

Abstract
Mice infected with relapsing fever (RF) spirochaetes survive recurrent waves of high-level bacteraemia with little, if any, clinical complications or tissue injury. In the absence of B-cells, peak bacteraemia does not resolve, resulting in multi-organ complications. During peak bacteraemia, large amounts of interleukin-10 (IL-10) are produced in blood and tissues. In mice unable to clear peak bacteraemia, exogenous IL-10 greatly reduced the clinical manifestations, serum levels of CXCL13, cerebral microgliosis, and the pathogen load. In contrast, IL-10 deficiency in mice unable to clear peak bacteraemia resulted in microvascular complications with distinct severities, depending on the serotype: serotype 2 (Bt2), which causes peak bacteraemia of c. 108/mL, resulted in rapid death from subarachnoid and intraparenchymal haemorrhage; in contrast, serotype 1, which causes peak bacteraemia of c. 107/mL, resulted in milder multi-organ haemorrhage and thrombosis. IL-10 deficiency also resulted in multi-organ haemorrhage and thrombosis with infarction in wild-type mice despite lower peak bacteraemia. Two mechanisms for pathogen control have been identified: antibody clearance of peak bacteraemia, and antibody-independent lowering of bacteraemia via phagocytosis in the spleen. IL-10 plays opposite roles in pathogen control, depending on the severity of bacteraemia: during persistent high bacteraemia, IL-10 helps to control it by protecting innate immune cells from apoptosis; in contrast, during transient peak bacteraemia, IL-10 slows down antibody-mediated clearance. A successful outcome from RF depends on a balanced immune response to clear bacteraemia while avoiding microvascular injury, in which production of IL-10, in response to the pathogen load, plays a critical role.
Keywords: Borreliosis, brain, haemorrhage, interleukin-10, microglia, relapsing fever, review
Introduction
Relapsing fever (RF) is a multisystemic spirochaetal infection caused by a variety of Borrelia species that occurs in two forms, epidemic and endemic. Epidemic RF is louse-borne and caused by Borrelia recurrentis, whereas endemic RF is tick-borne and caused by different Borrelia species in endemic areas throughout the world [1]. The clinical and pathological manifestations of RF borreliosis are diverse, depending on the Borrelia species and the genetic background of the host. The relapsing course of the disease is caused by antigenic variation of RF spirochaetes, which results in febrile periods at times of high bacteraemia, alternating with periods of relative wellbeing during low bacteraemia. IgM antibodies specific for outer membrane lipoproteins are responsible for resolution of bactaeremic peaks [2–6]. Untreated infection can result in multisystemic complications, and even death. Our laboratory studies the immunopathogenesis and tissue tropism of the North American RF spirochaetes Borrelia hermsii [2] and Borrelia turicatae [7–9] in mice.
Infection
All outbred stocks (Swiss Webster) and inbred strains (C57BL/6, B10, BALB/c, C3H/HeJ, and SWR) of the mice that we have tested are susceptible to infection with North American RF spirochaetes [2,10,11]. The magnitude of peak bacteraemia varies from 105/mL to 108/mL, with higher counts usually being observed during the first peak and when higher inocula are used, or in B-cell-deficient mice [2,10]. After inoculation of mice with a single RF spirochaete, peak bacteraemia usually occurs on day 4, after which clearance occurs, and the original inoculum is replaced by a mixed population that persists for 2–3 days before being replaced by newer serotypes [2,12]. RF spirochaetes are found free in the blood and extravascularly in the interstitium of multiple tissues, including the skin, the joints, the heart, the aorta, the leptomeninges, the subarachnoid space, the brain parenchyma, and the labyrinth [13,14]. Like the causative agent of Lyme disease, RF spirochaetes show a distinct tropism for collagenous tissues [13].
An interesting phenomenon in RF is that the brain often continues to be infected after the blood has ceased to be; this is referred to as residual brain infection [7]. In our laboratory, we observed residual brain infection in 20% of mice examined 1 month after intraperitoneal inoculation with B. turicatae [10]. Approximately 20% of mice develop persistent infection in the blood, and this was observed more frequently in TLR2-deficient mice [10]. Although, in the majority of cases, residual brain infection is caused by new serotypes, in at least one case it was caused by the serotype that was originally inoculated [10]. A study of residual brain infection due to different Borrelia spp. showed that it is more prevalent with the African species Borrelia duttonii [15]. The average pathogen load during residual brain infection was determined to be c. 2000 spirochaetes per gram of brain. RF spirochaetes causing residual brain infection can re-infect the blood if immunosuppression occurs [15].
