/ml penicillin/streptomycin (all from Mediatech).
Figure 1. MARCH1 is unstable in multiple cell types.

A. CD86 expression on DC2.4 cells stably transduced with a lentiviral vector encoding either wildtype N-terminal HA-tagged MARCH1 (WT) or a RING-CH mutant (W104A). The thin gray peaks indicate staining with an irrelevant isotype-control antibody. B. CD86-expressing WT3 fibroblasts were stably transduced with retroviral vectors encoding MARCH1 with an N-terminal HA tag. The thin gray peaks indicate staining with an irrelevant isotype-control antibody. C. DC2.4 cells stably expressing N-terminal HA-tagged MARCH1 were incubated for the indicated times with cycloheximide, then lysates were blotted for MARCH1 (HA tag). Incubation in diluent (DMSO) was used as a control, and actin blots were performed as loading controls. Graphs show the turnover of MARCH1 from three independent experiments. The signal intensity of the MARCH1 and actin bands was estimated using Quantity One software. For each time point, the actin-normalized values were averaged between the experiments and graphed as the percent-remaining relative to time zero (DMSO-treated samples) +/− SEM. D. Immunoblot for MARCH1 (HA tag) and actin was performed from lysates of MARCH1-expressing WT3 fibroblasts incubated with cycloheximide for the indicated times. E. Mouse bone marrow-derived DC (BMDC) were generated as described in Materials and Methods, and infected with a lentiviral vector encoding HA-MARCH1. Experiments were performed three days post-infection. Top panel = cychoheximide-chase and immunoblot of MARCH1 in BMDC, performed as described above. Immunoblot for GFP was also included; GFP is encoded by the lentivirus. Bottom panel = purity of the BMDC preparations (MHC class II versus CD11c staining, and isotype-matched control staining). F. DC2.4 cells stably expressing HA-tagged MARCH1 or the W104A mutant were incubated for the indicated times with cycloheximide, and treated as in panel C. Full-length MARCH1 is denoted with an asterisk. Graphs show the turnover of MARCH1 from three independent experiments. For each time point, the actin-normalized values were averaged between the experiments and plotted as the percent-remaining relative to time zero (DMSO-treated samples) +/− SEM. **p<0.01.
DNA constructs
The mouse MARCH1 cDNA was obtained by RT-PCR from splenocyte cDNA. A clone was obtained which encoded a protein that matched a MARCH1 sequence within GenBank (BAC29449). This clone was used as a template for subsequent constructs. An N-terminal HA tag (influenza hemagglutinin epitope) was added by PCR, encoding the following sequence: MAYPYDVPDYAPGPQFVS, immediately upstream of the MARCH1 start codon. Depictions of the MARCH1 mutant constructs generated for this study, as well as the specific residues mutated in each case, are provided in the relevant figures. The murine CD86 (B7.2) cDNA was generated by RT-PCR from C57BL/6 BMDC cDNA. Site-specific mutants were produced using Quick Change XL Mutagenesis Kit (Stratagene) according to the supplier’s instructions. Gene knockdown was accomplished using the lentiviral shRNA vector pLentiLox 3.7 (30) (American Type Culture Collection). Multiple shRNA oligos were designed for each target using Oligoengine or obtained from the RNAi consortium at the Broad Institute (http://www.broad.mit.edu/node/563) The sequences of the shRNA oligos (sense strands) are given below, with the 19-mer core sequence complementary to the target gene underlined. The remaining sequences create the hairpin and flanking sequences for ligation into Hpa1/Xho1-digested pLentiLox 3.7 as described (30).
CatL #1: TGCTTTCCAGTACATTAAGGTTCAAGAGACCTTAATGTACTGGAAAGCTTTTTTC;
CatL #2: TGTAGCGGTAATGAGGACTTTTCAAGAGAAAGTCCTCATTACCGCTACTTTTTTC
CatS #1: TGAGACCCTACCCTGGACTATTCAAGAGATAGTCCAGGGTAGGGTCTCTTTTTTC
CatS #2: TGAAGAAGTACGGCGTCTCATTCAAGAGATGAGACGCCGTACTTCTTCTTTTTTC
This design included homology searches for unwanted similarity to other genes. cDNA constructs were expressed from either retroviral or lentiviral bicistronic vectors. The retroviral vectors were all Murine Stem Cell Virus-derived non-replicating vectors. pMIG is bicistronic, with the gene of interest upstream of an IRES (internal ribosome entry site) element which precedes eGFP (pMSCV-IRES-GFP). pMIB is derived from pMIG, wherein the GFP cDNA was replaced with the bleomycin (zeocin) resistance genes (31). pMIH has been described (32) and encodes a hygromycin resistance gene as the second cistron. pMIH was used to express CD86. pCIG is a lentiviral vector derived from pCDH-CMV-MCS-EF1-copGFP (Systems Biosciences). It was generated by replacing the EF1 promoter-copGFP cassette with an IRES-eGFP cassette downstream from the CMV promoter. The correct sequence of all constructs was confirmed by DNA sequence analysis.
Transfections and transductions
Transient transfections were performed using FuGene 6 reagent (Roche Diagnostics) according to the supplier’s instructions. For retroviral vector production, PlatE cells (ecotropic; (33)) and Phoenix cells (amphotropic; (34)) were used to generate replication-defective viral particles following transient transfection, as described (35). Ecotropic virus was used to infect DC2.4, RAW 264.7, and WT-3 cells; amphotropic virus was used to infect A20 and MJDC cells. For stable lines, drug selection was done for >1 week, and maintained continuously during cell passage. For lentiviral vector infection of BMDC, replication-defective virus was packaged by transient transfection of 293T cells (36) with the ViraPower packaging plasmids (Invitrogen). Two and three days post-transfection, culture supernatants were harvested, centrifuged briefly at 200 × g, and filtered through a 0.2 μm filter. Then, virus-containing supernatants were used directly for infection, or the virus particles were concentrated by ultracentrifugation. This was done as described (37) using a Beckman XL-70 centrifuge. Supernatants were centrifuged in Optiseal tubes using an SW28 rotor for 2 hours at 4°C at 50,000 × g. Pellets were resuspended in complete medium at 1/10 the volume of the starting supernatant. For infection, bone marrow cultures were established as described above. After three days, virus was added along with 8 μg/ml hexadimethrine bromide (Polybrene; Sigma) and infection proceeded for 3 days. Lentiviral shRNA vectors (pLentiLox 3.7-based) were used for gene silencing experiments in DC2.4 cells. >3 days after adding virus, infected cells were sorted based on GFP-expression (encoded by pLentiLox 3.7) using a FACSAria sorter (BD Biosciences) and returned to culture for expansion.
Flow cytometry
The surface expression of CD80, CD86, and MHC class II was monitored on cells by flow cytometry. Cell staining was performed in staining buffer (1% BSA, 0.1% azide in Dulbecco’s-PBS; D-PBS). Fc receptors were blocked using Fc Block (anti-CD16/32) (clone 2.4G2, BD Biosciences). Primary antibodies were diluted in staining buffer and incubated with cells for at least 30 min. on ice. Then, cells were washed, resuspended in 1X D-PBS and fixed with an equal volume of 1% paraformaldehyde (in D-PBS). For intracellular staining, cells were fixed and permeabilized with 1% paraformaldehyde + 0.5% saponin (Calbiochem) in D-PBS for 20 min on ice. Cells were then washed twice and stained with the primary antibody for at least 30 min on ice. After two washes, cells were probed with fluorochrome-conjugated secondary antibodies on ice for 30 min, followed by washing. Cells were then fixed as above and samples collected using a FACScalibur cytometer (BD Biosciences) and data were analyzed using either CellQuest Pro software (BD Biosciences) or WinMidi (freeware available through the Scripps Research Institute).
