

Abstract
The redox-regulated chaperone Hsp33 is specifically activated upon exposure of cells to peroxide stress at elevated temperatures. Here we show that Hsp33 harbors two interdependent stress-sensing regions located in the C-terminal redox-switch domain of Hsp33: a zinc center sensing peroxide stress conditions and an adjacent linker region responding to unfolding conditions. Neither of these sensors works sufficiently in the absence of the other, making the simultaneous presence of both stress conditions a necessary requirement for Hsp33’s full activation. Upon activation, Hsp33’s redox-switch domain adopts a natively unfolded conformation, thereby exposing hydrophobic surfaces in its N-terminal substrate-binding domain. The specific activation of Hsp33 by the oxidative unfolding of its redox-switch domain makes this chaperone optimally suited to quickly respond to oxidative stress conditions that lead to protein unfolding.
Reactive oxygen species develop as unavoidable consequences of aerobic life. Their oxidizing effects on most cellular macromolecules can be deleterious to cells and organisms1. Cells have developed effective antioxidant systems to counteract this hazard (for review, see ref. 2). Disruption of the fine balance between oxidants and antioxidants, however, leads to the accumulation of reactive oxygen species and to a condition termed oxidative stress3. Oxidative stress conditions have been shown to develop in, and may even cause, numerous physiological and pathological conditions, such as aging, heart disease, diabetes and neurodegenerative diseases4.
A growing number of proteins have been identified that are not damaged by reactive oxygen species, but rather use the oxidation state of one or more reactive cysteines to modulate their activity in response to oxidative stress5. One such protein is the redox-regulated molecular chaperone Hsp33 from Escherichia coli. Like most E. coli cytosolic chaperones, Hsp33 is under the control of σ32. Its expression is induced at elevated temperatures and other stress conditions that lead to protein unfolding6. Activation of Hsp33’s chaperone function requires the presence of reactive oxygen species such as H2O2 and hydroxyl radicals7. These are sensed by the thiol-containing zinc center of Hsp33’s C terminus8. When not under stress conditions, the four invariant cysteine residues of Hsp33, which are arranged in a 232-CXC-234 and 265-CXXC-268 motif, are reduced and coordinate one zinc ion9,10. The presence of this zinc center apparently keeps Hsp33 monomeric and functionally inactive. Upon exposure of Hsp33 to oxidative stress, the nearby cysteines form intramolecular disulfide bonds, inducing zinc release7,11. Large conformational rearrangements and partial unfolding then lead to the dimerization of Hsp33, a crucial step that fully activates Hsp33’s chaperone function7,12. The overall activation process of Hsp33 is unusually temperature dependent7, and in vivo studies have shown that Hsp33 protects E. coli cells against H2O2 treatment only at elevated temperatures13. This specific activation of Hsp33 compensates for the loss of function of DnaK, a major ATP-dependent chaperone foldase and member of the Hsp70 family, which is reversibly inactivated by the same conditions activating Hsp33 (ref. 13).
Key questions concerning Hsp33’s oxidative activation mechanism include how and why Hsp33’s activation is restricted to conditions where H2O2 is present at elevated temperatures in vivo. Here we demonstrate that Hsp33’s redox switch consists of two interdependent stress sensors, which are both located in Hsp33’s C-terminal domain (residues 179–294; Fig. 1). These sensor regions confer activation of Hsp33 only when H2O2 stress is combined with protein-unfolding conditions such as heat. Hydrogen peroxide oxidizes the thiol-containing zinc center (residues 232–294), inducing zinc release and the unfolding of the zinc center. Heat is sensed by regions directly upstream of the zinc-binding domain (residues 179–231), which become thermolabile and open once Hsp33 no longer binds zinc. The unfolding of Hsp33’s C terminus exposes large hydrophobic surfaces in the N-terminal substrate-binding domain of Hsp33 and causes the activation of Hsp33. These hydrophobic surfaces are masked in the reduced, inactive state, where zinc association provides the intrinsic scaffold that confers structure and stability to the C-terminal redox-switch domain.
Figure 1.
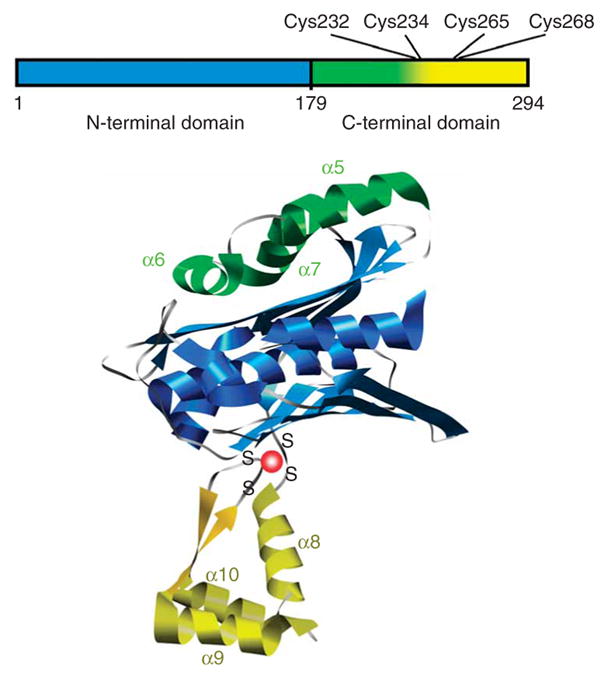
Domain structure of Hsp33. Linear representation (top) is color coded similarly to structural model of Hsp33 monomer (bottom), which is based on the crystal structure of the reduced Bacillus subtilis Hsp33 dimer (PDB 1VZY)10.