Clinical Complications
There is great heterogeneity in the clinical manifestations of RF in experimental animals, depending on the immune status of the host and the infecting organism. The severity of clinical disease is proportional to the pathogen load. Therefore, animals inoculated with large numbers of organisms tend to fall ill and even die early on [16]. Inoculation with B. recurrentis in grivet monkeys (Cercopithecus aethiops) uniformly caused high fever and resulted in 20% mortality, including two animals during a severe relapse [17]. Although we never observed clinical disease in various strains of inbred mice inoculated with B. turicatae, they did occur in all B-cell-deficient mice that we tested, independently of their genetic background. They include purulent eye discharge, ruffled skin, tibiotarsal joint reddening and swelling, and vestibular dysfunction [10]. Clinical complications have also been observed sporadically in outbred mice. Old World RF spirochaetes appear to be more pathogenic than New World species; inoculation of BALB/c mice with Borrelia crocidurae, but not with B. hermsii, resulted in clinical disease [11,18].
Tissue Tropism
During infection of wild-type mice with B. hermsii, we noticed, in several cases, the presence of serotypes in the blood that were not present in the brain [2]. This suggested that some serotypes of RF spirochaetes are more neurotropic than others. As serotype-specific antibodies rapidly clear RF spirochaetes from blood and tissues [2,12,19], we switched to B-cell-deficient mice to study serotype tropism. For this, we chose a strain of B. turicatae responsible for an outbreak of RF in Texas (USA) that resulted in prominent neurological complications, including meningitis and facial paralysis. Infection with this strain was well tolerated by SCID mice, which are B-cell-deficient and T-cell-deficient, without any mortality for at least 100 days [11]. Seven days after inoculation, we documented the presence of at least three serotypes, all of them present in both blood and brain [11]. Fifty days after inoculation, these three serotypes had been replaced by two new serotypes, one of which was predominant and appeared to be well tolerated. However, over the next 50 days, this serotype was gradually replaced by the second one, which was more virulent [11]. The two serotypes, originally named A and B, and since then renamed 1 and 2, have been extensively studied to examine their virulence and tissue tropism. The only discernible difference between them is the variable major protein that they express, 23-kDa variable small protein 1 in serotype 1 (Bt1), and 20-kDa variable small protein 2 in serotype 2 (Bt2) [20–22]. Although both serotypes have similar abilities to establish infection with as little as one spirochaete per mouse and to disseminate to distant tissues, clinical examinations in various B-cell-deficient mice, studied for up to 2 months, have revealed marked differences in their virulence and tissue tropism [11,14,23–27]. Only Bt2 kills infant mice. In adult mice, clinical disease manifested in the first week with purulent discharge from the eyes and ruffled skin, and this was followed in the second week by tibiotarsal joint swelling and redness, and by the third or fourth week by signs of vestibular dysfunction [11]. The differences in joint involvement between the two serotypes was most striking, with tibiotarsal and metatarsal joint reddening and swelling beginning earlier and becoming more severe with Bt2 [11]. Bt2 also caused more severe functional impairment in an equilibrium bar test. In contrast, Bt1 caused more severe vestibular disease but only after several weeks of persistent infection [28]. Bt1 also appeared to be more neurotropic than Bt2 [11]. When brain infection was studied by culture of whole brain homogenates, Bt1 was found in the brain much more frequently than Bt2 [11]. Furthermore, only Bt1 was apparent according to western blot on brain cultures from SCID mice that were co-infected with Bt1 and Bt2 [11]. As Bt2 had no problems infecting the brain after intracerebral inoculation [11], this suggests that Bt1 is better than Bt2 at crossing the blood–brain barrier or that the function of the blood–brain barrier is better preserved during infection with Bt2. Using immunohistochemistry, we found five times as many Bt1 as Bt2 spirochaetes in the leptomeninges in SCID mice examined 18 days after inoculation [13]. TaqMan RT-PCR showed that the mean number of spirochaetes per gram of brain RNA 1 month after inoculation was approximately ten-fold higher with Bt1 than with Bt2 [14]. The pathogen load in the brains of mice co-infected with Bt1 and Bt2 was intermediate between that of mice infected with either serotype alone [10], suggesting that co-infection with Bt2 decreases the entry of Bt1 into the brain. A comparison of mice co-infected with Bt1 and Bt2 also revealed that the severe vestibular dysfunction caused by Bt1 was prevented by co-infection with Bt2. In contrast, the severe arthritis and weight loss caused by infection with Bt2 alone were not influenced by co-infection with Bt1 [28].