Immunofluorescence
DC2.4 cells +/− MARCH1 and MARCH1 mutants (stable transductants) were stained for MARCH1 (HA tag) and various antibodies to define distinct cellular compartments. Staining was performed in suspension using a modified version of the intracellular flow cytometry protocol described above. After the final wash, nuclei were stained with Hoescht 33242 (Invitrogen) for 10 min at 4°C. After the final staining step, cells were suspended in D-PBS and deposited on microscope slides using Shandon Cytospin cytocentrifuge (Thermo Scientific) at 800 rpm for 2–3 min. Mounting medium was added (Prolong Gold; Invitrogen) and pre-cleaned coverslips were placed and sealed with fingernail polish. Images were taken using a Zeiss 510 Meta Confocal microscope using an NA1.4 aperture objective with 63X magnification. Image analysis was done with ImageJ (freeware available from: http://rsbweb.nih.gov/ij/), and the overlap between the MARCH1 signal and the various markers was estimated using the JACoP plug-in (38). Additional details are provided in the legend to Figure 6.
Figure 6. Distribution of wildtype versus mutant forms of MARCH1 within cells.
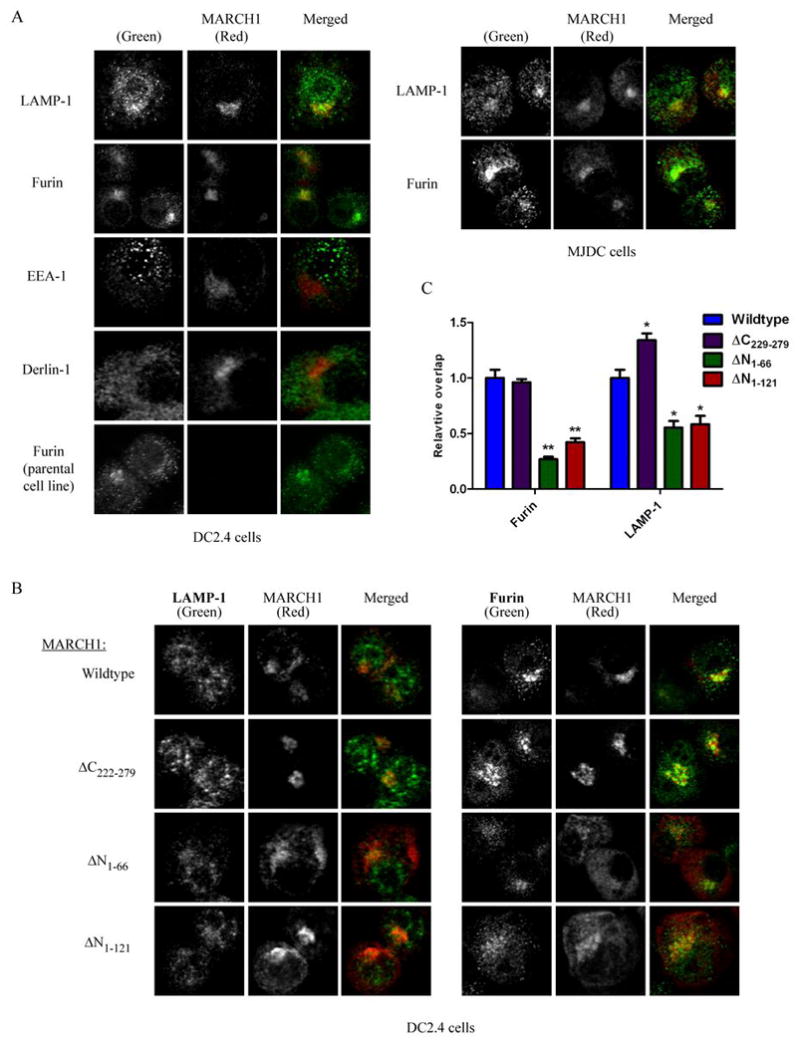
A. Immunofluorescence and confocal microscopy were performed with HA-MARCH1-expressing cells or non-expressing parental cells. Cells were co-stained for MARCH1 versus the indicated molecules and representative images are provided. The left panels show staining of DC2.4 cells, and right panels show staining of MJDC cells. In each set of images, staining of the indicated marker is shown as green in the merged image, and MARCH1 (HA) staining is shown in red in the merged image. Staining of the parental (no MARCH1) cell line is shown on the bottom left. B. Staining of each indicated HA-tagged MARCH1 mutant is shown versus furin and LAMP-1. Staining of additional markers (EEA-1 and Derlin-1) is provided for each mutant in Supplemental Figure 2. C. The relative distribution of each mutant as compared to wildtype MARCH1. Images representing single cells (n=15) taken from multiple fields were analyzed for each mutant stained together with furin or LAMP-1. The extent of overlap between the MARCH1 signal and the marker protein was determined using the JACoP (38) plug-in for NIH ImageJ with the default settings. Specifically, JACoP was used to determine the Manders coefficient (64) for MARCH1-positive staining which was also positive for the marker in question. This coefficient was given a value of 1 for wildtype MARCH1, and then the values for each mutant were normalized to wildtype. The relative Manders coefficients are plotted +/− SEM. Paired t-tests were used to determine whether the mutants were different from wildtype for each marker. *p<0.01, **p<0.0001. Note that for all images, a lower threshold was applied uniformly to each set of samples (imaged for a given marker) based on negative controls (background subtraction). The signal for MARCH1 (HA) and the marker protein in each set of images was normalized by setting the brightest pixel in each image (for each fluorescence channel) as the maximum signal (histogram-stretching) in order to facilitate visualization and comparison across fields. This was necessary to permit comparisons between wildtype and mutant MARCH1 constructs, which are expressed at different levels (see Figure 4).
SDS-PAGE and immunoblotting
Cell lysates were generated by lysis in 1% IGEPAL CA-630 (NP-40) (from Sigma) dissolved in 50 mM Tris, 150mM NaCl, pH 7.4 buffer (TBS), supplemented with 0.3 mM PMSF, 20 mM iodoacetamide, 10 μM MG132 (all from Sigma), and Protease Inhibitor Cocktail III (Calbiochem). Post nuclear lysates were mixed with LDS sample buffer (Invitrogen) and 2-mercaptoethanol (1% final concentration). Protein content was determined using the BCA protein assay from Thermo Scientific. Samples were separated by electrophoresis on 4–12% or 12% Nu-PAGE SDS polyacrylamide gels (Invitrogen). Proteins were then transferred to Immobilon-P PVDF membranes (Millipore). Blocking of membranes proceed for 1 hour with 5% dried milk, 0.1% Tween-20 (Sigma), and 0.01% SDS in D-PBS. After washing three times with 0.1% Tween-20, 0.01% SDS in D-PBS, membranes were incubated with the appropriate dilution of primary antibody for >1 h, washed three times, incubated with appropriate biotin-conjugated secondary antibodies for 1h, followed by incubation with streptavidin-conjugated horseradish peroxidase (Zymed) for 1 h. Membranes were incubated with ECL chemiluminescent substrate (GE Healthcare) and visualized using Blue Ultra Autorad film (from ISC BioExpress) or were incubated with SuperSignal West Femto (Thermo Scientific) and visualized using a ChemiDoc XRS (Bio-Rad) digital imaging system. Determination of the band intensities from immunoblots was done using Quantity One software (Bio-Rad) and plots were generated using GraphPad Prism software.