RESULTS
Hsp33’s activation requires H2O2 and elevated temperatures
Neither peroxide stress alone (4 mM H2O2) nor the exposure of E. coli cells to very high temperatures (>49 °C) causes Hsp33 activation in vivo13. We replicated this stringent requirement in vitro by comparing the kinetic profiles of Hsp33’s activation at 30 °C and 43 °C in the presence of H2O2, using chemically denatured citrate synthase (Fig. 2a) or firefly luciferase (data not shown) as the chaperone substrate protein. In both chaperone assays, Hsp33 was fully activated by H2O2 at 43 °C within 60 min of incubation, whereas Hsp33 remained virtually unresponsive in the presence of H2O2 at 30 °C (Fig. 2a) or in the absence of H2O2 at 43 °C (data not shown).
Figure 2.

In vitro activation of Hsp33 requires oxidative stress at elevated temperatures. (a) Activation of Hsp33 in vitro requires simultaneous presence of oxidants and elevated temperature. Hsp33red was incubated with H2O2 at either 30 °C (●) or 43 °C (○). At various time points, chaperone activity in aliquots was determined by measuring aggregation of chemically denatured citrate synthase at 30 °C. Light-scattering signal 4 min after addition of citrate synthase is plotted against incubation time; signal in the absence of Hsp33 was defined as 0% activity, and signal with fully active Hsp33DCCSSS mutant protein (which forms intermolecular dimers during its purification)7 as 100%. Inset, activity of Hsp33red, Hsp33ox30°C and Hsp33ox43°C with chemically denatured citrate synthase at 43 °C. (b) Temperature dependence of Hsp33’s oxidative zinc release. Reduced, zinc-reconstituted Hsp33(W212F Y267W) was incubated with H2O2 at 30 °C (triangles) or 43 °C (circles) and tryptophan fluorescence was monitored (open symbols, left axis; see Methods). Simultaneously, zinc-binding affinities of Hsp33(W212F Y267W) aliquots were analyzed using the PAR/PCMB assay (filled symbols, right axis). 100%, values for Hsp33(W212F Y267W)red; 0%, Hsp33(W212F Y267W)ox43°C (oxidized for 3 h). Inset, Hsp33(W212F Y267W) shows zinc-dependent tryptophan fluorescence. Fluorescence spectrum of zinc-reconstituted Hsp33(W212F Y267W) or zinc-depleted apo-Hsp33 was monitored under reducing conditions. AU, arbitrary units.
To investigate whether oxidized Hsp33 requires elevated temperatures for the binding of its unfolded substrate proteins, we removed any residual H2O2 still present after 180 min of oxidation at 30 or 43 °C, leaving protein preparations that we designated Hsp33ox30°C and Hsp33ox43°C. We then incubated both protein preparations at 43 °C and tested their influence on the aggregation of chemically unfolded citrate synthase or thermally unfolded luciferase at 43 °C (Fig. 2a, inset, and data not shown). Similar to the results of our chaperone assays at 30 °C, we found that Hsp33ox30°C had substantially lower activity at 43 °C than Hsp33ox43°C.
We have previously reported that oxidation of Hsp33 at its highly conserved cysteine residues leads to the release of zinc, which causes the unfolding of the zinc-binding domain12. To determine whether the lack of Hsp33 activation at 30 °C is due to a lack of zinc release, we used two different assays to monitor the zinc-binding status: a spectrophotometric PAR/PCMB assay, which depends on direct competition for zinc binding between Hsp33’s cysteines (Hsp33red, Ka = 2.5 × 1017 M−1) and the chelator PAR (Ka = 2 × 1012 M−1)14 and a fluorescence-based assay, which directly monitors zinc binding in Hsp33 (Fig. 2b). The fluorescence-based assay uses a newly generated Hsp33 mutant protein (Hsp33(W212F Y267W); Supplementary Methods online) with redox regulation and chaperone activity similar to those of wild-type Hsp33 (data not shown). The tryptophan residue is located between Cys265 and Cys268, where it serves as a highly sensitive zinc-binding sensor (Fig. 2b, inset). Both assays revealed that zinc is completely released from the high-affinity zinc center of Hsp33 within 180 min of peroxide treatment at 30 °C (Fig. 2b). These results demonstrate that oxidative zinc release is not sufficient to activate Hsp33 and suggest that oxidation at 30 °C leads to the formation of a zinc-free, inactive Hsp33 intermediate. A similar zinc-free, inactive oxidation intermediate might also transiently accumulate during Hsp33’s activation at 43 °C, according to the pronounced lag phase observed during Hsp33’s oxidative activation (Fig. 2a).
Identification of an oxidation intermediate at 30 °C
To analyze Hsp33’s thiol modification status upon oxidation at 30 °C and 43 °C, we performed thiol-trapping experiments with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonate (AMS). AMS covalently adds a 490-Da group to each free thiol in the protein. Incubation of Hsp33 in H2O2 at 30 °C with AMS led to the oxidation of Hsp33’s cysteines, but either the number of modified cysteines or their specific oxidative modifications seems to differ from those occurring upon incubation at 43 °C (Supplementary Data and Supplementary Fig. 1 online). We therefore decided to investigate the oxidative thiol modifications in more detail using mass spectrometry. Because Hsp33 mutants containing only the two N-terminal cysteines, Cys232 and Cys234, still function as redox-regulated chaperones (see below), we decided to primarily focus on the thiol status of these two cysteines in wild-type Hsp33. To visualize the reduced cysteines, Hsp33red and Hsp33 incubated in H2O2 at either 30 or 43 °C were denatured in urea and treated with the alkylating agent iodoacetamide (IAM). IAM irreversibly binds all reduced thiols and leads to the mass addition of 57 Da per cysteine. In the next step, all reversible thiol modifications present in the Hsp33 preparations, such as disulfide bonds or sulfenic acids, were reduced with DTT. These newly exposed thiol groups were then alkylated with the thiol-reactive reagent N-ethylmaleimide (NEM), which adds 125 Da per cysteine. After this differential labeling process, proteins were digested with trypsin and analyzed by mass spectrometry (Fig. 3). Cys232 and Cys234, which are present in a small tryptic peptide (residues 232–236; calculated m/z = 569.7), were modified by either IAM, NEM or both, depending on the original redox status of the proteins. As expected, in Hsp33red, both Cys232 and Cys234 were labeled with IAM, demonstrating the presence of reduced cysteines prior to differential thiol trapping (observed m/z = 683.8). After 1 h of oxidation at 30 °C, a substantial proportion of Hsp33 molecules were still in the fully reduced form, a result that agrees well with the slow kinetics of zinc release observed at 30 °C (Fig. 2b). Notably, this Hsp33 preparation also contained an appreciable population of peptides in which only one of the two cysteines was oxidized, while the other one was still reduced (m/z = 751.9). When we incubated this preparation in H2O2 at 43 °C, we found that oxidation at 43 °C rapidly and fully activated this species (data not shown). This result suggests the existence of an Hsp33 oxidation intermediate and not of a dead-end product. We were surprised to discover this intermediate, because H2O2 is thought to oxidize cysteine thiols to highly reactive sulfenic acids, which very rapidly react with nearby cysteines to form disulfide bonds15,16. Thus, the detection of a singly oxidized cysteine suggests that accessibility or reactivity of the nearby cysteine might limit the oxidation process at 30 °C. In contrast, no such intermediates were detectable at 43 °C; under these conditions, both Cys232 and Cys234 (m/z = 819.9) were fully oxidized. This result suggests that elevated temperatures might contribute to the accessibility and/or reactivity of these cysteines.
