

Abstract
Hydrogen bonding networks proximal to metal centers are emerging as a viable means for controlling secondary coordination spheres. This has led to the regulation of reactivity and isolation of complexes with new structural motifs. We have used the tridenate ligand bis[(N′-tert-butylureido)-N-ethyl]-N-methylaminato ([H21]2-) that contains two hydrogen bond donors to examine the oxidation of the FeII—acetate complex, [FeIIH21(η2-OAc)]- with dioxygen, amine N-oxides, and xylyl azide. A complex with FeIII—O—FeIII core results from the oxidation with dioxygen and amine N-oxides, in which the oxo ligand is involved in hydrogen bonding to the [H21]2- ligand. A distinctly different hydrogen bonding network was found in FeIII dimer isolated from the reaction with the xylyl azide: a rare FeIII—N(R)—FeIII core was observed that does not have hydrogen bonds to the bridging nitrogen atom. The intramolecular H-bond networks within these dimers appear to adjust to the presence of the bridging species and rearrange to its size and electron density.
1. Introduction
Oxo and imido complexes of transition metal ions are actively studied as species involved in the oxidative group transfer of oxo (O2-) and imido (NR2-) species to substrates such as alkanes and alkenes.i,ii,iii We have been investigating the effects of non-covalent interactions on the metal-mediated transformation involving oxo and imido units. As part of this program, we have examined structure-function relationships in synthetic systems with hydrogen bonds (H-bonds),iv which has led to the development of urea-based ligands that provide intramolecular H-bond donors proxmal to coordinatively unsaturated metal centers. Most of these systems are sterically-bulky tripodal ligands, such as the C3-symmetrical tris[(N′-tert-butylureido)-N-ethyl]aminato ligand [H3buea]3-, which favor formation of monomeric iron complexes with terminal chalcogenido,v amido,vi and hydroxo ligands.vii We recently introduce the related tridentate ligand bis[(N′-tert-butylureido)-N-ethyl]-N-methylaminato ([H21]2-),viii in which one of the urea arms has been replaced with a methyl group. The molecular structure of the FeII—OAc, [FeIIH21(η2-OAc)]- showed a five-coordinate structure with intramolecular H-bonds involving the bidentate acetate ligand. The H-bond cavity surrounding the Fe—acetate moiety is relatively open, suggesting that polynuclear species could be formed upon oxidation. Reported herein are examples of dimeric FeIII—(μ-O)—FeIII and a rare FeIII—(μ-NR)—FeIII complexes formed upon the oxidation of [FeII(H21)(η2-OAc)]- with O2 and xylyl azide, respectively. These complexes have different H-bond networks that reflect the nature of the bridging atom between the iron centers.
2. Experimental
2.1 General
All reagents were purchased from commercial sources and used as received, unless noted otherwise. Anhydrous solvents were purchased form Aldrich. The syntheses of all metal complexes were conducted in a Vacuum Atmospheres, Co. drybox under an argon atmosphere. Elemental analysis of all compounds was performed by Desert Analytics, Tucson, AZ. All samples were dried in vacuo before analysis. FT-IR spectroscopy was used to corroborate the presence of solvates. Xylyl azide was prepared according to the literature.ix
2.2 Physical Methods
Fourier transform infrared spectra were recorded on an ATI Mattson Genesis Series FTIR spectrometer and are reported in wavenumbers. Solid samples were prepared in mineral oil and run between KBr plates. Perpendicular-mode X-band electron paramagnetic resonance spectra were collected using a Bruker EMX spectrometer equipped with an ER4102ST cavity and ER041XG microwave bridge. The instrument was previously calibrated using DPPH. EPR spectra were collected using the following spectrometer settings: attenuation, 25 dB; microwave frequency, 9.46 GHz; microwave power, 0.638 mW; sweep width, 5000 G; modulation frequency, 100 kHz; modulation amplitude, 10.02 G; gain, 1.00 × 103; conversion time, 81.920 ms; time constant, 655.36 ms; resolution, 1024 points. Electronic spectra were collected on a Cary 50 spectrophotometer using 1.00 mm quartz cuvets.
