

Abstract
Waldenström’s macroglobulinemia (WM) is a distinct clinico-biological entity defined as a B-cell neoplasm characterized by a lymphoplasmacytic infiltrate in the bone marrow (BM) and immunoglobulin M paraprotein production. Cytogenetic analyses were historically limited by the difficulty in obtaining tumor metaphases and the genetic basis of the disease remains poorly defined. Here we performed a comprehensive analysis in 42 WM patients by using high-resolution, array-based comparative genomic hybridization approach to unravel the genetic mechanisms associated with WM pathogenesis. Overall, 83% of patients have chromosomal abnormalities, with a median of three abnormalities per patient. Gain of 6p was the second most common abnormality (17%) and its presence was always concomitant with 6q loss. A minimal deleted region, including MIRN15A and MIRN16-1, was delineated on 13q14 in 10% of patients. Of interest, we reported biallelic deletions and/or inactivating mutations with uniparental disomy in TRAF3 and TNFAIP3, two negative regulators of the NF-kB signaling pathway. Furthermore, we confirmed the association between TRAF3 inactivation and increased transcriptional activity of NF-kB target genes. Mutational activation of the NF-kB pathway, which is normally activated by ligand-receptor interactions within the BM microenvironment, highlights its biologic importance, and suggests a therapeutic role for inhibitors of NF-kB pathway activation in the treatment of Waldenström’s macroglobulinemia.
Keywords: Waldenström’s Macroglobulinemia, aCGH, TRAF3, TNFAIP3, NF-kB signaling pathways
Introduction
Waldenström’s macroglobulinemia (WM) is an incurable low-grade B-cell lymphoproliferative disorder characterized by bone marrow (BM) infiltration of a clonal population of small B-lymphocytes, plasmacytoid lymphocytes and plasma cells that secrete monoclonal IgM antibody (1). The etiology of the disease is still unknown. Although believed to be predominantly sporadic, familial cases suggest a possible predisposing genetic defect (2, 3).
The genetic basis of the disease remains poorly defined. Few recurrent chromosomal abnormalities have been reported in WM, reflecting the difficulty in obtaining tumor metaphases for karyotype studies. Deletion of 6q is the most common abnormality, identified in approximately half of the patients when analyzed by FISH (4). To a lesser extent, trisomy of chromosome 4 (5) and 13q14 and 17p13 deletions have been described, the last two being mainly associated with advance disease (6). On the other hand, no high-resolution whole-genome approaches have been used in the study of WM.
Nuclear factor kappa B (NF-kB) comprises a family of transcription factors that regulate the transcription of hundreds of genes involved in inflammation, innate immunity, cell growth and apoptosis (7). NF-kB transcription factors are homo and hetero-dimeric complexes formed by five members of the Rel family, NFKB1 (p50), NFKB2 (p52), RELA (p65), RelB and c-REL. Two signaling pathways are involved in the regulation of NF-kB complexes, canonical and non-canonical, which are respectively responsible for the activation of p50 and p52 from their inactive precursors p105 and p100 (8, 9). The activation of these pathways results in the translocation of p50/RELA and p52/RelB complexes into the nucleus and the subsequent transcriptional activation of target genes. Constitutive activation of the NF-kB pathways, either by inactivating mutations of negative regulators or upregulation of positive regulators has been linked to several tumor types (10, 11), but the molecular basis has remained largely unknown. Recently, we identified abnormalities affecting 11 regulator genes of the NF-kB pathways in at least 17% of multiple myeloma (MM) patients and 41.3% of human myeloma cell lines (HMCLs), resulting in the activation of NF-kB (12). TNF receptor-associated factor 3 (TRAF3), a negative regulator of the non-canonical NF-kB pathway, was the most commonly affected gene, with inactivating abnormalities identified in ~12% of patients (12). Additional inactivating abnormalities were identified in other negative NF-kB regulators (CYLD, cIAP1, cIAP2 and TRAF2) as well as gain-of-function mutations in positive regulators (MAP3K14/NIK, NFKB1, NFKB2, CD40, LTBR and TNFRSF13B/TACI) of NF-kB signaling, albeit less frequently (12, 13). Based on these observations and previous studies in other B-cell neoplasias (14–16), we hypothesized that abnormalities in genes that regulate the NF-kB pathways might also be present in WM.