Figure 3.

Thiol status of Hsp33’s tryptic peptide 232-CTCSR-236 (calculated m/z = 569.7), containing the first redox-active cysteine pair. Wild-type Hsp33red, Hsp33ox30°C or Hsp33ox43°C (oxidized for 1 h) was incubated with IAM, then reduced by DTT and treated with NEM. Mass spectra of the differentially labeled tryptic peptides were obtained by MALDI-MS. m/z value of 683.8 represents the fully reduced (IAM-labeled) peptide; 819.9, fully oxidized (NEM-labeled) peptide; 751.9, partially oxidized intermediate harboring one NEM and one IAM label. The identities of the peaks were confirmed by MS/MS analysis. Arrows mark expected theoretical mass of the modified peptide.
After 3 h of incubation at 30 °C, both cysteines were oxidized, although the nature of this reversible modification (data not shown) could not be identified because of poor peptide ionization, which precludes mass spectrometric detection. This has been observed for other thiol-containing peptides and agrees with our original mass spectrometry studies, where only a very small signal for the disulfide-bonded 232–236 peptide was detected11. Nevertheless, our identification of an oxidation intermediate shows that the oxidative thiol modifications occurring at 30 °C differ from those at 43 °C and suggests formation of an inactive oxidation intermediate at 30 °C.
Identification of a thermolabile region in Hsp33ox30°C
Ultracentrifugation analysis of Hsp33red, Hsp33ox30°C and Hsp33ox43°C revealed that Hsp33red and Hsp33ox30°C sediment as monomers, whereas Hsp33ox43°C sediments predominantly as a dimer (data not shown). These results suggest that temperature- and oxidant-specific rearrangements are necessary prerequisites for Hsp33 dimerization and activation. To obtain insights into the specific conformational changes that occur in Hsp33 upon its oxidation at 43 °C, but not at 30 °C, we compared the secondary structures of Hsp33red, Hsp33ox30°C and Hsp33ox43°C using CD spectroscopy and analyzed their surface hydrophobicity (Fig. 4a). As previously shown, Hsp33red contains both α-helices and β-sheets and has little to no hydrophobic surface that can interact with the fluorescent probe 4,4′-dianilino-1,1′-binaphtyl-5,5′-disulfonic acid (bis-ANS; Fig. 4a, inset). Hsp33ox43°C has substantially less α-helical content than Hsp33red. Secondary structure predictions by K2D17 show a large decrease in the α-helical content, from 37% in Hsp33red to 19% in Hsp33ox43°C (Supplementary Table 1 online). This unfolding causes either the formation or exposure of hydrophobic surfaces that are probably the substrate-binding site(s) in Hsp33 (Fig. 4a, inset)12,18. Hsp33ox30°C, in contrast, retains much of the α-helical content (30%) found in reduced Hsp33. The CD spectrum of Hsp33ox30°C was very similar to that of the cysteine-free variant of Hsp33 (Hsp33Cys−), which is zinc-free, contains an unfolded zinc-binding domain and is inactive at 30 °C12 (Fig. 4a, compare traces 2 and 3). These results suggest that the oxidative stress–induced zinc release at 30 °C indeed causes unfolding of Hsp33’s zinc-binding domain. This unfolding, however, is apparently not sufficient to expose hydrophobic surfaces in Hsp33 (Fig. 4a, inset), which is probably the reason for the low affinity of Hsp33ox30°C for unfolded substrate proteins. Furthermore, these results also demonstrate that Hsp33 activation involves major structural rearrangements beyond the unfolding of the redox-sensitive zinc-binding domain originally suggested12.
Figure 4.

Activation of Hsp33 is accompanied by major conformational rearrangements. (a) Far-UV CD spectra of Hsp33red (1, solid line), Hsp33ox30°C (2, dotted line), Hsp33Cys− (3, dotted and dashed line) and Hsp33ox43°C (4, dashed line) were recorded at 30 °C, whereas Hsp33Cys− (5, double dotted and dashed line) was recorded at 43 °C. Inset, bis-ANS fluorescence to monitor surface hydrophobicity in reduced and oxidized Hsp33. 10 μM bis-ANS was incubated with 3 μM Hsp33red, Hsp33ox30°C or Hsp33ox43°C. Emission spectra were recorded using λex of 370 nm at 30 °C. (b) Oxidative zinc release destabilizes additional regions in Hsp33. Analysis of temperature-induced conformational changes by monitoring the changes in molecular ellipticity at 197 nm. Hsp33red, Hsp33ox30°C, Hsp33Cys− or Hsp33ox43°C was heated to 50 °C (solid line), then cooled to 20 °C (dotted line).