The high number of spirochaetes found in blood and tissues of B-cell-deficient mice inoculated with B. turicatae provided an opportunity to study their dissemination to various tissues after intraperitoneal inoculation [13]. Whole decalcified heads were used to study multiple tissues simultaneously without disrupting important anatomical relationships. Spirochaetes were observed microscopically as early as 4 days after inoculation [13]. The first tissue in which spirochaetes were observed outside of the vasculature was the dura mater [13]. In the brain, the localization was primarily leptomeningeal, although, rarely, spirochaetes were also found in the brain parenchyma itself. The site of entry into the brain appeared to be the leptomeningeal microcirculation, where they were observed attached to, or in the process of crossing, brain–microvascular endothelial cell junctions [14]. The spirochaetes were not limited to the central nervous system. They were also observed in the middle and inner ear, endoneurium and perineurium of cranial and peripheral nerves, skin, bone marrow, and heart [13,25]. Clumps of spirochaetes were observed in the subarachnoid space, inner ear, and skin. Spirochaetes were also found in the extracellular matrix of skeletal and cardiac muscle. No spirochaetes were found in salivary glands or in the choroid plexus or intracellularly [13,14]. Whereas Bt2 was more abundant in the skin [13], the heart [25], and the tibiotarsal joints [29], Bt1 was more abundant in the leptomeninges. In all B-cell-deficient mice that we have tested, including SCID and RAG1, RAG2 and Igh6 null mice, the pathogen load in the blood was five to ten times higher with Bt2 than with Bt1 [14,23,24,26,30].
We studied the pathogen load during co-infection with various serotypes indirectly by comparing circulating levels of the B-cell chemokine CXCL13 in SCID mice persistently infected with Bt2 alone or in combination with Bt1 [28]. This approach was valid because the levels of this chemokine correlate very strongly with the pathogen load in Borrelia infections [23,31]. The results revealed that co-infected mice had significantly higher CXCL13 levels than mice infected with Bt2 alone; the mean (standard deviation) serum pg/mL CXCL13 was 72 625 (4750) in co-infected mice, as compared with 42 625 (5406) in mice infected with Bt2 alone (p <0.001) [28].
Pathology
Examination of the blood during acute RF revealed increased numbers of neutrophils, lymphocytes, and monocytes, and anaemia [18,32]. However, when mice were inoculated with very large numbers of spirochaetes, the opposite was observed: a decline in the number of circulating leukocytes that was proportional to the increase in bacteraemia [16]. Infection with RF spirochaetes also causes thrombocytopenia [16,18,32], which also correlates with the magnitude of bacteraemia [33]. Another consistent finding in infected mice is marked hepatosplenomegaly [16,18,27,32]. Infection with some RF species can lead to inflammation of the heart with perivascular mononuclear cell infiltrates in the pericardium and myocardium, with interstitial oedema and necrosis of cardiomyocytes [32,34,35]. Prominent vascular pathology with high mortality did occur after inoculation with large numbers of Old World RF spirochaetes [16]; microscopic examination revealed blood vessel fibrin thrombi in the kidneys, liver, spleen, brain, and lungs, prominent focal areas of endothelial cell swelling, and nuclear pyknosis and karyorrhexis in hepatocytes and Kupffer cells [16,18].
Another common pathological finding in mice during the course of RF is inflammation in the brain [7]. Wild-type and TLR2-deficient C57BL/6 mice inoculated with Bt1 developed mild leptomeningeal inflammation and brain parenchymal macrophage infiltration/microglial activation (upper panels in Fig. 1), with few, if any, lymphocytes, and absent neutrophils [10]. Inoculation of BALB/c mice with B. crocidurae resulted in increased numbers of perivascular macrophages, and microgliosis with small numbers of lymphocytes and granulocytes in the brain [36]. Mice inoculated with an RF strain from Spain developed perivascular mononuclear cell infiltration of the leptomeninges, accumulation of activated macrophages perivascularly, and plexitis [32,37]. Unlike during acute infection, no evidence of inflammation was found in the brains of mice experiencing residual brain infection [15].
FIG. 1.