RESULTS
MARCH1 protein stability
During the course of LPS-induced DC maturation, it has been shown that MARCH1 mRNA levels decrease as soon as 4 hours post-induction, and protein levels are also reduced at this time point (13). These relatively rapid changes suggest that MARCH1 protein levels closely mirror those of its mRNA, and this could occur if the MARCH1 protein is relatively unstable. To explore the properties of the MARCH1 protein, including stability, we began by expressing it in a variety of murine cell lines, as well as primary bone marrow-derived DC (BMDC). Figures 1A and 1B show the expression of CD86 at the cell surface of cell lines retrovirally-transduced with vectors encoding an epitope-tagged version of MARCH1. CD86 expression levels were used as an indicator of MARCH1 expression and function since it is a natural target of MARCH1 (13) and is expressed in the cell lines used here. In a DC-like cell line (DC2.4), expression of MARCH1 resulted in a significant decrease in CD86 expression (Figure 1A). Though these cells are derived from BMDC, they do not express endogenous MARCH1 as determined by quantitative RT-PCR (not shown). As a control, we expressed a mutant version of MARCH1 harboring a point mutation within the RING-CH domain. We mutated a tryptophan residue (W104 of MARCH1) conserved among RING-CH-containing ligases and many other RING-containing ligases, that is directly involved in E2 recruitment (39, 40). This RING-CH domain mutant was incapable of downregulating surface CD86 expression, as expected (Figure 1A), even though it was expressed at comparable levels to wildtype MARCH1 (see below). Similar to the DC2.4 cells, transduction of CD86-expressing fibroblasts with MARCH1 caused a drop in surface CD86 expression (Figure 1B). Thus, as has been reported previously in human cells (13, 14, 16), MARCH1 is functional in non-APC, indicating that no additional APC-specific factors are strictly required for its function.
We next examined MARCH1 protein levels in these cells lines over time following inhibition of translation using cycloheximide (cycloheximide-chase). Figure 1C shows an immunoblot for epitope-tagged MARCH1 from lysates of DC2.4 cells treated with cycloheximide for various times. We observed a rapid decrease in the steady-state levels of MARCH1 with an estimated half-life of <30 min. Similar decay rates were found in other APC-derived cell lines (A20 B cells and RAW 264.7 macrophages; data not shown), as well as a fibroblast line (B6/WT3; Figure 1D). Placing the epitope-tag at either end of MARCH1 did not make a difference in terms of stability (not shown). To determine if these results are representative of MARCH1 in primary APC, we examined MARCH1 turnover in BMDC. As mentioned above, endogenous MARCH1 is quite difficult to detect. Therefore, we used lentiviral vectors to transduce BMDC with an HA-tagged version of MARCH1. In these cells, MARCH1 expression decreased precipitously within the first 30 min after the addition of cycloheximide, confirming that MARCH1 is highly unstable in DC (Figure 1E).
These findings with MARCH1 are somewhat similar to MARCH7, which is also unstable. In the case of MARCH7, the instability is related to its autoubiquitination (41). We examined the ligase-deficient mutant of MARCH1 (W104A) and found that while its steady-state expression levels were comparable to wildtype MARCH1, it did exhibit a modest, but significant increase in stability (Figure 1F). Thus, the ubiquitin ligase activity of MARCH1 does make a minor contribution to its instability, which could reflect autoubiquitination or an indirect effect on stability through a ubiquitin-dependent process initiated by MARCH1. It is also possible that MARCH1 could be ubiquitinated by another E3 ligase. Curiously, we did notice a band in our anti-MARCH1 immunoblots which runs above the predicted full-length MARCH1, and its levels seemed to increase at later time points of the cycloheximide chase, (see Figures 1D–1F, for example). The size difference between this band and the full-length version of MARCH1 is not large enough to be explained by mono-ubiquitination, and we have been unable to detect ubiquitination of MARCH1 by immunoprecipitation and anti-ubiquitin blotting (not shown). The nature of this larger band is presently unclear, but could represent another type of post-translational modification such as phosphorylation; N-linked glycosylation is excluded since no potential glycosylation sites exist in the MARCH1 lumenal domain. Regardless, these results indicate that additional factors besides ubiquitination are likely involved in the regulation of MARCH1 levels.
MARCH1 turnover requires lysosome function
Having determined that MARCH1 is rapidly degraded, we wanted to define the mechanisms responsible for this regulation. Given that MHC class II molecules are degraded in a lysosome-dependent manner following MARCH1-mediated ubiquitination (13, 14), and the reported presence of MARCH1 in lysosomes (16), we tested the requirement for lysosome function in MARCH1 degradation. DC2.4 cells expressing MARCH1 were treated with Bafilomycin A (Baf A), which blocks lysosome acidification (42), and steady-state levels of MARCH1 were analyzed by immunoblot. Treatment of cells with Baf A led to an increase in the MARCH1 signal over time (Figure 2A). To determine if the increase in MARCH1 levels after Baf A treatment was due to stabilization of MARCH1, an experiment was performed in which DC2.4+MARCH1 cells were treated with Baf A and cycloheximide to follow the turnover of MARCH1 over time. Figure 2B shows that Baf A does stabilize MARCH1 in cells treated with cycloheximide. Though stabilization was not complete, MARCH1 turned over more slowly when lysosome function was blocked (as compared to control (no Baf A) cells), demonstrating that MARCH1 was more stable when lysosomal pH was increased. The fact that MARCH1 turnover was not completely abolished with Baf A treatment suggested an alternative mechanism for the turnover of a fraction of MARCH1 within the cells. Indeed, we found that MARCH1 could be partially stabilized by treatment of cells with a proteasome inhibitor (MG132), indicating that some MARCH1 turnover was mediated by proteasomes (data not shown). Proteasome-dependent turnover of membrane proteins is typically a result of ER-associated degradation during initial biogenesis (43, 44), and a portion of MARCH1 may be degraded via this pathway.
Figure 2. Lysosome-dependent turnover of MARCH1.

A. DC2.4+HA-MARCH1 cells were incubated with Bafilomycin A (Baf A) or diluent (DMSO) for 1, 2.5, or 5 hours, and then cell lysates were blotted for MARCH1 and actin. The band intensities were quantified from four independent experiments, normalized to actin, and graphed as the fold-increase over control (DMSO) +/− SEM. B. DC2.4+MARCH1 cells were pre-treated for 0.5 hours with Baf A (or DMSO). Then, cycloheximide-chase was performed as described in the Figure 1 legend. In this case, Baf A and cycloheximide were present during the chase period. Note: MARCH1 blot samples were run on the same gel, and the gel was split to remove irrelevant lanes. The band intensities were quantified, normalized to actin, and graphed as the percent-remaining as compared to time zero (DMSO-treated samples) +/− SEM. Results are representative of four experiments. C. BMDC were infected with lentiviral vectors encoding HA.MARCH1 or MARCH1.myc (C-terminal tag). Experiments were performed three days post-infection. Left panels = infected cells were treated with Baf A (or DMSO control) and blotted for the myc-tagged MARCH1 and for invariant chain (Ii). Right panels = BMDC were transduced with HA-MARCH1, and cycloheximide-chase and immunoblot of MARCH1 were performed as described in the legend of Figure 1. Right lane represents the same cells treated for 5 hours with Baf A.
We next sought to confirm our findings using primary APC. Immature BMDC were transduced with MARCH1-expressing lentiviral vectors. Treatment of these cells with Baf A led to increased levels of MARCH1, similar to the results obtained above with the cell lines. This was the case for MARCH1 with either an N-terminal (HA) or C-terminal (myc) epitope tag (Figure 2C). As a control for the effects of Baf A treatment, we examined the processing of the MHC class II invariant chain (Ii), where it is known that effective inhibition of lysosomal peptidases leads to the accumulation of the invariant-chain fragments (3); Baf A treatment led to the expected accumulation of p31 and p10 invariant-chain fragments in BMDC (Figure 2C). Overall, these experiments demonstrate that MARCH1 is stabilized when lysosome function is inhibited, and this holds true in multiple cell types, including BMDC. In addition, proteasome activity also regulates turnover of a fraction of MARCH1. Together, these two activities (lysosome- and proteasome-dependent degradation) limit the levels of MARCH1 which are present at steady-state.