To investigate which stage in Hsp33’s activation process might be affected by elevated temperature, we measured the thermal stability of Hsp33. Whereas Hsp33red is quite thermostable (Tm ~65 °C; data not shown) and begins to change conformation only at temperatures above 47 °C, Hsp33ox30°C starts to unfold substantially at temperatures above 40 °C, as evidenced by the loss in ellipticity at 197 nm (Fig. 4b). This result suggests that oxidative stress–induced unfolding of Hsp33’s zinc binding domain may destabilize other regions in the protein, which become thermolabile and unfold at physiologically relevant temperatures.
This destabilization is even more pronounced in the cysteine-free variant of Hsp33 (Hsp33Cys−), which lacks all of Hsp33’s highly conserved cysteines and does not coordinate zinc. We found that incubation of Hsp33Cys− at 43 °C leads to the same structural changes that were observed when wild-type Hsp33 was activated by H2O2 treatment at 43 °C (Fig. 4). These results explain previous findings that Hsp33Cys− exerts chaperone activity at 43 °C but not at 30 °C12. Furthermore, we observed that active Hsp33Cys− remains monomeric at elevated temperatures, as determined by analytical ultracentrifugation, and that the conformational changes in Hsp33Cys− can be fully reversed by decreasing the temperature. These properties contrast with Hsp33ox43°C, which dimerizes upon activation7 and maintains its conformational changes upon cooling to 20 °C (Fig. 4b, dotted line). The results suggest that an intrinsically unstable region in Hsp33 opens upon zinc release, whose folding status is crucial in the chaperone function of Hsp33. This region is apparently stably folded in reduced, zinc-coordinated Hsp33, where it covers hydrophobic surfaces in Hsp33 that are the likely binding site(s) for unfolded protein18. It becomes thermolabile once the redox sensor has been triggered and the zinc-binding domain is unfolded. However, formation of stable Hsp33 dimers, which maintain their crucial conformational rearrangements and activity upon decrease of the temperature, seems to require the correct oxidative cysteine modifications. These oxidative thiol modifications in turn depend on the presence of elevated temperatures during the oxidation process.
Activation requires rearrangements in two distinct regions
To further dissect the structural rearrangements necessary for Hsp33 activation, we analyzed the structural and functional properties of an Hsp33 truncation mutant, Hsp331–235. This mutant spans the first pair of cysteines, Cys232 and Cys234, but lacks more than 95% of the zinc-binding domain. Hsp331–235 has been described as a constitutively active chaperone19, but our functional analysis revealed that Hsp331–235 still functions as a redox-regulated chaperone that can be inactivated in the presence of reducing conditions and is readily activated by the exposure to H2O2 at 43 °C (Fig. 5a). We observed that the remaining cysteine pair rapidly air-oxidizes, which suggests that zinc coordination is important in reducing the reactivity of the cysteines. In the study that reported constitutive Hsp331–235 activity, an absence of reducing agents in combination with elevated temperatures (43 °C) probably caused the activation of Hsp33 and led to the conclusion that the truncation mutant is constitutively active19.
Figure 5.

C-terminal truncation mutant Hsp331–235 functions as a redox-regulated chaperone. (a) Activation kinetics of Hsp331–235 upon oxidation at 43 °C. 50 μM Hsp331–235(red) was incubated with H2O2 at 43 °C and chaperone activity was determined as in Figure 2a. Inset, influence of Hsp331–235(red) (in 1 mM DTT), Hsp331–235(ox30°C) or Hsp331–235(ox43°C) on aggregation of chemically unfolded citrate synthase measured at 43 °C. (b) Activation of Hsp331–235 is accompanied by major conformational changes, shown by far-UV CD spectra of Hsp331–235(red), Hsp331–235(ox30°C) or Hsp331–235(ox43°C) at 30 °C. Inset, bis-ANS binding to monitor hydrophobic surfaces in Hsp331–235(red), Hsp331–235(ox30°C) and Hsp331–235(ox43°C) as in Figure 4a. AU, arbitrary units.
We found that activation of Hsp331–235 is very rapid, lacking the lag phase observed for wild-type Hsp33 (Fig. 2a). This result agrees with our conclusion above that the lag phase in Hsp33’s activation reaction presumably is caused by the oxidation of Hsp33’s zinc center, which generates a zinc-free, inactive Hsp33 intermediate. Absence of the zinc-binding domain apparently removes this step from the activation process.
Activation of Hsp331–235 was accompanied by marked losses in secondary structure elements and a large increase in surface hydrophobicity (Fig. 5b). These results indicate that the crucial conformational rearrangements necessary for full activation of Hsp33 take place adjacent to the zinc redox center. Functional analysis of Hsp331–235 confirmed the interdependent nature of the dual stress-sensing mechanisms, as incubation of reduced Hsp331–235 at elevated temperatures did not activate the reduced Hsp331–235 mutant protein (Fig. 5a, inset). Thus, the zinc center per se does not stabilize the upstream region of Hsp33; rather, presumably, one or both of the thiol groups that remain in the truncation mutant interact with residue(s) in this upstream region. Similarly, incubation of Hsp331–235 in H2O2 at 30 °C did not fully activate the mutant protein (Fig. 5), and we conclude from these data that the two crucial cysteines are not readily accessible unless elevated temperatures unfold additional regions in Hsp33.
Hsp33’s linker region might function as a thermosensor
Our structural analysis of the Hsp331–235 mutant protein, together with the published NMR spectra of Hsp33 (ref. 12), suggest that the conformational changes required for the activation of Hsp33 might involve a linker region (179–231) that connects the N-terminal substrate-binding domain with the redox-sensitive zinc center. In inactive Hsp33, this linker region folds on top of Hsp33’s N terminus and covers ~3,800 A ° 2 of largely hydrophobic (~75%) surface area10,12. In active Hsp33, in contrast, most of the C terminus, including the linker region, seems to be unfolded12. The α-helical contents of our three Hsp33 preparations are consistent with the hypothesis that unfolding of the zinc-binding domain as well as of the adjacent linker region is necessary for full activation (Supplementary Table 1).