2.3 Preparation of the Complexes
2.3.1. Bis{bis[(N′-tert-butylureaylato)-N-ethyl]-N-methylaminatoferrate(III)}(μ-oxo), [(FeIIIH21)2(μ-O)]
A solution of K[FeIIH21(η2-OAc)]viii (100.0 mg, 0.214 mmol) in anhydrous N,N-dimethylacetamide (DMA, 5 mL) was treated with trimethylamine N-oxide (8.1 mg, 0.108 mmol) under an Ar atmosphere. The initially light yellow solution rapidly turned red-orange and was stirred for 1 h. The DMA was removed under vacuum and the residue dissolved in anhydrous methylene chloride. The resulting orange solution was then filtered to remove insoluble KOAc (20 mg, 0.20 mmol). Anhydrous pentane was layered on the methylene chloride solutions in two vials. The orange solid that precipitated was collected by filtration and washed with pentane to yield 67 mg (83%). Anal. Calcd (found) for [(FeIIIH21)2(μ-O)], C30H62Fe2N10O5: C, 47.75 (47.44); H, 8.28 (7.74); N, 18.56 (18.08). FTIR (Nujol, cm-1): v 3301s, 3087ms (NH), 1593s, 1564s, 1500s, 1349s, 1304s, 1217s, 1070s, 905m, 777m, 658w, 580w λmax/nm (CH2Cl2, ε, M-1 cm-1): 316 (7700), 361 (6900).
2.3.2 Bis{Bis[(N′-tert-butylureaylato)-N-ethyl]-N-methylaminatoferrate(III)}-(μ-xylylimido), [(FeIIIH21)2(μ-N-Xylyl)]
A solution of K[FeIIH21(η2-OAc)] (99.4 mg, 0.213 mmol) in anhydrous N,N-dimethylacetamide (DMA, 5 mL) was treated with xylyl azide (18.2 mg, 0.124 mmol) under an Ar atmosphere. The initially light yellow solution rapidly turned dark purple with evolution of gas and was stirred for 1 h. The DMA was removed under vacuum and the residue dissolved in anhydrous methylene chloride. The resulting purple solution was then filtered to remove insoluble KOAc (19 mg, 0.19 mmol). Methylene chloride was removed under vacuum and solid isolated crude to yield 85 mg (93%). Anal. Calcd (found) for [(FeIIIH21)2(μ-N-Xylyl)], C38H71Fe2N11O4: C, 53.21 (52.85); H, 8.34 (8.30); N, 17.96 (17.71). FTIR (Nujol, cm-1): v 3290m, 3181m (NH), 1590s, 1557s, 1377s, 1294s, 1210s, 1061m, 900m, 760m, 659w, 593w. λmax/nm (CH2Cl2, ε, M-1 cm-1): 508 (5400), 417 (5000).
2.4 Crystal Structure Determination
Intensity data for the compounds were collected using a Bruker APEX CCD area detectorx mounted on a Bruker D8 goniometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The data were collected at 100(2) K. For [(FeIIIH21)]2(μ-O) the intensity data were measured as a series of ω oscillation frames each of 0.25° for 90 s / frame. Coverage of unique data was 99.4% complete to 25.00 degrees in θ. For [(FeIIIH21)]2(μ-N-Xylyl)·CH2Cl2 the intensity data were measured as a series of ω oscillation frames each of 0.25° for 20 s / frame. Coverage of unique data was 94.1% complete to 26.00 degrees in θ.
2.4.1 [(FeIIIH21)2(μ-O)]
crystallized in the monoclinic space group P21/c, which was determined by systematic absences and statistical methods and verified by subsequent refinement. Cell parameters were determined from non-linear least squares fit of 2276 peaks in the range 2.18 < θ < 24.18°. A total of 21286 data were measured in the range 2.00 < θ < 25.00°. The data were corrected for absorption by the semi-empirical methodxi from equivalent reflections giving minimum and maximum transmission factors of 0.7151 and 0.9839. The structure was solved by direct methods and refined by full-matrix least-squares methods on F2.xii The carbons (C2B, C12B and C22B) around N1B were disordered and modeled in two orientations with refined occupancies of 0.833(6) and 0.167(6) for the unprimed and primed atoms, respectively. Restraints on the positional and diplacement parameters of the disordered atoms were included in the refinement. The displacement ellipsoids were drawn at the 50% probability level. Hydrogen atom positions were initially determined by geometry and refined by a riding model. Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atom displacement parameters were set to 1.2 (1.5 for methyl) times the displacement parameters of the bonded atoms. A total of 4552 parameters were refined against 28 restraints and 6609 data to give wR(F2) = 0.1279 and S = 0.999 for weights of w = 1/[σ2(F2) + (0.0600 P)2], where P = [Fo2 + 2Fc2] / 3. The final R(F) was 0.0572 for the 4196 observed, [F > 4σ(F)], data. The largest shift/s.u. was 0.001 in the final refinement cycle. The final difference map had maxima and minima of 1.130 and -0.318 e/Å3, respectively.