The aim of this study was to perform a comprehensive, high-resolution, array-based comparative genomic hybridization (aCGH) analysis to identify genomic abnormalities present in WM, specially focusing on the status of NF-kB pathway key regulators. We identified biallelic deletions and loss-of-heterozygosity with inactivating mutations in two negative regulators of the NF-kB signaling pathways, TRAF3 and tumor necrosis factor, alpha-induced protein 3 (TNFAIP3/A20), highlighting the role of these pathways in WM pathogenesis.
Materials and Methods
Patients
Fifty-seven WM patients were included in this study. Clinical and laboratory features of the patients are shown in Supplementary Table S1. Bone marrow (BM) samples and lymph node biopsies were collected after informed consent was obtained in accordance with the Declaration of Helsinki. The Mayo Clinic Institutional Review and DFCI review boards approved the study. For FISH experiments from BM samples, cytospin slides were prepared from ACK lysis buffer treated BM aspirates to remove red blood cells. For aCGH and gene expression profiling (GEP), tumor cells were enrichment with anti-CD19+ immunomagnetic beads or by concomitant positive selection using anti-CD19+ and CD138+ (AutoMACS; Miltenyi-Biotec, Auburn, CA) and stored in TRIZOL reagent. RNA and DNA were extracted as recommended by the manufacture with subsequent clean-up using the RNAeasy Mini kit (Qiagen; Valencia, CA) and standard phenol-chloroform extraction methods, respectively.
aCGH
High-resolution aCGH was performed in 42 patients with the Human Genome 244A microarray (Agilent Technologies; Palo Alto, CA). Additionally, in order to reach the maximum available resolution on 6q arm, a custom array was used in eleven patients with 6q monoallelic deletions (Supplementary Table S2). Therefore, we designed a 4x44K custom array (Agilent Technologies), covering all the exons plus 700 bp of each adjacent intronic region with an average resolution of 200 bp from 6q15 to 6q telomere. The remaining intronic and intragenic regions were covered on an average resolution of 15 Kb. The digestion, labeling and hybridization steps were done according to the manufactures protocols, with some modifications. Briefly, 1.2 μg of tumor and reference DNAs were separately digested with Bovine DNaseI (Ambion; Austin, TX) for 12 minutes at room temperature. The normal human reference DNA comprised of a mixture of DNA derived from multiple female donors (Promega; Madison, WI). In patients with low DNA yields, a linear whole genome amplification procedure was incorporated before the DNA digestion step (GenomiPhi DNA Amplification Kit; GE Healthcare, United Kingdom). Next, random primers and exo-Klenow fragment were used to differentially label tumor (Cy5) and reference (Cy3) genomic DNA samples (Agilent Technologies). Labeled genomic reactions were cleaned-up with Microcon YM-30 columns (Millipore; Billerica, MA) and hybridized at 65° C for 40 hours. Microarrays were scanned in a DNA Microarray Scanner (Agilent Technologies). Feature extraction was performed with Feature extraction Software, version 9.5 (Agilent Technologies). Data was analyzed using CGH Analytics 3.5.1 software (Agilent Technologies).
The abnormalities were identified using two-probe filter and aberration detection module 1 (ADM-1) and ADM-2 algorithms (17) with thresholds of 7.5 and 5.5, respectively. Based on the study done in 50 healthy Caucasian males by de Smith et al. (18), we identified and eliminated copy number variation regions (CNVRs) existing in at least 5% of individuals. Using this high-resolution genome-wide data (average resolution of 500 bp) only 5% of the genome was eliminated of the analysis, compared with up to 30% when other public databases were used. An in house algorithm was developed to smooth the data (JJ Keats, unpublished data). Unsupervised clustering analyses were done with Genespring 7 software (Agilent Technologies).
cIgM-FISH
FISH DNA probes to validate numerical and structural abnormalities were selected for BAC and fosmid clones using the UCSC genome browser. The specificity of each probe at chromosome and gene level was confirmed by hybridization to normal metaphase preparations and by gene specific PCR, respectively. A list of probes used and chromosomal localization is provided in Supplementary Table S3. WM cells were identified with cytoplasmic anti-IgM staining concomitantly with the FISH technique (cIgM-FISH), as previously described (19).