To test this model, we decided to directly monitor the folding state of the linker region using tryptophan fluorescence. We substituted Phe187, which is located at the N-terminal boundary of the linker region, with tryptophan and generated a mutant that contains only one tryptophan, Hsp33(F187W W212F). In the crystal structure of inactive Hsp33, this nonconserved Phe187 residue sits on top of the N terminus with its side chain buried in a nonpolar protein environment10. Unfolding of the linker region would probably expose this side chain to the polar solvent, resulting in a red shift in the wavelength of the tryptophan fluorescence20.
The purified Hsp33(F187W W212F) mutant had chaperone function similar to that of the wild-type protein (data not shown). Comparison of the fluorescence spectra of Hsp33(F187W W212F)red and Hsp33(F187W W212F)ox43°C revealed that oxidation at 43 °C greatly decreased fluorescence intensity and led to an 11-nm red shift, from 335 nm to 346 nm (Fig. 6a). Oxidation of Hsp33(F187W W212F) at 30 °C induced only a ~2-nm shift in the emission spectra, suggesting that the conformation of the linker region does not substantially change during oxidation at 30 °C (Fig. 6a). Incubation of reduced Hsp33(F187W W212F) at 43 °C did not change the fluorescence spectrum (data not shown). This is in agreement with our previous activation experiments (Fig. 2a, inset) and confirms our assumption that the zinc center needs to be unfolded to destabilize Hsp33’s linker region.
Figure 6.

Unfolding of the linker region is crucial for Hsp33’s activation. (a) Fluorescence spectra of Hsp33(F187W W212F)red, Hsp33(F187W W212F)ox30°C and Hsp33(F187W W212F)ox43°C. AU, arbitrary units. (b) Correlation between zinc release and conformational changes in Hsp33’s linker region upon oxidation of Hsp33 at 30 and 43 °C. Hsp33(F187W W212F)red was incubated with H2O2 at 43 °C (larger chart) or 30 °C (inset). Emission maxima of fluorescence spectra are plotted against incubation time (○, right axis). At indicated times, zinc release in aliquots was also measured using the PAR-PCMB assay (●, left axis). (c) Stability of Hsp33(F187W W212F) incubated in Gdn-HCl for 24 h (supplemented with 5 mM DTT). Tryptophan fluorescence was measured; wavelength of the emission maximum (λmax) is plotted against Gdn-HCl concentration. Arrow indicates λmax of active Hsp33(F187W W212F) after 3 h of incubation in 2 mM H2O2 at 43 °C. Inset, oxidative activation of Hsp33 in 1.5 M Gdn-HCl at 30 °C. Hsp33red was incubated with H2O2 at 30 °C in the absence (ox30°C) or presence of 1.5 M Gdn-HCl (ox30°C/Gdn) or at 43 °C (ox43°C). As control, Hsp33red was incubated in 1.5 M Gdn-HCl in the absence of oxidizing reagents (red/Gdn). The activity of Hsp33 was determined as in Figure 2a.
To gain insight into the kinetics of linker unfolding, we then followed the changes in the tryptophan emission maximum during the oxidation process at 30 °C and 43 °C (Fig. 6b) and compared this to the kinetics of zinc release and activation. At 43 °C, we found that the structural rearrangements of the linker region are substantially slower than the zinc release (Fig. 6b) and closely mirror the activation kinetics monitored at 43 °C (Fig. 2a), suggesting that linker unfolding occurs only after oxidative stress–induced zinc release. This became even more apparent at 30 °C, where zinc was released with only minor structural changes in the linker region (Fig. 6b, inset). Both the kinetics and the extent of structural rearrangements correlated very well with the slow and inefficient activation of Hsp33 at this temperature (Fig. 2a). This result highlights the conclusion that activation of Hsp33 involves conformational changes in two distinct regions of Hsp33, which depend on heat and oxidative stress.
Activation by H2O2 and Gdn-HCl–induced linker unfolding
To analyze the extent of unfolding of Hsp33’s linker region, we compared the tryptophan fluorescence of Hsp33(F187W W212F) after oxidation at 43 °C with the fluorescence of reduced Hsp33(F187W W212F) upon chemical unfolding in guanidine hydrochloride (Gdn-HCl). Incubation of Hsp33(F187W W212F) in 1.5 M Gdn-HCl led to a red shift in fluorescence emission spectra, resembling the fluorescence shift observed with Hsp33ox43°C (Fig. 6c). Further increase in the Gdn-HCl concentration only slightly increased the emission maximum, suggesting that 1.5 M Gdn-HCl is sufficient to expose Trp187 to the polar environment but does not fully denature Hsp33 (Fig. 6c). We therefore investigated whether we could use these denaturing conditions to study the role of protein unfolding in Hsp33’s activation process independently of temperature. We incubated wild-type Hsp33 (Fig. 6c, inset) or the Hsp33(F187W W212F) mutant protein (data not shown) in 1.5 M Gdn-HCl at 30 °C in the absence or presence of H2O2. After 1 h of incubation, Gdn-HCl and excess H2O2 were removed and the activity of Hsp33 was compared with Hsp33 oxidized with H2O2 for 1 h at either 30 or 43 °C (Fig. 6c, inset). As before, oxidation at 30 °C did not activate Hsp33, whereas incubation at 43 °C led to considerable activation. Incubation of Gdn-HCl–treated Hsp33 in H2O2 at 30 °C was even more efficient than oxidation at 43 °C. Zinc-binding analysis showed that unfolding of the linker region also increased the rate of oxidative zinc release (data not shown). This result excludes the possibility that elevated temperatures indirectly affect H2O2 function—for instance, by changing its redox potential. However, unfolding of the linker region by either elevated temperature or chemical denaturation may increase the accessibility of the crucial cysteines, whose correct oxidative modifications might represent the true rate-determining step in Hsp33 activation.