2.4.2 [(FeIIIH21)2(μ-N-xylyl)]·CH2Cl2
crystallized in the triclinic space group P 1, which was determined by statistical tests and verified by subsequent refinement. Cell parameters were determined from non-linear least squares fit of 5542 peaks in the range 2.50 < θ < 30.00°. A total of 19783 data were measured in the range 1.94 < θ < 30.03°. The data were corrected for absorption by the semi-empirical methodxi from equivalent reflections giving minimum and maximum transmission factors of 0.7974 and 0.9413. The structure was solved by direct methods and refined by full-matrix least-squares methods on F2.xii One side arm, N7A-C11 A, of one ligand was disordered and was modeled in two orientations with refined occupancies of 0.628(9) and 0.372(9) for the unprimed and primed atoms. The solvent molecule was disordered and was modeled in two orientations with refined occupancies of 0.570(11) and 0.430(11) for the unprimed and primed atoms. Restraints on the positional and displacement parameters of the disordered atoms were required. The displacement ellipsoids were drawn at the 50% probability level. Hydrogen atom positions were initially determined by geometry and refined by a riding model. Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atom displacement parameters were set to 1.2 (1.5 for methyl) times the displacement parameters of the bonded atoms. A total of 597 parameters were refined against 64 restraints and 12512 data to give wR(F2) = 0.1292 and S = 1.022 for weights of w = 1/[σ2(F2) + (0.0760 P)2], where P = [Fo2 + 2Fc2] / 3. The final R(F) was 0.0480 for the 9371 observed, [F > 4σ(F)], data. The largest shift/s.u. was 0.001 in the final refinement cycle. The final difference map had maxima and minima of 0.744 and -0.368 e/Å3, respectively.
3. Results and Discussion
3.1 Synthesis and Solution Properties
Scheme 1 outlines the preparative routes to [(FeIIIH21)2(μ-X)] (X = O2- and N-xylyl2-). Addition of either 0.25 equivalent (equiv) of O2 or 0.5 equiv of trimethylamine-N-oxide to a dry dimethylacetamide (DMA) solution of K[FeIIH21(η2-OAc)] resulted in an immediate color change form light yellow to deep orange. Removal of the solvent after 1 h and dissolving the residue CH2Cl2 before filtering roomed 1 equiv of KOAc. [(FeIIIH21)2(μ-O)] was isolated as orange crystals in 80% yield after layering the CH2Cl2 solution of the compound with pentane. The bridged imido complexes [(FeIIIH21)2(μ-N-xylyl)] was prepared analogously by treating K[FeIIH21(η-OAc)] in DMA with 0.5 equiv of xylylazide. This immediately resulted in a color change from light yellow to dark purple with concomitant evolution of N2. The dark purple bridging imido complex, [(FeIIIH21)2(μ-N-xylyl)], was obtained in its purest form by simply removing volatiles after dissolving and filtering the crude product in CH2Cl2. Error! Bookmark not defined.b Single crystals could be obtained from CH2Cl2/pentane or THF/pentane mixtures, but prolonged standing in the recystallization media led to impure product.
Scheme 1.
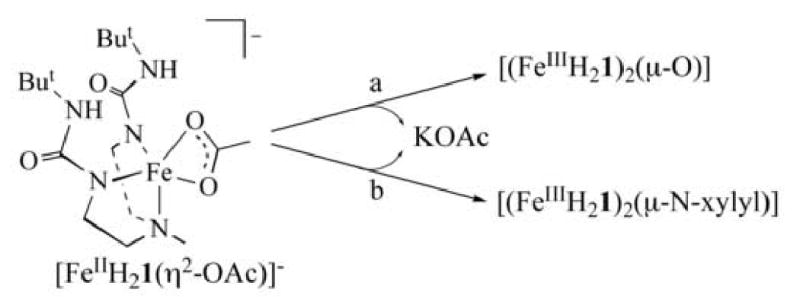
Conditions: (a) 0.5 equiv. TMANO or 0.25 equiv. O2, DMA, rt, Ar; (b) 0.5 equiv. N3Xylyl, DMA, rt, Ar.