GEP
RNA isolation, purification, and microarray hybridization was performed in 22 patients using Affymetrix U133A arrays (Affymetrix; Santa Clara, CA) as previously reported (20). The complete data set has been deposited in the NCBI Gene Expression Omnibus (GEO) and is accessible through GEO series accession number GSE12668. In eleven of them we also possess aCGH data (see Supplementary Table S2). Gene-expression intensities were MAS5 transformed and median normalized values were analyzed using GeneSpring 7.
A NF-kB index, a measure of NF-kB transcriptional activity, was calculated based on a gene expression index developed from HMCLs, as previously described (12). Briefly, we used an ANOVA test using the error estimate from a cross gene error model with multiple testing correction to identify probe sets differentially expressed in HMCLs with identified mutations compared to those without identified NF-kB pathway abnormalities (p < 0.05). The presence of mutations in the NF-kB pathways was clearly associated with a higher level of NF-kB transcription activity in the HMCLs. The mean expression level of four-probe set corresponding to CD74, IL2RG & TNFAIP3 (2) was used to calculate the index. The probes used were 209619_at (CD74), 204116_at (IL2RG), 202643_s_at and 202644_s_at (TNFAIP3).
DNA sequencing
Genome sequencing was performed on the PAX5 (N=16), PRDM1 (N=9), TNFAIP3 (N=24) and TRAF3 (N=24) coding exons and adjacent intron-exon junctions. All the coding regions were amplified using 10 ng of genomic DNA in 25 μl reactions. The specific primers used in this study are listed in Supplementary Table S4. Capillary electrophoresis was performed on an ABI3730 sequencer (Applied Biosystems; Foster City, CA). DNA sequences were analyzed using Sequencher V4.5.
Immunofluorescence
NFKB2 subcellular localization was determined by immunofluorescence (IF) staining. Cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton-100 and incubated with a 1:50 dilution of anti-NFKB2 (Cell Signaling Technology; Beverly, MA) and subsequently stained with 1:100 Alexa-Fluor 647 (Invitrogen; Carlsbad, CA). Cytoplasmic staining with anti-Ig kappa/lambda identified WM cells. Confocal imaging was performed on a Zeiss LSM510 microscope using a 63x objective.
Results
General overview of comparative genomic hybridization
We performed aCGH in 42 WM patients. The complete data set is accessible through GEO series accession number GSE12668. Overall, 35 of 42 (83.3%) cases showed chromosomal abnormalities, with a median of three abnormalities per patient (range 0 to 27). In total, 187 abnormalities were found, with 110 deletions and 77 gains. All the abnormalities identified are detailed in Supplementary Table S5. We identified 16 recurrent regions of copy number change found in >5% (identified in three or more patients); ten deleted and six amplified regions. Minimal deleted regions (MDRs) and minimal amplified regions (MARs) are detailed in Table 1.
Table 1.
Delineation of MDRs and MARs based on recurrent abnormalities identified in >5% of patients. In regions smaller than 20 Mb were listed the CIG (cancer implicated genes) expressed in at least 50% of WM patients. CIG include tumor suppressor genes and/or genes with inactivated mutations already described.