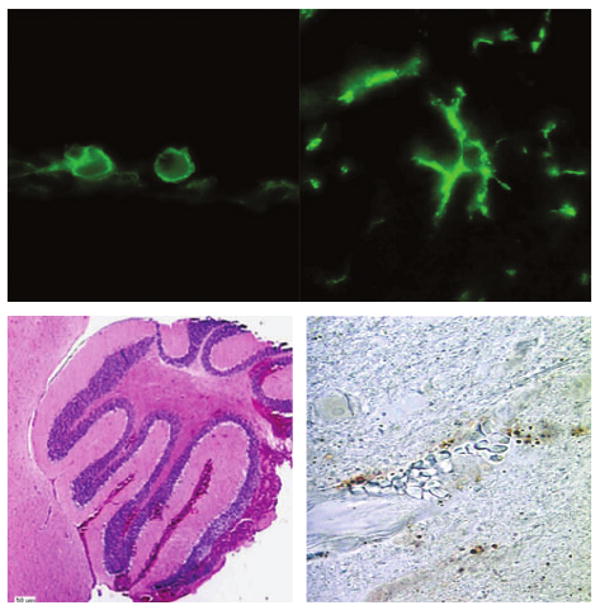
Spectrum of brain pathology in relapsing fever (RF). Infection with the RF spirochaete Borrelia turicatae can result in prominent changes in the brain parenchyma that vary from macrophage infiltration/microglial activation (top panels, ×1000 magnification) of varying degrees in wild-type and B-cell-deficient mice to severe intracerebral haemorrhage (left lower panel) with prominent brain microvascular endothelial cell apoptosis (right lower panel) in RAG2-deficient mice infected with serotype 2. The upper panels show immunofluorescence staining with rat monoclonal antibody anti-mouse F4/80. The left lower panel shows haematoxylin and eosin staining of the cerebellum, and the right lower panel shows terminal deoxynucleotidyl transferase dUTP nick end labelling staining of the cerebral cortex.
Studies in B-cell-deficient mice persistently infected with Bt1 or Bt2 have shown marked differences in their pathogenicity [11,14,23–28]. SCID mice inoculated with Bt1 or Bt2 develop frank arthritis after 2 weeks that is more severe with Bt2 [11]. Haematoxylin and eosin-stained sections revealed mixed inflammation of the synovial lining of both joints and tendon sheaths with mononuclear and polymorphonuclear cells, and prominent thickening of the synovium and leukocyte exudate in the joint space. There was also prominent inflammation and tissue injury in the heart. Cardiac inflammation involved both the myocardium and pericardium, and was characterized by a prominent mononuclear leukocytic infiltrate between myocardial fibres, with exudate in the pericardium. Cardiac inflammation was more severe in the atria and around the great vessels in the mediastinum [11,25], similar to what has been seen in Lyme borreliosis [38].
The loss of cardiomyocytes, macrophage infiltration and upregulation of interleukin-6 were all more prominent in the atrium and more severe with Bt2 than with Bt1 [25]. Morphological features of apoptosis, including fragmented nuclei and condensed chromatin, terminal deoxynucleotidyl transferase dUTP nick end labelling positivity, and BAX immunostaining, were readily apparent in areas of cardiomyocyte injury. Analysis of caspase activation showed significant upregulation of several caspases, most notably caspase 1 and caspase 11, with strong expression in areas of injury that co-localized with activated macrophages. Similar cardiomyocyte apoptosis was observed in RAG1 and Igh6 null mice, with prominent upregulation of tumour necrosis factor (TNF) and interleukin-10 (IL-10) of similar degree with Bt1 and Bt2 [23,25].
The severity of inflammation in the skin was similar with either serotype [13]. Significant inflammation was also found in the vestibular and auditory labyrinths with either serotype, which probably explains the prominent vestibular dysfunction that characterizes this model [28]. Mild inflammation was also found in the leptomeninges, with no obvious difference between Bt1 and Bt2.
Although haematoxylin and eosin staining in SCID mice did not reveal any apparent abnormalities in the brain parenchyma, immunostaining for F4/80, a marker of activated microglia/macrophages, revealed that infection resulted in widespread infiltration by activated macrophages/microglia with a 30-fold increase with Bt1, as compared with uninfected controls [14]. Microgliosis with Bt2 was only four times greater than that in uninfected controls. The morphology of F4/80-stained cells was heterogeneous, with some cells being amoeboid (upper left panel in Fig. 1) and some ramified (upper right panel in Fig. 1). Similar findings were observed in RAG1 and Igh6 null mice, with prominent upregulation of TNF and IL-10 by 2 weeks, but only upregulation of IL-10 remaining after 4 weeks [23]. Fully activated microglia were the main source of IL-10 in the brain. A comparison of gene expression in Igh6 null mice persistently infected with Bt1 with uninfected controls revealed upregulation of many genes known to be upregulated during microglial activation [23]. The gene encoding CXCL13 was the most highly upregulated gene in infected brains, and CXCL13 was also the most abundant of the chemokines that we measured in the blood [23]. An analysis of functional categories within the gene ontology database showed that the majority of categories with the highest EASE scores referred to functions of the immune response to pathogens, including proteins involved in antigen processing and presentation, chemotaxis, adhesion, and migration, lipopolysaccharide/lipoprotein toll-like receptor ligation and signalling, and pathogen control and host defence [23]. A surprising finding of these studies was the lack of any evidence, either morphological or according to gene arrays, of brain injury, despite persistent infection.