Solutions of both complexes are intensely colored: the absorbance spectrum of [(FeIIIH21)2(μ-N-xylyl)] in CH2Cl2 has bands at λmax (εM) = 508 (5400) and 417 (5000) nm, while bands for [(FeIIIH21)2(μ-O)] in CH2Cl2 are at 361 (6900) and 316 (7700) nm. Frozen solution of [(FeIIIH21)2(μ-N-xylyl)] and [(FeIIIH21)2(μ-O) were EPR silent at 4 and 77 K.
3.2 Crystal Structures
X-ray diffraction measurements reveal dimeric structures for each complex. Table 1 contains crystallographic details, Table 2 pertinent metrical results, and Figures 1 and 2 contain the thermal ellipsoid plots and for [(FeIIIH21)2(μ-O)] and [(FeIIIH21)2(μ-N-xylyl)]·CH2Cl2, respectively. Each of the metal ions has a trigonal bipyramidal coordination geometry. Donor atoms from the [H21]2- ligands occupy four of the coordination sites on the iron ions; the bridging ligand occupies the fifth site. One urea arm from each [H21]2- ligand bridges between the iron centers to form two μ-1,3-(κN:κO)-ureate linkages. In this binding mode, the deprotonated urea N- atom coordinates to one iron ion, while its carbonyl oxygen atom binds to the other metal center.
Table 1.
Crystallographic data for [(FeIIIH21)2(μ-O)] and [(FeIIIH21)2(μ-N-xylyl)]·CH2Cl2
| [(FeIIIH21)2(μ-O)] | [(FeIIIH21)2(μ-N-xylyl)]·CH2Cl2 | |
|---|---|---|
| molecular formula | C30H62Fe2N10O5 | C39H73Cl2Fe2N11O4 |
| formula weight | 754.60 | 942.68 |
| T (K) | 100(2) | 100(2) |
| space group | P21/c | P1̄ |
| a (Å) | 11.9219(10) | 11.9975(7) |
| b (Å) | 20.3802(17) | 13.5221(8) |
| c (Å) | 15.9341(13) | 16.3671(9) |
| α(deg) | 90 | 78.507(2) |
| β(deg) | 103.349(2) | 73.002(2) |
| γ(deg) | 90 | 72.102(2) |
| Z | 4 (Z′ = 1) | 2 (Z′ =1) |
| V (Å3) | 3781.9(5) | 2398.9(2) |
| δ calc (Mg/m3) | 1.325 | 1.305 |
| Ra | 0.0572 | 0.00480 |
| Rwb | 0.1279 | 0.1292 |
| GOFc | 0.999 | 1.022 |
R = [Σ|ΔF|/Σ|Fo|].
Rw = [Σω(ΔF)2/ΣωFo2].
goodness of fit on F2.
Table 2.
Selected bond distances (Å) and angles (°) for [(FeIIIH21)2(μ-O)] and [(FeIIIH21)2(μ-N-xylyl)]·CH2Cl2.a
| Bond lengths or angles | [(FeIIIH21)2(μ-O)] | [(FeIIIH21)2(μ-N-xylyl)]·CH2Cl2 |
|---|---|---|
| Fe1A—X | 1.818(3) | 1.872(2) |
| Fe1B—X | 1.827(3) | 1.867(3) |
| Fe1A—N1A | 2.260(3) | 2.266(2) |
| Fe1A—N4A | 2.045(3) | 2.034(2) |
| Fe1A—N14A | 2.007(3) | 2.011(2) |
| Fe1A—O6A | 1.973(3) | 2.057(1) |
| Fe1B—N1B | 2.302(3) | 2.288(2) |
| Fe1B—N4B | 2.047(4) | 2.037(2) |
| Fe1B—N14B | 1.997(3) | 2.013(20 |
| Fe1B—O6A | 1.984(3) | 2.051(2) |
| N17A⋯X | 3.062(4) | — |
| N17B⋯X | 2.935(5) | — |
| Fe1A⋯Fe1B | 3.005(3) | 2.9741(4) |
| Fe1A-X-Fe1B | 111.02(2) | 105.37(8) |
| N1A-Fe1A-X | 174.4(1) | 101.91(7) |
| N1A-Fe1A-N4A | 76.9(1) | 74.84(7) |
| N1A-Fe1A-N14A | 77.7(1) | 79.01(7) |
| N1A-Fe1A-O6B | 89.5(1) | 151.34(6) |
| N4A-Fe1A-X | 97.9(1) | 105.99(7) |
| N4A-Fe1A-N14A | 129.9(1) | 133.96(7) |
| N4A-Fe1A-O6B | 114.8(1) | 89.27(7) |
| N14A-Fe1A-X | 104.3(1) | 116.15(7) |
| N14A-Fe1A-O6B | 110.7(1) | 96.45(7) |
| O6B-Fe1A-X | 94.6(1) | 105.37(6) |
| N1B-Fe1B-X | 172.7(1) | 105.81(7) |
| N1B-Fe1B-N4B | 77.1(1) | 75.83(7) |
| N1B-Fe1B-N14B | 79.0(1) | 79.25(7) |
| N1B-Fe1B-O6A | 88.7(1) | 147.13(6) |
| N4B-Fe1B-X | 95.7(1) | 105.53(7) |
| N4B-Fe1B-N14B | 126.1(1) | 136.67(7) |
| N4B-Fe1B-O6A | 123.3(1) | 86.09(7) |
| N14B-Fe1B-X | 106.5(1) | 115.14(7) |
| N14B-Fe1B-O6A | 103.4(1) | 96.65(6) |
| O6A-Fe1B-X | 94.5(1) | 105.37(7) |
X denotes the bridging atom for each species: O1 for [(FeIIIH21)2(μ-O)] and N1C for [(FeIIIH21)2(μ-N-xylyl)]·CH2Cl2.