Proteolytic processing of MARCH1
The stabilizing effect of inhibitors of lysosome acidification on MARCH1 suggested a role for lysosomal hydrolases in MARCH1 turnover. In fact, we noted the presence of apparent breakdown fragments of MARCH1 in some cells, consistent with proteolysis (data not shown). We reasoned that cysteine proteases, and cathepsins in particular, were good candidates to affect MARCH1, since these proteases are directly involved in the processing of invariant-chain during MHC class II biogenesis within APC (3, 45). The cathepsin family contains many members which exhibit cell type-specific expression and activity (45, 46), as well as cross-regulation (47, 48). We focused on the leupeptin-sensitive cathepsins, including cathepsin S and cathepsin L, which are important in MHC class II antigen presentation (46, 49). Figure 3A shows that treatment of various cell lines with leupeptin for five hours resulted in an increase in MARCH1 levels in each of the cell lines tested. Hence, cysteine/serine proteases affect MARCH1 expression.
Figure 3. Lysosomal proteases affect MARCH1 turnover.

A. DC2.4, WT3, and RAW 264.7 (macrophage-like) cells were incubated for five hours with the indicated inhibitors: leupeptin, cathepsin L inhibitor (Z-FF-FMK), and Baf A. Cell lysates were then blotted for MARCH1 and actin. The band intensities were determined, normalized to actin, and displayed below the blots as fold-increase over control (DMSO). B. DC2.4 and RAW cells +/− HA-MARCH1 were incubated for five hours with the indicated cathepsin inhibitors and then cell lysates were blotted for MARCH1 and the invariant-chain (p31 and p10 fragments indicated). C. Cycloheximide-chase in HA-MARCH1-expressing DC2.4 cells +/− the cathepsin L inhibitor. Graphs show the turnover of MARCH1 from three independent experiments, where the signal intensity of the MARCH1 bands was quantified, normalized to actin, and graphed as the percent-remaining relative to time zero (DMSO-treated samples) +/− SEM. The inset graph shows quantitation of the same samples, except that it begins with the 30 min chase point, with all samples normalized to the 30 min value so that turnover of the MARCH1 remaining at 30 min could be evaluated. *p<0.05, **p<0.01. D. DC2.4 cells + HA-MARCH1 were infected with shRNA lentiviral vectors to knockdown expression of cathepsin S (left panel) or cathepsin L (right panel). For each gene, two different shRNA oligos were designed. At least three days or more after infection, lysates from the different cell lines were blotted with the indicated antibodies.
We next sought to determine which of the leupeptin-sensitive protease(s) are involved in MARCH1 regulation. Here, we took a candidate approach and tested inhibitors specific for different cathepsins; results for cathepsin L and S inhibitors are shown in Figure 3A and 3B. We consistently observed an increase in the steady-state levels of MARCH1 when using an inhibitor of cathepsin L in APC cell lines. This was due to an increase in the stability of MARCH1 as determined by cycloheximide-chase in the presence of the inhibitor (Figure 3C). This stabilization was similar to what we observed when MARCH1-expressing cells were treated with Baf A (see Figure 2B), being most apparent at the later chase time points. By contrast, drug inhibition of cathepsin S did not increase MARCH1 levels in any of the cell lines. Invariant-chain blots were performed to confirm that the drug treatments were effective, since invariant-chain processing involves both cathepsin L and S (Figure 3B). This analysis showed a consistent increase in the p10 fragment and in full-length invariant chain using the cathepsin L inhibitor, as expected (50). The anticipated effect of cathepsin S inhibition on invariant-chain processing was more difficult to show, raising the possibility that the drug inhibition was ineffective. Therefore, we utilized a complementary approach to assess the possible role of cathepsin S in MARCH1 regulation. RNA interference was employed to knockdown the expression of cathepsin S and L. In this case, lentiviral shRNA vectors were designed encoding oligos specific for both cathepsin S and L. As shown in Figure 3D, knockdown of cathepsin S, while effective in reducing cathepsin S levels (with oligo #2), did not stabilize MARCH1 in DC2.4 cells. In contrast, knockdown of cathepsin L did lead to an increase in MARCH1 steady-state levels (Figure 3D). For cathepsin L, the efficiency of knockdown was not as great, explaining the relatively modest increase in MARCH1 protein (with oligo #1; Figure 3D). However, when considered along with the inhibitor experiments, it seems that cathepsin L does contribute, in part, to the inherent instability of MARCH1. Cathepsin L inhibition had no obvious effect on MARCH1 in the fibroblast line (Figure 3A), even though these cells express cathepsin L (not shown) and MARCH1 is unstable in these cells (Figure 1D). In addition, the cathepsin L inhibitor did not consistently increase MARCH1 levels to the same extent as Baf A in each of the cell types tested, so we conclude that multiple lysosomal proteases contribute to the turnover of MARCH1, in addition to cathepsin L. The combined findings for treatment with Baf A, leupeptin, and the cathepsin L inhibitor argue that endo-lysosomes are an important site of MARCH1 turnover.
Domains of MARCH1 involved in stability and function
To define important functional domains of MARCH1, including domains which might influence stability, we generated a series of MARCH1 mutants. Several MARCH1 variants were created, deleting either N-terminal or C-terminal sequences, as well as point mutations (depicted in Figure 4A). The various mutants were expressed in APC cell lines following transduction with retroviral vectors. Initially, the expression of each mutant was characterized by immunoblot for the epitope tag. Figure 4B reveals that the steady-state levels of mutants with N-terminal truncations were significantly higher than wildtype MARCH1. By contrast, the C-terminal deletion mutants were expressed at slightly lower levels that wildtype MARCH1, and the internal deletion mutant (ΔN120-141) was barely detectable. The increased levels of the N-terminal truncation mutants correlated with an increase in the half-life of these mutants as determined by cycloheximide-chase (Figure 4C and data not shown). In addition, intracellular staining/flow cytometry was used to compare the expression levels of each mutant construct, and these results correlated well with the immunoblot results. Specifically, truncations of the N-terminus resulted in higher expression than wildtype MARCH1. We noted that the staining peaks for the N-terminal truncations ΔN1-40 and ΔN1-66 were biphasic, suggesting loss of these constructs within some cells with continued cell passage (discussed below). The C-terminal truncations were similar in expression to wildtype (Figure 4D). These results indicate the presence of a stability determinant within the N-terminus of MARCH1, and in its absence, MARCH1 is expressed at higher levels.
Figure 4. Expression and stability of MARCH1 mutants.

A. Depiction of the various mutations made within MARCH1. All of the mutants possess an N-terminal HA tag. B. Immunoblot of DC2.4 cell lines stably expressing the indicated MARCH1 constructs following retroviral transduction. C. Cycloheximide-chase of DC2.4 cells expressing wildtype MARCH1 or the ΔN1–66 mutant. Two exposures of the MARCH1 (HA) blot are shown. The band intensities were quantified, normalized to actin, and graphed as the percent-remaining from time zero (DMSO). D. Intracellular staining and flow cytometry of DC2.4 cells expressing the indicated MARCH1 constructs. Cells were stained for MARCH1 (HA tag). The gray peak in each histogram represents staining of the parental DC2.4 cells, which represents background staining.