DISCUSSION
Here, we describe Hsp33 as a molecular chaperone whose full activation requires the simultaneous presence of oxidative and protein-unfolding conditions (for example, heat). This dual stress sensing is conferred by Hsp33’s C-terminal redox-switch domain, which consists of a thermolabile linker region and a redox-sensitive zinc center (Fig. 7). Upon reaction with peroxide at low temperatures, zinc is released and Hsp33’s zinc-binding domain unfolds12. Under these conditions, Hsp33 remains in its monomeric, inactive state with a closed linker region. To fully activate Hsp33, the simultaneous input of H2O2 and unfolding conditions is necessary. Thiol-trapping experiments suggest that unfolding conditions contribute to the formation of the activating oxidative modifications and that Hsp33ox30°C lacks this oxidative signature. This suggests that either the reactivity or the accessibility of at least one of the cysteines becomes rate determining in the activation process at 30 °C but not at 43 °C. Unfortunately, all solved crystal structures of full-length Hsp33 show Hsp33 as a dimeric protein10,19,21. Therefore, we are unable to predict the structural environment of these cysteines in monomeric Hsp33. We can also not fully exclude the possibility that modification of other, non-thiol amino acids might contribute to Hsp33’s activation. However, the observation that oxidative activation of wild-type Hsp33 is largely reversible upon treatment with thiol-reducing agents makes this possibility highly unlikely.
Figure 7.

Schematic model of Hsp33’s activation process. Upon oxidation of Hsp33 at 30 °C, zinc is released and the zinc center (α-helices 8–10) unfolds. This unfolding is apparently not sufficient to activate the chaperone function, but generates an oxidation intermediate of Hsp33 (shown in brackets), which is presumably only very transiently populated during oxidation at 43 °C. Upon exposure to oxidizing and unfolding conditions—for example, H2O2 at elevated temperature—the complete C terminus (α-helices 5–10) converts to a natively unfolded protein. These extensive conformational rearrangements lead to the exposure of hydrophobic surfaces, presumably on the N-terminal substrate-binding domain of Hsp33, and allow Hsp33 to dimerize.
On the basis of our structural and functional findings, we developed a model that explains the interdependent nature of Hsp33’s dual stress sensing functions in vitro and in vivo (Fig. 7). In the absence of oxidants, the zinc center is fully folded (closed state) and the linker region is thermostable (Tm ≥ 65 °C). Reduced Hsp33 will not unfold the linker region at heat-shock temperatures and hence will not activate its chaperone function. This emphasizes the importance of the zinc center for the thermostability of the reduced, inactive protein. Conversely, oxidative stress causes zinc release and primes the protein for activation by destabilizing the linker region (open state). However, as long as no other stress conditions arise that cause protein unfolding, the linker region remains buried in the hydrophobic surface. Upon exposure to unfolding conditions, the conformational changes in the linker provide access to the first two cysteines of Hsp33’s zinc-binding site. Oxidative modification of these cysteines fully opens the linker region, which allows Hsp33 to dimerize and remain stably activated even at low temperatures, until restoration of normal conditions (Fig. 7).
What prevents Hsp33’s unfolded C terminus from binding to its own substrate-binding sites and thereby competing out other unfolded substrate proteins? We assume that is the hydrophilic character of the redox-switch domain, which contains only a few hydrophobic residues and a large number of charged residues (pI = 4.16). These are classical features of proteins that adopt disordered structures in vitro and presumably also in absence of their folding scaffolds in vivo22. Fold-prediction programs such as FoldIndex23 agree with this assumption and predict that the C-terminal 110 residues in Hsp33 are disordered in the absence of zinc (Supplementary Fig. 2 online). Natively unfolded (disordered) proteins constitute a growing class of proteins, which are involved in regulation of important cellular processes, including cell cycle and signal transduction (for review, see ref. 24). Whereas protein unfolding serves as trigger and activator of Hsp33’s chaperone function, most other proteins are inactive when unfolded. Their folding requires interaction with specific partner proteins, nucleic acids or membranes, which provides the necessary binding enthalpy to drive the entropically costly folding process22. In Hsp33, folding of the C terminus is apparently triggered by reduction of its cysteines. It is conceivable that Hsp33’s high-affinity zinc binding (Ka = 2.5 × 1017 M−1) provides the energy to drive the folding of the zinc center25. Reassociation of zinc would then generate reduced Hsp33 dimers, which are still active and stabilized by substrate binding26. Once the substrate proteins were released, the linker region would refold using its own N-terminal substrate-binding region as a folding scaffold, returning the chaperone to its inactive state.
The sensor regions present in Hsp33 have individual uses in other redox-regulated proteins (for example, RsrA) or temperature-regulated chaperones (for example, GrpE and Hsp26)27–29. In contrast to Hsp33, however, these proteins are regulated by only a single stress signal and therefore require only a single stress sensor. Hsp33 apparently needs to sense two signals simultaneously and therefore has developed two stress sensors that act interdependently. This model seems to make sense in a physiological context, because peroxide stress induced by H2O2 concentrations as high as 4 mM does not cause any appreciable protein aggregation in vivo (Supplementary Data and Supplementary Fig. 3 online). Therefore, activation of such a potent chaperone holdase as Hsp33 is not required under these conditions. Once oxidative stress is combined with elevated temperatures, however, protein aggregates rapidly accumulate in bacteria (Supplementary Fig. 3). Under these conditions, the presence of activated Hsp33 as ATP-independent chaperone holdase becomes essential, because ATP-dependent chaperones such as DnaK are incapacitated by the oxidative stress–induced decrease in intracellular ATP levels22. At the same time, Hsp33 transcription is massively upregulated as part of the heat-shock response, further contributing to Hsp33’s protection of unfolding proteins6,22.
METHODS
Strains, plasmids and proteins
Details of plasmid and strain generation can be found in Supplementary Methods. Wild-type Hsp33, Hsp331–235, Hsp33(F187W W212F) and Hsp33(W212F Y267W) were all purified in the absence of reducing agents as described for wild-type Hsp33 in ref. 8. All reactions were carried out in 40 mM potassium phosphate buffer (pH 7.5) unless mentioned otherwise.