Figure 1.
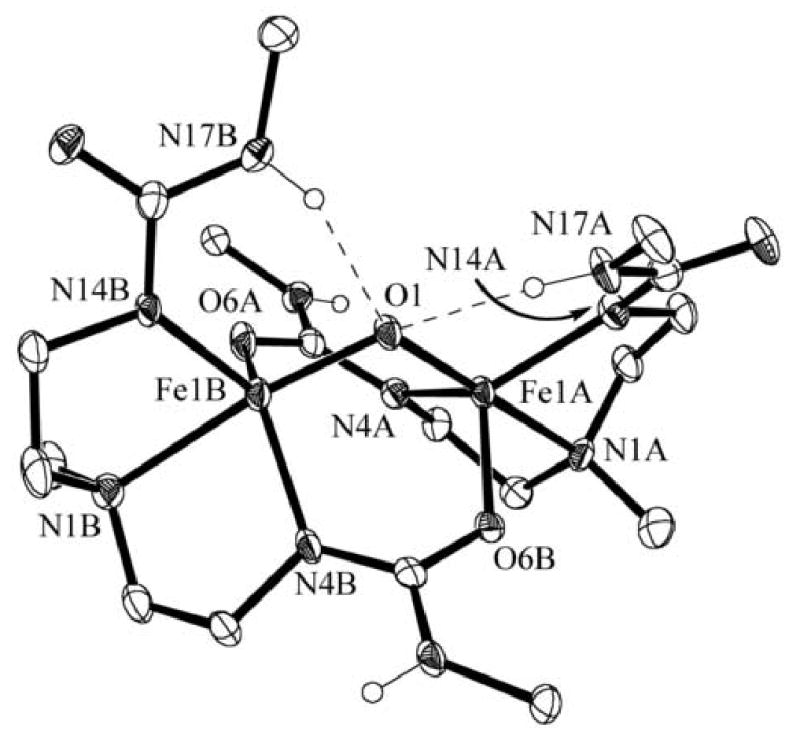
Molecular structure of [(FeIIIH21)2(μ-O)]. The ellipsoids are drawn at the 50% probability level. Non-urea hydrogen atoms and the methyl groups of the tert-butyl functionalities have been removed for clarity.
Figure 2.