Function of MARCH1 mutants
We next evaluated the function of these MARCH1 mutants, with function defined as the ability to regulate the surface expression of MARCH1 targets. To this end, we transiently transfected each mutant into WT3 fibroblasts (that express CD86) to assess the ability of the mutants to regulate the surface expression of CD86. Figure 5A shows flow cytometric analysis of each transfectant. Transient transfection permits the analysis of function within a cohort of cells that express a range of different levels of MARCH1. Here, a GFP-expressing plasmid was co-transfected to denote transfected cells. The shorter C-terminal truncation (ΔC257-279) retained its ability to decrease CD86 at the cell surface, whereas the larger deletion (ΔC222-279) did not, though both mutants had comparable expression to wildtype MARCH1 (as seen in Figure 4B and 4D). This result is consistent with studies of viral E3 RING-CH ligases such as mK3, kK3, and kK5, where the C-terminal domain is critical for function (51–56), and where examined, for substrate interaction (52, 54). Somewhat surprisingly, deletion of the first 40, or first 66, residues from the N-terminus of MARCH1 did not abolish function toward CD86. This was only accomplished by deletion of the entire N-terminus, including the RING-CH domain (ΔN1-121). We also tested the activity of MARCH1 variants with mutations in potential tyrosine-based sorting motifs, which are typically involved in trafficking within the endo-lysosomal system (YXXΦ; (57, 58)). Three such motifs exist in MARCH1 – one in the N-terminus and two in the C-terminus. Simultaneous mutation of both motifs in the C-terminus (L215S, Y222F) eliminated most of the ability of MARCH1 to regulate CD86 surface levels (Figure 5A), without affecting steady-state levels (Figure 4D). Mutation of each motif, separately, revealed that the L215-containing motif was required for full activity of MARCH1 (data not shown). Mutation of the N-terminal motif (Y118F) had no effect on MARCH1’s ability to downregulate CD86 (Figure 5A).
Figure 5. Comparison of the activity of the MARCH1 mutants.

A. WT3 fibroblasts that stably express CD86 were transiently co-transfected with MARCH1 expressing constructs and a GFP-expressing plasmid. Flow cytometry was performed to analyze surface CD86 levels two days post-transfection. The shaded peak indicates staining of cells transfected with the GFP plasmid alone, gated on the GFP-positive fraction. The heavy trace represents CD86 staining of the MARCH1 co-transfected cells (gated on GFP-positive cells). B. DC2.4 cell lines stably transduced with the indicated MARCH1 mutants were analyzed for surface CD80 and CD86 levels. C. MJDC cells were analyzed for CD86, MHC class II, and CD80 levels after stable retroviral transduction with the indicated MARCH1 mutant constructs. Cells were treated with 100 units/ml of mouse IFN-γ for 18 hours prior to analysis to increase transcription of MHC class II genes. The gray peaks in B and C represent staining of the respective parental cell lines (lacking MARCH1) and the shaded peaks represent staining of parental cells with an isotype-control antibody.
To complement these data, we also expressed MARCH1 mutants stably in APC cell lines. This was done in part to determine whether each mutant could regulate MHC class II surface expression, and if this correlated with the ability of that particular mutant to also regulate CD86. In other words, did mutation of certain regions affect MARCH1’s function equally toward both targets? In addition, this approach provides a means to evaluate the function of each mutant when expressed at comparable levels (comparable transcription) following retroviral transduction. First, we stably transduced MARCH1 variants into DC2.4 cells and found that each mutant which failed to regulate surface CD86 levels in fibroblasts also failed to regulate its expression in DC2.4 cells (Figure 5B). In order to assess the ability of the mutants to regulate MHC class II, we employed an additional cell line, since the levels of MHC class II were quite low on the DC2.4 parental cells, in spite of the fact that they do not express endogenous MARCH1. To circumvent this problem, we generated a “DC-like” cell line (MJDC) by immortalization of BMDC with the J2 retrovirus (28, 29). Subclones of the transduced line were screened for a DC-like phenotype, which included expression of DEC205, CD11c, costimulatory molecules, and most importantly for our analyses, MHC class II (see Supplemental Figure 1). This cell line permits simultaneous analysis of MARCH1 function against CD86 and MHC class II. As shown in Figure 5C, MARCH1 mutants that failed to regulate CD86 surface expression also failed to regulate MHC class II surface expression, such as the larger C-terminal truncation (ΔC229-279). Similarly, the N-terminal truncations (ΔN1-44 and ΔN1-66) that retained activity toward CD86 were also able to downregulate MHC class II (Figure 5C and data not shown). None of the variants of MARCH1, including wildtype, affected CD80 (B7.1) cell surface expression, as reported (16).
In the course of our characterization of the N-terminal truncation mutants lacking either the first 40 or 66 N-terminal residues, we noted that stable cell lines lost protein expression over several cell passages, even in the presence of selecting antibiotics. This is evident from our data, where intracellular MARCH1 staining and flow cytometry revealed two distinct peaks of expression with these mutants (Figure 4D). Similarly, surface CD86 expression in cells expressing these mutants also showed a biphasic pattern wherein a fraction of the stable transductants express wildtype levels of CD86 (Figure 5B and 5C). In these cells, the levels of the MARCH1 mutants continue to drop over time. This pattern was reproducible in independent transductions of multiple cell lines, and argues that these MARCH1 variants are detrimental to the cells, probably by affecting transport pathways. Nonetheless, we can conclude that the N-terminal deletion which retains the RING-CH domain (ΔN1-66) retains its ability to downregulate CD86 and MHC class II surface expression, a fact which is confirmed by the short-term transient transfection assays with this construct where toxicity is not an issue (Figure 5A). On the basis of these results, we conclude that the N-terminal region of MARCH1 contains sequence elements that affect the stability of the protein, but do not directly affect function. Therefore, this N-terminal region is not involved in substrate recognition or recruitment of effector molecules. Conversely, the C-terminal 50 residues of MARCH1 are required for function, but do not influence the stability of MARCH1. A summary of the properties of each mutant is provided in Table 1.
Table I.
Properties of the MARCH1 mutants
| MARCH1 constructs: |
WT | RING mutant (W104A) |
ΔN1-40 | ΔN1-66 | ΔN1-121 | ΔN120-141 | ΔC257-279 | ΔC229-279 | Y118F | L215S, Y222F |
|---|---|---|---|---|---|---|---|---|---|---|
| Steady-state levels | + | + | ++ | +++ | ++++ | − | + | + | + | + |
| Functiona (CD86, MHC II) | +++ | − | +++ | +++ | − | − | ++ | − | +++ | +/− |
| Cellular localizationb | LAMP-1+, Furin +++ | N/Ac | N/A | Furin+, LAMP-1+ & other | Furin+, LAMP-1+ & other | N/A | N/A | LAMP-1++ Furin+++ | N/A | N/A |
Function – determined by analyzing the surface expression of either CD86 or MHC class II in the presence of each MARCH1 construct using flow cytometry (“+” indicates a functional MARCH1 construct; “−” represents a non-functional MARCH1 construct; see Figure 5)
Localization – determined by co-staining with HA antibody against MARCH1 and either Derlin-1, LAMP-1, Furin, or EEA-1 (see Figure 6 and Figure S2 for complete immunofluorescence analysis)
Not available
Subcellular distribution of MARCH1 mutants
Analysis of the MARCH1 mutants defined regions of MARCH1 that affect its expression levels and function. To what extent can these changes be explained by alterations in the trafficking of the MARCH1 mutants? For example, could the increase in the steady-state levels of the MARCH1 N-terminal truncation mutants be explained by redistribution of these mutants to different cellular compartments? To address these questions, we used confocal microscopy to examine the localization of wildtype MARCH1 and the mutants. It has been reported that human MARCH1 resides, in part, in late endosome/lysosomes, due to its partial co-localization with LAMP-1 in HeLa cells (16). In addition, a GFP-fused form of human MARCH1 in HeLa cells was detected in early endosomes (transferrin receptor-positive; (13)). Figure 6A shows the distribution of mouse MARCH1 in DC2.4 cells, where MARCH1 staining is compared to that of representative markers for various cellular compartments including ER (Derlin-1), trans-Golgi (furin), early endosomes (EEA-1), and late endosomes/lysosomes (LAMP-1). Staining for wildtype MARCH1 revealed modest, partial overlap with LAMP-1-positive compartments. This was most apparent in the perinuclear fraction of LAMP-1-positive structures. Perinuclear clustering of endo/lysosomes is not uncommon and is related to microtubules organizing centers (59). Interestingly, it has been shown that MHC class II molecules are preferentially found in perinuclear endo/lysosomes in immature DC, and LPS-induced DC maturation causes redistribution of MHC class II from these structures to the cell surface (8, 9). Additionally, we observed substantial overlap between MARCH1 and the trans-Golgi marker, furin. This staining pattern for wildtype MARCH1 was also seen with MJDC cells (Figure 6A, right panels). Limited co-localization was seen with an ER marker (Derlin-1) and an early endosome marker (EEA-1) (Figure 6A). The presence of MARCH1 within the secretory pathway and within endo-lysosomes is consistent with the trafficking of the MARCH1 target, MHC class II.