Protective Role of IL-10
During the studies on persistent infection in B-cell-deficient mice, we were intrigued by the observation that, despite the absence of specific antibody to clear the infection, many clinical manifestations either resolved completely (purulent eye discharge) or significantly decreased (ruffled fur and arthritis) over days to weeks [11]. Similarly intriguing was the observation that persistent infection with the more virulent serotype Bt2 protected mice from the severe and fatal vestibular dysfunction that characterized persisting infection with Bt1 [28]. The first clue as to a possible explanation for these observations came from a study comparing the severity of clinical disease between RAG1 null mice, which are B-cell-deficient and T-cell-deficient, and Igh6 null mice, which lack only B-cells. This study showed that although RAG1 null mice developed a higher pathogen load, they had much less severe clinical disease and produced more IL-10 [23,24]. This was an important observation, in view of the previous report by Cooper et al. [39] that humans infected with the louse-borne RF spirochaete B. recurrentis have extraordinarily high levels of circulating IL-10 at times when they appear to be relatively well. In fact, IL-10 had the highest levels among all the cytokines that we measured, with a strong positive correlation with the pathogen load. The increase in IL-10 levels was associated with a concomitant decrease in circulating levels of TNF and all other proinflammatory cytokines and many chemokines [24,27]. The upregulation of IL-10 was found not only in blood, but also in other tissues, including the heart and the brain [24].
The first direct evidence that IL-10 was responsible for the previously observed amelioration of clinical disease came from the observation that giving infected Igh6 null mice exogenous IL-10 reduced bacteraemia, clinical disease, cerebral microgliosis, and serum CXCL13 levels [23,24,27]. The lowering of the pathogen load in Igh6 null mice was opposite to the finding in wild-type mice infected with RF [30] or Lyme disease [40] spirochaetes that IL-10 deficiency lowered the pathogen load via improved specific-antibody production. Definite proof of the fundamental protective role of IL-10 in RF came from the observation that in Bt2-infected RAG2 null mice, IL-10 deficiency resulted in increased bacteraemia with rapid death due to severe intracerebral haemorrhage and apoptosis of brain microvascular endothelial cells (lower panels in Fig. 1) [26,27]. The protective role of IL-10 was not restricted to RAG2 null mice infected with Bt2, as IL-10 deficiency also resulted in multi-organ haemorrhage and microvascular thrombosis in Bt1-infected RAG2 null mice [26] and in wild-type mice that had been inoculated with Bt2 [30].
During RF in wild-type mice, serum levels of IL-10 increased but only at times of peak bacteraemia (Fig. 2). Remarkably, although mice examined during peaks of bacteraemia had high serum levels of IL-10, IL-10 was absent on the day following resolution of peak bacteraemia. This suggests that peak bacteraemia in RF triggers transient production of IL-10 in the circulation. A clue to the role of IL-10 production came from the finding that IL-10-deficient mice developed microvascular injury and thrombosis with infarction in the liver, even though their peak bacteraemia, c. 104/mL [30], was much lower, and the original serotype was cleared faster (unpublished observation). The lower peak bacteraemia was probably the result of stronger production of serotype-specific antibodies [30], which was probably facilitated by strong production of the B-cell chemokine CXCL13 in the absence of IL-10 (Fig. 3). These results indicate that a fundamental protective mechanism during RF is the production of IL-10 at times of peak bacteraemia to prevent vascular injury, which may be fatal. This observation has important implications for our understanding of RF in humans.
FIG. 2.