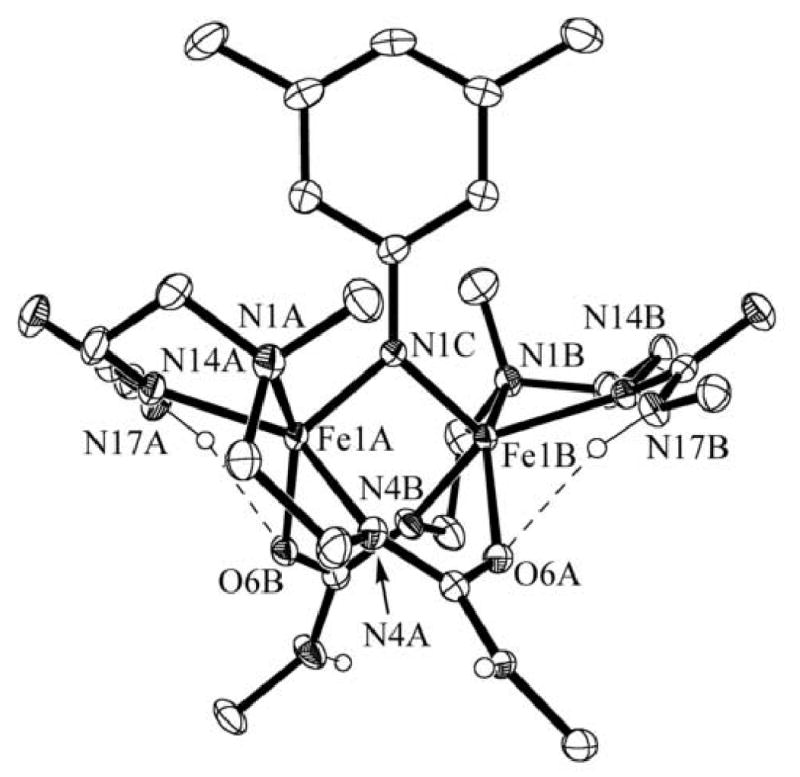
Molecular structure of [(FeIIIH21)2(μ-N-xylyl)]. The ellipsoids are drawn at the 50% probability level. Non-urea hydrogen atoms and the methyl groups of the tert-butyl functionalities have been removed for clarity.
These similarities in structural orientation are also reflected in their metrical parameters. In [(FeIIIH21)2(μ-O)], the average Fe—Namine distance is 2.281(3) Å, significantly longer than the average Fe—Nureate distance of 2.024(3) Å. For [(FeIIIH21)2(μ-N-xylyl)], these average distances are 2.277(2) and 2.024(2) Å, respectively. This difference in bond length between amine and deprotonated urea nitrogen atoms to the metal center is similar to that seen in the Fe(II) precursor, FeIIH21(η2-OAc)]-. The average Fe—Oureate distances are 1.979(3) Å for [(FeIIIH21)2(μ-O)] compared with 2.054(2) Å in the case of [(FeIIIH21)2(μ-N-xylyl)].
In [(FeIIIH21)2(μ-O)], the average Fe-Ooxo bond length is 1.823(3) Å. This value lies towards the longer side for known μ-oxo dimers of FeIII where the range is 1.73–1.82 Å and the average is 1.77 Å.xiii This may be an effect of the H-bond donation by the two urea arms. The Fe-Fe separation is 3.005(3) Å and is slightly shorter compared to distances found in other di- and tri-bridged μ-oxo dimers that range from 3.05–3.39 Å. Similarly, the Fe1A-O1-Fe1B angle is 111.0(2)°, falling at the acute end of the range for known μ-oxo dimers (114-180°).6
In [(FeIIIH21)2(μ-N-xylyl)], the average Fe—Nimido bond length is 1.870(2) Å. Imido bridge FeIII dimers are rare: the only other known example of is (μ-p-tolylimido)bis[(N,N′-ethane-1,2-diyl-bis(salicylaldiminato))iron(III)], [(FeIIIsalen)2(μ-N-p-tolyl)].xiv The average bond length for [(FeIIIsalen)2(μ-N-p-tolyl)] is comparable at 1.88(1) Å. Note that FeIII complexes with terminal organoimido ligands have considerably shorter Fe—N lengths (less than 1.7 Å), reflecting their multiple bond character that is absent in the dimer complexes.xv The Fe—Fe separation in [(FeIIIH21)2(μ-N-xylyl)] is 2.974(4) Å while that in [(FeIIIsalen)2(μ-N-p-tolyl)] is significantly longer at 3.399(3) Å. The shorter distance in [(FeIIIH21)2(μ-N-xylyl)] is most likely a result of three bridging groups versus the single bridging group in [(FeIIIsalen)2(μ-N-p-tolyl)]. The Fe1A—N1C—Fe1B angle is 105.37(8)° compared with 129.6(6)° in [(FeIIIsalen)2(μ-N-p-tolyl)].