The distribution pattern for wildtype MARCH1 was compared against the indicated MARCH1 mutants. Representative images are shown for MARCH1 versus furin and LAMP-1 (Figure 6B), since these markers showed consistent differences between wildtype MARCH1 and some of the mutants. Staining data for the other markers tested is provided in Supplemental Figure 2. To compare the various MARCH1 constructs, multiple individual cells (n=15) were analyzed for the accumulation of each MARCH1 variant in a given compartment, relative to wildtype MARCH1. The extent of co-staining of each form of MARCH1 with a given marker was estimated and the values were normalized to those observed with wildtype MARCH1 to determine whether the mutants were altered in their relative localization with respect to wildtype. The C-terminal truncation (ΔC229-279) mutant displayed a pattern very comparable to wildtype. Golgi staining was pronounced and there was a modest, but significant increase in the lysosomal fraction (Figure 6C). Given the similar distribution of this mutant (ΔC229-279) to wildtype, and the fact that it exhibits similar stability to wildtype, it appears that deletion of the C-terminal 50 residues does not grossly alter the folding or trafficking of MARCH1. Therefore, the inability of this mutant to function (regulate expression of its targets) must result from an inability to recruit its substrates and/or interact with downstream effector molecules.
Each of the N-terminal truncation mutants tested (ΔN1-66 and ΔN1-121) was altered in its localization, when compared to wildtype MARCH1. Most noticeably, these mutants showed less relative accumulation in the Golgi and endo-lysosomes (Figure 6B and 6C). Instead, in addition to the Golgi and endo-lysosome staining, both mutants exhibited abundant staining throughout the cells which did not correlate strongly to ER, or early endosomes (Figure 6B and Supplemental Figure 2). It is currently not clear what vesicular structures harbor these mutant proteins, but since both variants are expressed at higher levels than wildtype MARCH1 (see Figure 4B and 4D), it is possible that the altered distribution of these proteins prevents their rapid degradation. Regardless, it is notable that the ΔN1-66 mutant (but not the RING domain-deleted ΔN1-121 mutant) retains the ability to efficiently downregulate both CD86 and MHC class II (Figure 5). Thus, the altered localization of MARCH1 ΔN1-66 does not dramatically affect its basic function.
DISCUSSION
MARCH1 has emerged as a critical regulator of antigen presentation, serving to suppress the ability of immature APC to activate T cells through its effects on MHC and costimulatory molecules. The transition of APC from the immature to the mature state is essential for effective T cell priming by APC, and is necessarily subject to tight regulation. Though MARCH1 is clearly a component of this regulatory system, much remains to be determined regarding its involvement and control. MARCH1 transcription decreases in response to DC maturation with LPS (13), and increases after stimulation of monocytes with the anti-inflammatory cytokine IL-10 (21). Thus, MARCH1 activity is regulated, at least in part, through changes in gene transcription. Since MARCH1 protein levels appear to be balanced such that relatively small changes significantly alter the cell surface display of MHC class II, then the instability of MARCH1 is an essential component of its regulation. Even in immature DC where MARCH1 is active, MARCH1 turns over quite rapidly. It should be noted that we have not observed any appreciable increase in MARCH1 turnover following maturation with LPS (data not shown), which is known to enhance lysosomal activity in DC (60). It is interesting that the more stable N-terminal mutants appear to be somewhat more potent than wildtype MARCH1 at downregulating CD86 (Figure 5B), consistent with the idea that the inherent stability of MARCH1 maintains its levels at a critical functional threshold.
Lysosomal acidification significantly affects the levels of MARCH1, and this implies proteolysis of MARCH1 within endo-lysosomal compartments, where MHC class II ubiquitination appears to occur (11, 12). However, the topology of MARCH1 presents a potential obstacle to this model. MARCH1 possesses two transmembrane domains, with a short, connecting lumenal domain; the bulk of the molecule resides on the cytosolic face of the membrane (16). How could lysosomal enzymes access MARCH1? It is possible that the lumenal domain is targeted, but replacement of this domain with unrelated sequences did not stabilize MARCH1 (not shown). Further, the size of the fragments of MARCH1 which we detect is not consistent with cleavage within the lumenal domain (data not shown). The most likely explanation is that the cytosolic domains of MARCH1 are exposed to proteases within multivesicular bodies (61). Indeed, electron microscopy has shown an abundance of MHC class II molecules within these structures (3), and this requires ubiquitination of the MHC class II beta-chain (11, 12). Therefore, it is probable that MARCH1 also traffics through multivesicular bodies where it is exposed to the myriad proteases present in this compartment. Among these proteases, cathpesins are likely candidates to affect MARCH1, given their established roles in antigen processing (3, 46). Most of the cathepsins are cysteine proteases (46, 49) and we found that the cysteine/serine protease inhibitor leupeptin was able to stabilize MARCH1. Further, inhibition of cathepsin L increased MARCH1 levels through stabilization, but not in all cell types. We failed to observe any stabilizing effect of cathepsin S inhibition/knockdown on MARCH1. From our results, we conclude that MARCH1 protein levels are regulated by multiple, redundant proteases within lysosomes, including cathepsin L. A similar finding has emerged for the processing of Toll-like Receptor 9, which must be cleaved within endo-lysosomes prior to signaling, and this cleavage event can be mediated by different proteases (62, 63).
Our characterization of mutant forms of MARCH1 complemented our studies of the factors which affect MARCH1 protein levels. In particular, we identified a region of MARCH1 which influenced its stability. Removal of as few as the first 40 residues of the N-terminus affected the stability of MARCH1, without compromising activity. The increased stability of the N-terminal truncations was correlated with a change in the subcellular distribution of the mutants relative to wildtype. Most notably, in addition to the Golgi and lysosomal staining seen with wildtype MARCH1, these mutants exhibited abundant vesicular staining throughout the cell. The distribution of MARCH1 raises questions about its subcellular site of action. At steady-state, we observed the most pronounced co-localization between MARCH1 and a trans-Golgi marker, with relatively little MARCH1 present within endo-lysosomes. However, since MARCH1 turnover requires lysosomal activity, it must traffic through this compartment. In the case of MHC class II, it seems that the effects of MARCH1 are manifested in a post-Golgi compartment, after processing of the invariant-chain (11, 13), resulting in rapid endocytosis of MHC class II from the cell surface (12–14). Consistent with this finding, it was shown that human MARCH1 can be detected in early endosomes (transferrin-receptor-positive), but not later, HLA-DM-positive compartments (13). In this instance, MARCH1 was expressed as a fusion with GFP, which could affect its localization, perhaps causing some accumulation in early endosomes where it could continue to ubiquitinate MHC class II. Though MHC class II molecules are quickly internalized in the presence of MARCH1, it is possible that ubiquitination occurs prior to the initial arrival of MHC class II at the cell surface or during recycling within early endosomes. Upon reaching the cell surface, ubiquitin could then exert its effects on MHC class II. Whatever the case, MARCH1 itself appears to traffic through the late endocytic compartment, at least transiently, in order to be degraded.