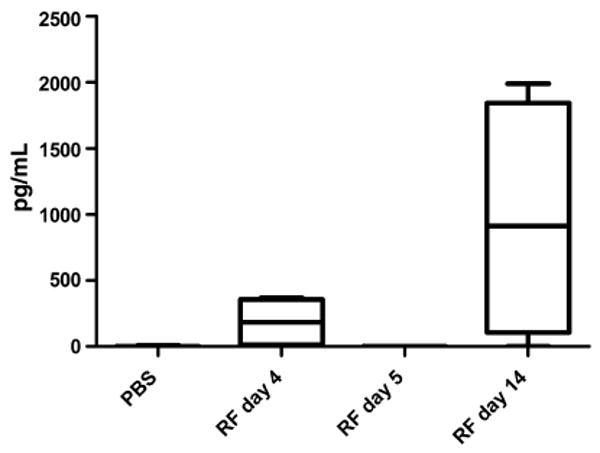
Serum levels of interleukin-10 (IL-10) in relapsing fever (RF) in mice. Groups of four to eight wild-type mice were necropsied at various times after inoculation of serotype 2 of the RF spirochaete Borrelia turicatae (Bt2), and the serum levels of IL-10 were measured by ELISA in comparison with uninfected control mice (phosphate-buffered saline (PBS)). Infected mice were necropsied during the first peak of bacteraemia (day 4), immediately after resolution of peak bacteraemia (day 5), or on day 14, when some mice were experiencing another peak of bacteraemia. Note that IL-10 was found in serum only during peak bacteraemia.
FIG. 3.
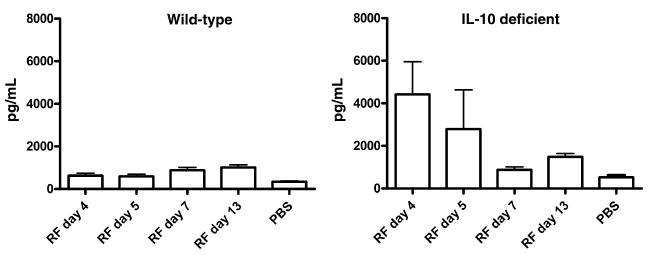
Serum levels of the B-cell chemokine CXCL13 in wild-type and interleukin-10 (IL-10)-deficient mice. Groups of four to eight wild-type (left panel) or IL-10-deficient C57BL/10 (right panel) mice were necropsied at various times after inoculation of serotype 2 of Borrelia turicatae (Bt2), and the serum levels of CXCL13 were measured by ELISA in comparison with uninfected control mice (phosphate-buffered saline (PBS)). Whereas peak bacteraemia occurred in wild-type mice on day 4 and had resolved by day 5, peak bacteraemia was of lower magnitude and occurred earlier in IL-10-deficient mice. Note that production of CXCL13 was ten times higher early on in IL-10-deficient mice (right panel). RF, relapsing fever.
Acknowledgments
We thank H. Gelderblom and A. Marques for their contributions to the studies described in this review.
Transparency Declaration
The research described in this review was supported by award number R21NS057545 from the National Institute of Neurological Disorders and Stroke (NINDS) to D. Cadavid. The content is solely the responsibility of the authors, and does not necessarily represent the views of the NINDS or the National Institutes of Health. D. Cadavid is currently a full-time employee of Biogen Idec. This review is not related to his employment at Biogen Idec.
References
- 1.Barbour AG, Hayes SF. Biology of Borrelia species. Microbiol Rev. 1986;50:381–400. doi: 10.1128/mr.50.4.381-400.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cadavid D, Bundoc V, Barbour AG. Experimental infection of the mouse brain by a relapsing fever Borrelia species: a molecular analysis. J Infect Dis. 1993;168:143–151. doi: 10.1093/infdis/168.1.143. [DOI] [PubMed] [Google Scholar]
- 3.Connolly SE, Benach JL. Cutting edge: the spirochetemia of murine relapsing fever is cleared by complement-independent bactericidal antibodies. J Immunol. 2001;167:3029–3032. doi: 10.4049/jimmunol.167.6.3029. [DOI] [PubMed] [Google Scholar]
- 4.Alugupalli KR, Gerstein RM, Chen J, Szomolanyi-Tsuda E, Woodland RT, Leong JM. The resolution of relapsing fever borreliosis requires IgM and is concurrent with expansion of B1b lymphocytes. J Immunol. 2003;170:3819–3827. doi: 10.4049/jimmunol.170.7.3819. [DOI] [PubMed] [Google Scholar]
- 5.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity. 2004;21:379–390. doi: 10.1016/j.immuni.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 6.Belperron AA, Dailey CM, Bockenstedt LK. Infection-induced marginal zone B cell production of Borrelia hermsii-specific antibody is impaired in the absence of CD1d. J Immunol. 2005;174:5681–5686. doi: 10.4049/jimmunol.174.9.5681. [DOI] [PubMed] [Google Scholar]
- 7.Cadavid D, Barbour AG. Neuroborreliosis during relapsing fever: review of the clinical manifestations, pathology, and treatment of infections in humans and experimental animals. Clin Infect Dis. 1998;26:151–164. doi: 10.1086/516276. [DOI] [PubMed] [Google Scholar]
- 8.Schuhardt V, O'Bryan B. Effect of intracranial penicillin therapy on brain involvement in experimental relapsing fever. J Bacteriol. 1945;49:312–313. doi: 10.1128/jb.49.3.312-313.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trape JF, Duplantier JM, Bouganali H, et al. Tick-borne borreliosis in west Africa. Lancet. 1991;337:473–475. doi: 10.1016/0140-6736(91)93404-w. [DOI] [PubMed] [Google Scholar]