3.3 H-Bonding Networks
The nonbridging urea arms from each ligand form intramolecular hydrogen bonds in each of the two complexes. For [(FeIIIH21)2(μ-O)], the two arms containing N17A and N17B are H-bonded to the bridging oxo group. The N⋯O distances are 2.935(5) and 3.062(4) Å, which are consistent with H-bonds. This mode of interaction is not observed for [(FeIIIH21)2(μ-N- xylyl)] where intramolecular H-bonds involving the bridging imido ligand might be anticipated. However, the nitrogen atom of the bridging arylimido ligand is not expected to have as much electron density as a μ-oxo ligand. Moreover, the larger xylyl imido group may affect the intramolecular H-bond network. Therefore, the urea arms are found to H-bond with the oxygen atoms O6A and O6B of the bridging ureate, forming NH⋯O=C H-bonds at distances of 2.975(3) and 3.023(2) Å. In addition, the methyl groups of the tertiary amines, N1A and N1B, have different orientations in the two complexes: in [(FeIIIH21)2(μ-O)] they are positioned anti to the dimer core, whereas in [(FeIIIH21)2(μ-N-xylyl)] the methyls are syn, flanking the xylyl group. Taken together, these results suggest that during complex formation, the H-bond donating arms can adjust to the nature of the bridging species within the molecule.
The oxidation chemistry observed for [FeIIH21(η-OAc)]- contrasts that found for iron(II) complexes of the tripodal ligand, [H3buea]3-, which yield monomeric Fe(III) complexes with terminal oxo or hydroxo ligands. The more protective cavity of the tripodal ligands in conjunction with the extensive H-bond network surrounding the Fe(III)—O(H) units stabilizes the monomeric species. Similarily, we have found that monomeric Fe(III)-amido complexes are formed when Fe(II) tripodal complexes are treated with aryl azides. Our studies with the iron tripodal complexes suggest that high valent FeIV species are the likely intermediates during the activation of dioxygen or arylazidesxvi For [FeII(H21)(η2-OAc)]-, where the metal center is more exposed, a dimerization process would be likely and lead to the formation of the observed products. Numerous examples of heme and non-heme complexes with FeIII—O—FeIII unit are know and proceed through a similar dimerization route.xvii In addition, a analogous pathway with arylazides has been proposed in the formation of [(FeIIIsalen)2(μ-N-p-tolyl)].xiv
4. Summary
We have reported the preparations and structural properties for [(FeIIIH21)2(μ-X)] complexes formed upon the oxidation of [FeII(H21)(η2-OAc)]- with dioxygen and xylyl azide. Both dimers display bridging μ-1,3-(κN:κO)-ureate linkages, which are becoming more prevalent in coordination chemistry.xviii [(FeIIIH21)2(μ-N-xylyl)] exhibits an unusual bridging imido ligand with this being only the second example reported. The μ-oxo FeIII dimer contains two intramolecular H-bonds to the bridging oxo, which is rare in synthetic complexes, but is common in metalloproteins with Fe—(O)n—Fe cores.xix The intramolecular H-bond donors within these dimers can “sense” the presence of a bridging species and rearrange to its size and electron density.
Supplementary Material
Acknowledgments
We thank the NIH (GM50781) for financial support of this work. The National Science Foundation (CHE-0079282) and the University of Kansas funded the purchase of the X-ray instrumentation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- i.a) Jensen MP, Mehn MP, Que L., Jr Angew Chem Int Ed. 2003;42:4357. doi: 10.1002/anie.200351605. [DOI] [PubMed] [Google Scholar]; b) Feichtinger D, Plattner DA. Chem Eur J. 2001;7:591. doi: 10.1002/1521-3765(20010202)7:3<591::aid-chem591>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]; c) Du Bois J, Tomooka CS, Hong J, Carreira EM. Acc Chem Res. 1997;30:364. [Google Scholar]; d) Groves JT, Lee J, Marla SS. J Am Chem Soc. 1997;119:6229. [Google Scholar]; e) Brandt P, Södergren MJ, Anderson PG, Norrby PO. J Am Chem Soc. 2000;122:8013. [Google Scholar]