Collectively, the available data suggest a dynamic pattern of trafficking along the endocytic pathway for MARCH1, between the trans-Golgi, early endosomes, and late endosomes. Our results indicate that this trafficking requires information within the N-terminus. However, disruption of this pathway does not necessarily abolish function. Rather, it affects the stability of MARCH1, which appears to be important for maintaining the proper levels of MARCH1. Deletion of the C-terminal 50 residues (ΔC229-279) did not dramatically affect localization or stability of MARCH1. Notably, this mutant retains both of the C-terminal YXXΦ motifs (Figure 4A). However, mutation of these motifs affected MARCH1 function, but not expression levels, arguing that they are not required for trafficking into endo-lysosmes. Rather, these residues may be important for substrate interaction or recruitment of downstream effector molecules. While it remains to be determined exactly where and when MHC class II and CD86 encounter MARCH1 during biogenesis, and how this process is influenced by APC maturation, it seems likely that these two targets of MARCH1 share some common steps in their trafficking. This may help explain how these two unrelated proteins can both be targeted by the same E3 ligase. A curious feature of the viral RING-CH E3 ligases is the ability of some to target multiple, unrelated substrates (18). As a whole, the basis of substrate recognition by viral RING-CH molecules is not well understood. Our characterization of the viral mK3 protein has demonstrated a clear requirement for “adapter-type” proteins in substrate recruitment (31, 52). MARCH1 may also require cofactors to assist in recruitment of its distinct substrates, and such cofactors would likely tie into the trafficking pathways utilized by MARCH1 and its substrates. Elucidation of the full spectrum of molecules involved in MARCH1-dependent regulation of antigen presentation will be essential to understand how MARCH1 may contribute to both immune activation and tolerance.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. David Elliott and Dr. Jean Wilson for assistance with image analysis, to Kathleen Kunke and Carl Boswell for technical support, to Dr. Kathleen Corcoran and Maria Ordaz for critical reading of the manuscript, to Dr. Marilyn Halonen and Dr. Felicia Goodrum for providing access to critical equipment, to Paula Campbell and Barb Carolus for flow cytometry assistance, and to Dr. Scott Boitano and Dr. Howard Young for providing reagents.
ABBREVIATIONS
- MARCH1
Membrane-associated RING-CH protein
- RING
Really Interesting New Gene
- BMDC
Bone marrow-derived dendritic cell
Footnotes
This work was supported by grants from the National Institutes of Health (AI060723) and the Arizona Biomedical Research Commission (8-123).
References
- 1.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 4.Berger AC, Roche PA. MHC class II transport at a glance. Journal of cell science. 2009;122:1–4. doi: 10.1242/jcs.035089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, Inaba K, Steinman RM, Mellman I. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388:787–792. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 6.Villadangos JA, Cardoso M, Steptoe RJ, van Berkel D, Pooley J, Carbone FR, Shortman K. MHC class II expression is regulated in dendritic cells independently of invariant chain degradation. Immunity. 2001;14:739–749. doi: 10.1016/s1074-7613(01)00148-0. [DOI] [PubMed] [Google Scholar]
- 7.Turley SJ, Inaba K, Garrett WS, Ebersold M, Unternaehrer J, Steinman RM, Mellman I. Transport of peptide-MHC class II complexes in developing dendritic cells. Science (New York, N Y) 2000;288:522–527. doi: 10.1126/science.288.5465.522. [DOI] [PubMed] [Google Scholar]
- 8.Chow A, Toomre D, Garrett W, Mellman I. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature. 2002;418:988–994. doi: 10.1038/nature01006. [DOI] [PubMed] [Google Scholar]
- 9.Boes M, Cerny J, Massol R, Op den Brouw M, Kirchhausen T, Chen J, Ploegh HL. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 2002;418:983–988. doi: 10.1038/nature01004. [DOI] [PubMed] [Google Scholar]
- 10.Ohmura-Hoshino M, Matsuki Y, Aoki M, Goto E, Mito M, Uematsu M, Kakiuchi T, Hotta H, Ishido S. Inhibition of MHC class II expression and immune responses by c-MIR. J Immunol. 2006;177:341–354. doi: 10.4049/jimmunol.177.1.341. [DOI] [PubMed] [Google Scholar]
- 11.van Niel G, Wubbolts R, Ten Broeke T, Buschow SI, Ossendorp FA, Melief CJ, Raposo G, van Balkom BW, Stoorvogel W. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity. 2006;25:885–894. doi: 10.1016/j.immuni.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Shin JS, Ebersold M, Pypaert M, Delamarre L, Hartley A, Mellman I. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature. 2006;444:115–118. doi: 10.1038/nature05261. [DOI] [PubMed] [Google Scholar]
- 13.De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, Gatti E. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3491–3496. doi: 10.1073/pnas.0708874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, Uematsu M, Hasegawa T, Koseki H, Ohara O, Nakayama M, Toyooka K, Matsuoka K, Hotta H, Yamamoto A, Ishido S. Novel regulation of MHC class II function in B cells. The EMBO journal. 2007;26:846–854. doi: 10.1038/sj.emboj.7601556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young LJ, Wilson NS, Schnorrer P, Proietto A, ten Broeke T, Matsuki Y, Mount AM, Belz GT, O’Keeffe M, Ohmura-Hoshino M, Ishido S, Stoorvogel W, Heath WR, Shortman K, Villadangos JA. Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nature immunology. 2008;9:1244–1252. doi: 10.1038/ni.1665. [DOI] [PubMed] [Google Scholar]
- 16.Bartee E, Mansouri M, Hovey Nerenberg BT, Gouveia K, Fruh K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. Journal of virology. 2004;78:1109–1120. doi: 10.1128/JVI.78.3.1109-1120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goto E, Ishido S, Sato Y, Ohgimoto S, Ohgimoto K, Nagano-Fujii M, Hotta H. c-MIR, a human E3 ubiquitin ligase, is a functional homolog of herpesvirus proteins MIR1 and MIR2 and has similar activity. J Biol Chem. 2003;278:14657–14668. doi: 10.1074/jbc.M211285200. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Herr RA, Hansen T. Viral and cellular MARCH ubiquitin ligases and cancer. Seminars in cancer biology. 2008;18:441–450. doi: 10.1016/j.semcancer.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohmura-Hoshino M, Goto E, Matsuki Y, Aoki M, Mito M, Uematsu M, Hotta H, Ishido S. A novel family of membrane-bound E3 ubiquitin ligases. J Biochem. 2006;140:147–154. doi: 10.1093/jb/mvj160. [DOI] [PubMed] [Google Scholar]
- 20.Goto E, Ishido S, Sato Y, Ohgimoto S, Ohgimoto K, Nagano-Fujii M, Hotta H. c-MIR, a Human E3 Ubiquitin Ligase, Is a Functional Homolog of Herpesvirus Proteins MIR1 and MIR2 and Has Similar Activity. J Biol Chem. 2003;278:14657–14668. doi: 10.1074/jbc.M211285200. [DOI] [PubMed] [Google Scholar]
- 21.Thibodeau J, Bourgeois-Daigneault MC, Huppe G, Tremblay J, Aumont A, Houde M, Bartee E, Brunet A, Gauvreau ME, de Gassart A, Gatti E, Baril M, Cloutier M, Bontron S, Fruh K, Lamarre D, Steimle V. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol. 2008;38:1225–1230. doi: 10.1002/eji.200737902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- 23.Pretell J, Greenfield RS, Tevethia SS. Biology of simian virus 40 (SV40) transplantation antigen (TrAg). V In vitro demonstration of SV40 TrAg in SV40 infected nonpermissive mouse cells by the lymphocyte mediated cytotoxicity assay. Virology. 1979;97:32–41. doi: 10.1016/0042-6822(79)90370-2. [DOI] [PubMed] [Google Scholar]