Preparation of Hsp33red, Hsp33ox30°C, Hsp33ox43°C and apo-Hsp33
Reduced, zinc-reconstituted Hsp33 (Hsp33red) was prepared as described7. To prepare oxidized Hsp33, 50 μM Hsp33red was incubated in 2 mM H2O2 either at 30 °C for 3 h (Hsp33ox30°C) or at 43 °C for 1 or 3 h (Hsp33ox43°C). No appreciable difference was observed between the 1-h and 3-h oxidation reactions at 43 °C. Oxidants were removed using NAP-5 columns. To investigate the influence of Gdn-HCl on the activation of Hsp33, 7 μM Hsp33red was incubated for 60 min in 1.5 M Gdn-HCl and 2 mM H2O2 at 30 °C. Oxidants and Gdn-HCl were removed using a NAP-5 column. Zinc-free (apo) Hsp33 was prepared as described25 in 40 mM potassium phosphate buffer (pH 7.5).
Hsp33 chaperone activity assay
Citrate synthase (12 μM; Roche Applied Sciences) was denatured in 4.5 M Gdn-HCl in 40 mM HEPES-KOH (pH 7.5) for 90 min at room temperature. To initiate aggregation, denatured citrate synthase was diluted to a final concentration of 75 nM into prewarmed HEPES-KOH (pH 7.5) buffer at either 30 or 43 °C under constant stirring. To test the activity of Hsp33, the buffer was supplemented with 0.3 μM Hsp33 at 30 °C or 0.45 μM at 43 °C. Light scattering was monitored at excitation (λex) and emission (λem) wavelengths of 360 nm using a Hitachi F4500 fluorescence spectrophotometer equipped with thermostated cuvette holder and stirrer.
Determination of oxidation-induced zinc release
To monitor fluorescence quenching during zinc release, 50 μM Hsp33(W212F Y267W)red was incubated with 2 mM H2O2 at either 30 or 43 °C. λex was set to 295 nm and the relative intensity of emission at 347 nm was recorded every 3 min using a Hitachi F4500 fluorescence spectrophotometer. The excitation and emission slit widths were set to 2.5 and 5 nm, respectively. At various time points, aliquots were removed and the proportion of free and protein-bound zinc was determined using the PAR/PCMB assay very similar to one described7, except that thiol bound zinc was released with para-chloromercuribenzoic acid (PCMB, ICN Biomedicals) instead of PMPS, which is no longer commercially available. To prepare zinc-free apo-Hsp33(W212F Y267W), 3 μM Hsp33(W212F Y267W) was incubated with 1 mM N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) at 30 °C for 3 h.
Mass spectrometric analysis of the thiol status of Hsp33
Thirty microliters of 50 μM Hsp33red, Hsp33ox30°C or Hsp33ox43°C was precipitated with 10% (w/v) trichloroacetic acid. Pellets were resuspended in 20 μl of DAB buffer (6 M urea, 200 mM Tris-HCl (pH 8.5), 10 mM EDTA, 0.5% (w/v) SDS) containing 100 mM IAM for 1 h under vigorous shaking. The proteins were then precipitated again with TCA (10% w/v) and resuspended in 20 μl DAB buffer supplemented with 10 mM DTT to reduce all existing oxidative thiol modifications. After 1 h at 25 °C, 20 μl of 50 mM NEM was added and all newly accessible thiol groups were alkylated for 1 h at 25 °C. All small molecules were removed with C-18–reversed phase material deposited in gel-loader pipette tips (Millipore). Tryptic digests of Hsp33 and MALDI-MS/MS analysis of the peptides were conducted at the Michigan Proteome consortium (http://www.proteomeconsortium.org).
Bis-ANS fluorescence
To test for the presence of hydrophobic surfaces in wild-type Hsp33 and Hsp331–235, the fluorescence probe bis-ANS (Molecular Probes) was used as described12.
Fluorescence measurement and zinc release of Hsp33(F187W W212F)
To monitor changes in fluorescence emission of Hsp33(F187W W212F) upon H2O2-induced oxidation, 50 μM reduced Hsp33(F187W W212F) was incubated in 40 mM potassium phosphate (pH 7.5) at 30 or 43 °C. The reaction was started by the addition of 2 mM H2O2. λex was set to 295 nm and emission spectra were recorded from 300 to 400 nm every 3 or 6 min, respectively (slit widths: 2.5 and 5 nm). The shutter was closed between measurements to minimize photobleaching and UV-induced hydroxyl radical formation. At various time points, aliquots were removed and the proportion of free and protein-bound zinc was determined using the PAR/PCMB assay (described above).
Equilibrium denaturation experiment
To monitor the stability of Hsp33(F187W W212F), 3 μM of Hsp33(F187W W212F) was incubated in 0–6 M Gdn-HCl and 40 mM potassium phosphate (pH 7.5) for up to 24 h at 25 °C (supplemented with 5 mM DTT). Equilibrium was reached within 60 min of incubation. λex was set to 295 nm and fluorescence emission spectra were recorded from 305–390 nm. The emission and excitation slit widths were 5 and 10 nm, respectively. The emission maximum was calculated for each concentration of denaturant by applying a Gaussian fit to each fluorescence emission spectra (SigmaPlot).
Far-UV circular dichroism spectroscopy
Far-UV CD spectra of Hsp33 and its derivatives (7 μM) were recorded in 20 mM potassium phosphate (pH 7.5) at either 30 or 43 °C using a Jasco J-810 spectropolarimeter as described12. To determine the thermostability of Hsp33, the CD signal at 197 nm was recorded at increasing temperatures. A scan rate of 1° min−1 was used. The temperature was controlled with a Jasco Peltier device.
Supplementary Material
Acknowledgments
We thank S. VanHaerents for excellent technical assistance, J. Bardwell, L. Leichert and T. Tapley for critically reading the manuscript, and Leopoldina Gesellschaft Deutscher Naturforscher for a postdoctoral fellowship to J.W. P.C.F.G. was supported by a Rackham Predoctoral Fellowship. US National Institutes of Health grant GM065318 supported this work.