- ii.a) Wigley DE. Prog Inorg Chem. 1994;42:239. [Google Scholar]; b) Eikey RA, Abu-Omar MM. Coord Chem Rev. 2003;243:83. [Google Scholar]
- iii.Recent examples:; a) Mindiola DJ, Hillhouse GL. J Am Chem Soc. 2001;123:4623. doi: 10.1021/ja010358a. [DOI] [PubMed] [Google Scholar]; b) Jenkins DM, Betley TA, Peters JC. J Am Chem Soc. 2002;124:11238. doi: 10.1021/ja026852b. [DOI] [PubMed] [Google Scholar]; c) Thyagarajan S, Shay DT, Incarvito CD, Rheingold AL, Theopold KH. J Am Chem Soc. 2003;125:4440. doi: 10.1021/ja028267g. [DOI] [PubMed] [Google Scholar]; d) Eikey RA, Khan SI, Abu-Omar MM. Angew Chem, Int Ed. 2002;41:3592. doi: 10.1002/1521-3773(20021004)41:19<3591::AID-ANIE3591>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; e) Dai X, Kapoor P, Warren TH. J Am Chem Soc. 2004;126:4798. doi: 10.1021/ja036308i. [DOI] [PubMed] [Google Scholar]; f) Thomas CM, Mankad NP, Peters JC. J Am Chem Soc. 2006;128:4957. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Jenson MP, Mehn MP, Que L., Jr Angew Chem, Int Ed. 2003;42:4357. doi: 10.1002/anie.200351605. [DOI] [PubMed] [Google Scholar]; h) Nazif AK, Nazif TN, Achim C, Lee SC. J Am Chem Soc. 2000;122:11013. [Google Scholar]
- Borovik AS. Acc Chem Res. 2005;38:54. doi: 10.1021/ar030160q. references therein. [DOI] [PubMed] [Google Scholar]
- v.a) MacBeth CE, Golombek AP, Young VG, Jr, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]; b) MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Jr, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. J Am Chem Soc. 2004;126:2556. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]
- vi.Lucas RL, Powell DR, Borovik AS. J Am Chem Soc. 2005;127:11597. doi: 10.1021/ja052952g. [DOI] [PubMed] [Google Scholar]
- vii.a) Hammes BS, Young VG, Jr, Borovik AS. Angew Chem Int Ed Engl. 1999;38:666. doi: 10.1002/(SICI)1521-3773(19990301)38:5<666::AID-ANIE666>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; b) MacBeth CE, Hammes BS, Young VG, Jr, Borovik AS. Inorg Chem. 2001;40:4733. [PubMed] [Google Scholar]
- viii.Zart MK, Sorrell TN, Powell D, Borovik AS. Dalton Trans. 2003:1986. [Google Scholar]
- ix.Murata S, Abe S, Tomioka H. J Org Chem. 1997;62:3055. doi: 10.1021/jo9622901. [DOI] [PubMed] [Google Scholar]
- x.a) Data Collection: SMART Software Reference Manual. Bruker-AXS; Madison, WI: 1994. [Google Scholar]; b) wData Reduction: SAINT Software Reference Manual. Bruker-AXS; Madison, WI: 1995. [Google Scholar]
- xi.Sheldrick GM. SADABS: Program for Empirical Absorption Correction of Area Detector Data. University of Göttingen; Germany: 2000. [Google Scholar]
- xii.a) Sheldrick GM. SHELXTL Version 5 Reference Manual. Bruker-AXS; Madison, WI: 1994. [Google Scholar]; b) International Tables for Crystallography. C Kluwer Academic Publishers; Norwell, MA: 1995. [Google Scholar]
- xiii.Kurtz DM. Chem Rev. 1990;90:585. [Google Scholar]
- xiv.Nichols PJ, Fallon GD, Murray KS, West BO. Inorg Chem. 1988;27:2795. [Google Scholar]
- xv.a) Verma AK, Nazif TN, Achim C, Lee SC. J Am Chem Soc. 2000;122:11013. [Google Scholar]; b) Brown SD, Betley TA, Peters JC. J Am Chem Soc. 2003;125:322. doi: 10.1021/ja028448i. [DOI] [PubMed] [Google Scholar]
- xvi.Lucas RL, Powell DR, Borovik AS. J Am Chem Soc. 2005;127:11597. doi: 10.1021/ja052952g. [DOI] [PubMed] [Google Scholar]
- xvii.Chin DH, LaMar GN, Balch AL. J Am Chem Soc. 1980;102:4344. [Google Scholar]
- xviii.a) Meyer F, Pritzkow H. Chem Commun. 1998:1555. [Google Scholar]; b) Kryativ SV, Nazarenko AY, Robinson PD, Rybak-Akimova EV. Chem Commun. 2000:921. [Google Scholar]; c) Gupta R, Zhang ZH, Powell DR, Hendrich MP, Borovik AS. Inorg Chem. 2002;41:5100. doi: 10.1021/ic0202918. [DOI] [PubMed] [Google Scholar]
- xix.a) Du Bois J, Mizoguchi TJ, Lippard SJ. Coord Chem Rev. 2000;443:200–202. [Google Scholar]; b) Que L, Jr, Tolman WB. Angew Chem Int, Ed. 2002;41:1114. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.