- 24.Ralph P, Nakoinz I. Antibody-dependent killing of erythrocyte and tumor targets by macrophage-related cell lines: enhancement by PPD and LPS. J Immunol. 1977;119:950–954. [PubMed] [Google Scholar]
- 25.Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. Establishment and characterization of BALB/c lymphoma lines with B cell properties. J Immunol. 1979;122:549–554. [PubMed] [Google Scholar]
- 26.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. The Journal of experimental medicine. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. The Journal of experimental medicine. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blasi E, Radzioch D, Merletti L, Varesio L. Generation of macrophage cell line from fresh bone marrow cells with a myc/raf recombinant retrovirus. Cancer Biochem Biophys. 1989;10:303–317. [PubMed] [Google Scholar]
- 29.Blasi E, Mathieson BJ, Varesio L, Cleveland JL, Borchert PA, Rapp UR. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature. 1985;318:667–670. doi: 10.1038/318667a0. [DOI] [PubMed] [Google Scholar]
- 30.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 31.Corcoran K, Wang X, Lybarger L. Adapter-mediated Substrate Selection for Endoplasmic Reticulum-associated Degradation. J Biol Chem. 2009;284:17475–17487. doi: 10.1074/jbc.M808258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Connors R, Harris MR, Hansen TH, Lybarger L. Requirements for the selective degradation of endoplasmic reticulum-resident major histocompatibility complex class I proteins by the viral immune evasion molecule mK3. Journal of virology. 2005;79:4099–4108. doi: 10.1128/JVI.79.7.4099-4108.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene therapy. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 34.Swift S, Lorens J, Achacoso P, Nolan GP. Rapid Production of Retroviruses for Efficient Gene Delivery to Mammalian Cells Using 293T Cell–Based Systems. In: Coligan JE, Kruisbeek A, Margulies DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. Wiley; New York: 1999. pp. 1–17. [DOI] [PubMed] [Google Scholar]
- 35.Lybarger L, Wang X, Harris MR, Virgin HW, Hansen TH. Virus subversion of the MHC class I peptide-loading complex. Immunity. 2003;18:121–130. doi: 10.1016/s1074-7613(02)00509-5. [DOI] [PubMed] [Google Scholar]
- 36.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolte S, FPC A guided tour into subcellular colocalization analysis in light microscopy. Journal of Microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 39.Dodd RB, Allen MD, Brown SE, Sanderson CM, Duncan LM, Lehner PJ, Bycroft M, Read RJ. Solution structure of the Kaposi’s sarcoma-associated herpesvirus K3 N-terminal domain reveals a Novel E2-binding C4HC3-type RING domain. The Journal of biological chemistry. 2004;279:53840–53847. doi: 10.1074/jbc.M409662200. [DOI] [PubMed] [Google Scholar]
- 40.Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102:533–539. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 41.Nathan JA, Sengupta S, Wood SA, Admon A, Markson G, Sanderson C, Lehner PJ. The ubiquitin E3 ligase MARCH7 is differentially regulated by the deubiquitylating enzymes USP7 and USP9X. Traffic (Copenhagen, Denmark) 2008;9:1130–1145. doi: 10.1111/j.1600-0854.2008.00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- 43.Kostova Z, Wolf DH. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003;22:2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsing LC, Rudensky AY. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol Rev. 2005;207:229–241. doi: 10.1111/j.0105-2896.2005.00310.x. [DOI] [PubMed] [Google Scholar]
- 46.Zavasnik-Bergant T, Turk B. Cysteine cathepsins in the immune response. Tissue Antigens. 2006;67:349–355. doi: 10.1111/j.1399-0039.2006.00585.x. [DOI] [PubMed] [Google Scholar]
- 47.Beers C, Honey K, Fink S, Forbush K, Rudensky A. Differential regulation of cathepsin S and cathepsin L in interferon gamma-treated macrophages. The Journal of experimental medicine. 2003;197:169–179. doi: 10.1084/jem.20020978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honey K, Duff M, Beers C, Brissette WH, Elliott EA, Peters C, Maric M, Cresswell P, Rudensky A. Cathepsin S regulates the expression of cathepsin L and the turnover of gamma-interferon-inducible lysosomal thiol reductase in B lymphocytes. The Journal of biological chemistry. 2001;276:22573–22578. doi: 10.1074/jbc.M101851200. [DOI] [PubMed] [Google Scholar]
- 49.Chapman HA. Endosomal proteases in antigen presentation. Curr Opin Immunol. 2006;18:78–84. doi: 10.1016/j.coi.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 50.Honey K, Nakagawa T, Peters C, Rudensky A. Cathepsin L Regulates CD4+ T Cell Selection Independently of Its Effect on Invariant Chain: A Role in the Generation of Positively Selecting Peptide Ligands. J Exp Med. 2002;195:1349–1358. doi: 10.1084/jem.20011904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Means RE, Ishido S, Alvarez X, Jung JU. Multiple endocytic trafficking pathways of MHC class I molecules induced by a Herpesvirus protein. EMBO J. 2002;21:1638–1649. doi: 10.1093/emboj/21.7.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Lybarger L, Connors R, Harris MR, Hansen TH. Model for the interaction of gammaherpesvirus 68 RING-CH finger protein mK3 with major histocompatibility complex class I and the peptide-loading complex. J Virol. 2004;78:8673–8686. doi: 10.1128/JVI.78.16.8673-8686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mansouri M, Douglas J, Rose PP, Gouveia K, Thomas G, Means RE, Moses AV, Fruh K. Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood. 2006;108:1932–1940. doi: 10.1182/blood-2005-11-4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boname JM, Stevenson PG. MHC class I ubiquitination by a viral PHD/LAP finger protein. Immunity. 2001;15:627–636. doi: 10.1016/s1074-7613(01)00213-8. [DOI] [PubMed] [Google Scholar]
- 55.Sanchez DJ, Coscoy L, Ganem D. Functional organization of MIR2, a novel viral regulator of selective endocytosis. J Biol Chem. 2002;277:6124–6130. doi: 10.1074/jbc.M110265200. [DOI] [PubMed] [Google Scholar]
- 56.Coscoy L, Sanchez DJ, Ganem D. A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J Cell Biol. 2001;155:1265–1273. doi: 10.1083/jcb.200111010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 58.Lizee G, Basha G, Jefferies WA. Tails of wonder: endocytic-sorting motifs key for exogenous antigen presentation. Trends Immunol. 2005;26:141–149. doi: 10.1016/j.it.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 59.Matteoni R, Kreis TE. Translocation and clustering of endosomes and lysosomes depends on microtubules. The Journal of cell biology. 1987;105:1253–1265. doi: 10.1083/jcb.105.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science (New York, N Y) 2003;299:1400–1403. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- 61.Piper RC, Katzmann DJ. Biogenesis and function of multivesicular bodies. Annual review of cell and developmental biology. 2007;23:519–547. doi: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nature immunology. 2008;9:1407–1414. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. Journal of cell science. 1992;103(Pt 3):857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.