- 10.Cadavid D, Sondey M, Garcia E, Lawson CL. Residual brain infection in relapsing-fever borreliosis. J Infect Dis. 2006;193:1451–1458. doi: 10.1086/503367. [DOI] [PubMed] [Google Scholar]
- 11.Cadavid D, Thomas DD, Crawley R, Barbour AG. Variability of a bacterial surface protein and disease expression in a possible mouse model of systemic Lyme borreliosis. J Exp Med. 1994;179:631–642. doi: 10.1084/jem.179.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoenner HG, Dodd T, Larsen C. Antigenic variation of Borrelia hermsii. J Exp Med. 1982;156:1297–1311. doi: 10.1084/jem.156.5.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cadavid D, Pachner AR, Estanislao L, Patalapati R, Barbour AG. Isogenic serotypes of Borrelia turicatae show different localization in the brain and skin of mice. Infect Immun. 2001;69:3389–3397. doi: 10.1128/IAI.69.5.3389-3397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sethi N, Sondey M, Bai Y, Kim KS, Cadavid D. Interaction of a neurotropic strain of Borrelia turicatae with the cerebral microcirculation system. Infect Immun. 2006;74:6408–6418. doi: 10.1128/IAI.00538-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsson C, Andersson M, Pelkonen J, Guo BP, Nordstrand A, Bergstrom S. Persistent brain infection and disease reactivation in relapsing fever borreliosis. Microbes Infect. 2006;8:2213–2219. doi: 10.1016/j.micinf.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 16.Wright DJ, Woodrow DF. Terminal changes in mice experimentally infected with Borrelia duttoni. J Pathol. 1980;130:83–90. doi: 10.1002/path.1711300204. [DOI] [PubMed] [Google Scholar]
- 17.Judge DM, La Croix JT, Perine PL. Experimental louse-borne relapsing fever in the grivet monkey, Cercopithecus aethiops. I. Clinical course. Am J Trop Med Hyg. 1974;23:957–961. doi: 10.4269/ajtmh.1974.23.957. [DOI] [PubMed] [Google Scholar]
- 18.Shamaei-Tousi A, Martin P, Bergh A, Burman N, Brannstrom T, Bergstrom S. Erythrocyte-aggregating relapsing fever spirochete Borrelia crocidurae induces formation of microemboli. J Infect Dis. 1999;180:1929–1938. doi: 10.1086/315118. [DOI] [PubMed] [Google Scholar]
- 19.Barbour AG, Dai Q, Restrepo BI, Stoenner HG, Frank SA. Pathogen escape from host immunity by a genome program for antigenic variation. Proc Natl Acad Sci USA. 2006;103:18290–18295. doi: 10.1073/pnas.0605302103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cadavid D, Pennington PM, Kerentseva TA, Bergstrom S, Barbour AG. Immunologic and genetic analyses of VmpA of a neurotropic strain of Borrelia turicatae. Infect Immun. 1997;65:3352–3360. doi: 10.1128/iai.65.8.3352-3360.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pennington PM, Cadavid D, Barbour AG. Characterization of VspB of Borrelia turicatae, a major outer membrane protein expressed in blood and tissues of mice. Infect Immun. 1999;67:4637–4645. doi: 10.1128/iai.67.9.4637-4645.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pennington PM, Cadavid D, Bunikis J, Norris SJ, Barbour AG. Extensive interplasmidic duplications change the virulence phenotype of the relapsing fever agent Borrelia turicatae. Mol Microbiol. 1999;34:1120–1132. doi: 10.1046/j.1365-2958.1999.01675.x. [DOI] [PubMed] [Google Scholar]
- 23.Gelderblom H, Londono D, Bai Y, et al. High production of CXCL13 in blood and brain during persistent infection with the relapsing fever spirochete Borrelia turicatae. J Neuropathol Exp Neurol. 2007;66:208–217. doi: 10.1097/01.jnen.0000248556.30209.6d. [DOI] [PubMed] [Google Scholar]