Footnotes
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
AUTHOR CONTRIBUTIONS
M.I. performed the majority of the experiments, including the activation studies, zinc-release analysis, mass spectrometry, fluorescence spectroscopy and Gdn-HCl experiments. J.H. constructed the mutant proteins and performed the fluorescence experiments. S.A. analyzed the Hsp331–235 mutant protein. J.W. performed the two-dimensional gel analysis. P.C.F.G. performed the thermostability experiments and initiated many of the experiments. H.L. conducted the ultracentrifugation experiments and contributed to data evaluation. U.J. contributed ideas, evaluated and discussed data and prepared the manuscript.
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 2.Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal. 2000;2:811–820. doi: 10.1089/ars.2000.2.4-811. [DOI] [PubMed] [Google Scholar]
- 3.Sies H. Oxidative stress: from basic research to clinical application. Am J Med. 1991;91:31S–38S. doi: 10.1016/0002-9343(91)90281-2. [DOI] [PubMed] [Google Scholar]
- 4.Aliev G, et al. The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer’s disease. Brain Pathol. 2002;12:21–35. doi: 10.1111/j.1750-3639.2002.tb00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paget MS, Buttner MJ. Thiol-based regulatory switches. Annu Rev Genet. 2003;37:91–121. doi: 10.1146/annurev.genet.37.110801.142538. [DOI] [PubMed] [Google Scholar]
- 6.Chuang SE, Blattner FR. Characterization of twenty-six new heat shock genes of Escherichia coli. J Bacteriol. 1993;175:5242–5252. doi: 10.1128/jb.175.16.5242-5252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graumann J, et al. Activation of the redox-regulated molecular chaperone Hsp33– a two-step mechanism. Structure. 2001;9:377–387. doi: 10.1016/s0969-2126(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 8.Jakob U, Muse W, Eser M, Bardwell JC. Chaperone activity with a redox switch. Cell. 1999;96:341–352. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- 9.Won HS, et al. The zinc-dependent redox switch domain of the chaperone Hsp33 has a novel fold. J Mol Biol. 2004;341:893–899. doi: 10.1016/j.jmb.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 10.Janda I, et al. The crystal structure of the reduced, Zn2+-bound form of the B. subtilis Hsp33 chaperone and its implications for the activation mechanism. Structure. 2004;12:1901–1907. doi: 10.1016/j.str.2004.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbirz S, Jakob U, Glocker MO. Mass spectrometry unravels disulfide bond formation as the mechanism that activates a molecular chaperone. J Biol Chem. 2000;275:18759–18766. doi: 10.1074/jbc.M001089200. [DOI] [PubMed] [Google Scholar]
- 12.Graf PC, et al. Activation of the redox-regulated chaperone Hsp33 by domain unfolding. J Biol Chem. 2004;279:20529–20538. doi: 10.1074/jbc.M401764200. [DOI] [PubMed] [Google Scholar]
- 13.Winter J, Linke K, Jatzek A, Jakob U. Severe oxidative stress causes inactivation of DnaK and activation of the redox-regulated chaperone Hsp33. Mol Cell. 2005;17:381–392. doi: 10.1016/j.molcel.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 14.Hunt JB, Neece SH, Ginsburg A. The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal Biochem. 1985;146:150–157. doi: 10.1016/0003-2697(85)90409-9. [DOI] [PubMed] [Google Scholar]
- 15.Davis FA, Jenkins LA, Billmers RL. Chemistry of sulfenic acids. 4 The first evidence for the involvement of sulfenic acids in the oxidation of thiols. J Am Chem Soc. 1986;103:7016–7018. [Google Scholar]
- 16.Claiborne A, Miller H, Parsonage D, Ross RP. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 1993;7:1483–1490. doi: 10.1096/fasebj.7.15.8262333. [DOI] [PubMed] [Google Scholar]
- 17.Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 18.Raman B, Siva Kumar LV, Ramakrishna T, Mohan Rao C. Redox-regulated chaperone function and conformational changes of Escherichia coli Hsp33. FEBS Lett. 2001;489:19–24. doi: 10.1016/s0014-5793(01)02074-9. [DOI] [PubMed] [Google Scholar]
- 19.Kim SJ, Jeong DG, Chi SW, Lee JS, Ryu SE. Crystal structure of proteolytic fragments of the redox-sensitive Hsp33 with constitutive chaperone activity. Nat Struct Biol. 2001;8:459–466. doi: 10.1038/87639. [DOI] [PubMed] [Google Scholar]
- 20.Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vijayalakshmi J, Mukhergee MK, Graumann J, Jakob U, Saper MA. The 2.2 A crystal structure of Hsp33: a heat shock protein with redox-regulated chaperone activity. Structure. 2001;9:367–375. doi: 10.1016/s0969-2126(01)00597-4. [DOI] [PubMed] [Google Scholar]
- 22.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 23.Prilusky J, et al. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 24.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Jakob U, Eser M, Bardwell JC. Redox switch of Hsp33 has a novel zinc-binding motif. J Biol Chem. 2000;275:38302–38310. doi: 10.1074/jbc.M005957200. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann JH, Linke K, Graf PC, Lilie H, Jakob U. Identification of a redox-regulated chaperone network. EMBO J. 2004;23:160–168. doi: 10.1038/sj.emboj.7600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang JG, et al. RsrA, an anti-sigma factor regulated by redox change. EMBO J. 1999;18:4292–4298. doi: 10.1093/emboj/18.15.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grimshaw JP, Jelesarov I, Schonfeld HJ, Christen P. Reversible thermal transition in GrpE, the nucleotide exchange factor of the DnaK heat-shock system. J Biol Chem. 2001;276:6098–6104. doi: 10.1074/jbc.M009290200. [DOI] [PubMed] [Google Scholar]
- 29.Haslbeck M, et al. Hsp26: a temperature-regulated chaperone. EMBO J. 1999;18:6744–6751. doi: 10.1093/emboj/18.23.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.