- 24.Gelderblom H, Schmidt J, Londono D, et al. Role of interleukin 10 during persistent infection with the relapsing fever Spirochete Borrelia turicatae. Am J Pathol. 2007;170:251–262. doi: 10.2353/ajpath.2007.060407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Londoño D, Bai Y, Zuckert WR, Gelderblom H, Cadavid D. Cardiac apoptosis in severe relapsing fever borreliosis. Infect Immun. 2005;73:7669–7676. doi: 10.1128/IAI.73.11.7669-7676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Londono D, Carvajal J, Arguelles-Grande C, Marques A, Cadavid D. Interleukin 10 protects the brain microcirculation from Spirochetal injury. J Neuropathol Exp Neurol. 2008;67:976–983. doi: 10.1097/NEN.0b013e318187a279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Londono D, Marques A, Hornung RL, Cadavid D. IL-10 helps control pathogen load during high-level bacteraemia. J Immunol. 2008;181:2076–2083. doi: 10.4049/jimmunol.181.3.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cadavid D, Garcia E, Gelderblom H. Coinfection with Borrelia turicatae serotype 2 prevents the severe vestibular dysfunction and earlier mortality caused by serotype 1. J Infect Dis. 2007;195:1686–1693. doi: 10.1086/516783. [DOI] [PubMed] [Google Scholar]
- 29.Pennington PM, Allred CD, West CS, Alvarez R, Barbour AG. Arthritis severity and spirochete burden are determined by serotype in the Borrelia turicatae-mouse model of Lyme disease. Infect Immun. 1997;65:285–292. doi: 10.1128/iai.65.1.285-292.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Londoño Diana MA, Hornung Ronald L, Cadavid D. Relapsing fever borreliosis in IL-10 deficient mice. Infect Immun. 2008;77:5508–5513. doi: 10.1128/IAI.00587-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narayan K, Dail D, Li L, et al. The nervous system as ectopic germinal center: CXCL13 and IgG in Lyme neuroborreliosis. Ann Neurol. 2005;57:813–823. doi: 10.1002/ana.20486. [DOI] [PubMed] [Google Scholar]
- 32.Gebbia JA, Monco JC, Degen JL, Bugge TH, Benach JL. The plasminogen activation system enhances brain and heart invasion in murine relapsing fever borreliosis. J Clin Invest. 1999;103:81–87. doi: 10.1172/JCI5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alugupalli KR, Michelson AD, Joris I, et al. Spirochete–platelet attachment and thrombocytopenia in murine relapsing fever borreliosis. Blood. 2003;102:2843–2850. doi: 10.1182/blood-2003-02-0426. [DOI] [PubMed] [Google Scholar]
- 34.Judge DM, La Croix JT, Perine PL. Experimental louse-borne relapsing fever in the grivet monkey, Cercopithecus aethiops. II. Pathology. Am J Trop Med Hyg. 1974;23:962–968. doi: 10.4269/ajtmh.1974.23.962. [DOI] [PubMed] [Google Scholar]
- 35.Nordstrand A, Shamaei-Tousi A, Ny A, Bergstrom S. Delayed invasion of the kidney and brain by Borrelia crocidurae in plasminogen-deficient mice. Infect Immun. 2001;69:5832–5839. doi: 10.1128/IAI.69.9.5832-5839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andersson M, Nordstrand A, Shamaei-Tousi A, Jansson A, Bergstrom S, Guo BP. In situ immune response in brain and kidney during early relapsing fever borreliosis. J Neuroimmunol. 2007;183:26–32. doi: 10.1016/j.jneuroim.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Monco JC, Miller NS, Backenson PB, Anda P, Benach JL. A mouse model of Borrelia meningitis after intradermal injection. J Infect Dis. 1997;175:1243–1245. doi: 10.1086/593681. [DOI] [PubMed] [Google Scholar]
- 38.Barthold SW, de Souza MS, Janotka JL, Smith AL, Persing DH. Chronic Lyme borreliosis in the laboratory mouse. Am J Pathol. 1993;143:959–971. [PMC free article] [PubMed] [Google Scholar]
- 39.Cooper PJ, Fekade D, Remick DG, Grint P, Wherry J, Griffin GE. Recombinant human interleukin-10 fails to alter proinflammatory cytokine production or physiologic changes associated with the Jarisch–Herxheimer reaction. J Infect Dis. 2000;181:203–209. doi: 10.1086/315183. [DOI] [PubMed] [Google Scholar]
- 40.Brown JP, Zachary JF, Teuscher C, Weis JJ, Wooten RM. Dual role of interleukin-10 in murine Lyme disease: regulation of arthritis severity and host defense. Infect Immun. 1999;67:5142–5150. doi: 10.1128/iai.67.10.5142-5150.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]