| Cytoband | Chromsome position (bp) | Loss (%) | Gain (%) | Size (Mb) | Expressed CIG |
|---|---|---|---|---|---|
| 3q13.3-q28 | chr3: 121238508-199379625 | 9.5 | 78.1 | ||
| 4q13.1-q35.2 | chr4: 65598026-191173837 | 11.9 | 125.6 | ||
| 6p12.2-p25 | chr6: 1-39874314 | 16.6 | 39.8 | ||
| 6q16.1 | chr6: 93266833-97766491 | 33.3 | 4.5 | MANEA | |
| 6q21-q22.1 | chr6: 105811723-107183767 | 38.1 | 1.4 | PRDM1, AIM1 | |
| 6q23 | chr6: 138093510-141518652 | 38.1 | 3.4 | TNFAIP3 | |
| 6q25.2-q25.3 | chr6: 155012960-159967461 | 33.3 | 4.9 | No CIG | |
| 7q22.1-q22.2 | chr7: 99534923-103321939 | 7.1 | 3.8 | STAG3, CUTL1 | |
| 8p | chr8: 1-43647063 | 7.1 | 43.6 | ||
| 8q | chr8: 76726872-136133311 | 9.5 | 59.4 | ||
| 11q22-q23.3 | chr11: 102986825-117384510 | 7.1 | 14.4 | ATM, DDX10, POU2AF1, SDHD, KIAA0999 | |
| 11q23.2-q24 | chr11: 121042811- 128566270 | 7.1 | 7.5 | FLI1 | |
| 13q14 | chr13: 49414571-50454033 | 9.5 | 1.0 | MIRN15a, MIRN16-1* | |
| 17p11.2-p13.3 | chr17: 1-17570789 | 7.1 | 17.5 | NXN, SKIP, KIAA0664, ITGAE, MINK1, ZNF232, USP6, CENTB1, TNK1, TP53, PER1, PIK3R5, DNAH9, MAP2K4, NCOR1, COPS3 | |
| 18 | chr18: 16793910-58926021 | 16.6 | 42.1 | ||
| Xq27.1-q28 | chrX: 149024400-154582473 | 9.5 | 5.6 | BGN, IRAK1, FLNA, F8, MTCP1, BRCC3 |
No CIG were found in 13q14, but microRNAs with proposed cancer implicated function were identified. MDRs: minimal deleted regions; MARs: minimal amplified regions.
A penetrance plot and a hierarchical clustering including all the chromosomal abnormalities are shown in Figures 1a and 1b, respectively. The most frequent abnormality was the entire or partial deletion of the 6q arm, identified in 17 of 42 patients (40.4%; Table 1, Figure 1a) and being the sole abnormality in three of 17 cases (Figure 1b). Other recurrent deletions identified by aCGH include 13q14 in four of 42 patients (10%; Table 1 and Supplementary Figure S1), and deletions on 7q22, 8p, 11q22-23, 11q23.2-24 and 17p11.2-p13.3 in three patients each (7.1%; Table 1).
Figure 1.

Overview of the copy number abnormalities identified in WM. Red blocks represent chromosome gains; blue blocks represent chromosome losses. A) Penetrance plot summarizing the copy number imbalances per chromosome. The amplitude of each abnormality corresponds to its prevalence. B) Hierarchical unsupervised clustering done from the genomic imbalances detected by aCGH per chromosome.
Partial or whole gains on chromosome 18 and 6p arm were the most common gains (7 of 42 cases each, 16.6%), followed by chromosome 4 (5/42, 11.9%), 3 (4/42, 9.5%), 8q (9.5%) and Xq27-q28 (9.5%)(Table 1). Whole or partial gains on chromosome 3 and 18 were concomitantly identified in three patients, none of them with 6q deletion. On the other hand, gains on chromosome 4 were identified irrespective of 6q deletion (Figure 1b).
Gain of 6p was always concomitant with 6q loss (concomitant 6q loss/6p gain was found in seven patients, but no 6p gain/6q normal cases were identified) (Figure 1b), thus suggesting that 6p gain would be a cytogenetic consequence of 6q loss. We analyzed five patients with 6p gain/6q loss and three different cytogenetic abnormalities were identified (Figure 2a–b). By using a three-color FISH strategy, we observed two of five patients with three aqua (CEP6), three red (CCND3 probe at 6p21) and three green (6p telomeric probe) signals, thus indicating the gain of an extra 6p arm (Figure 2a–b, left panels). Alternatively, two patients showed 2A/3R/3G signals, suggesting the presence of an isochromosome 6p (Figure 2a–b, center panels). The remaining patient has a partial 6p gain and our findings are compatible with the presence of an interstitial duplication (2A/2R/3G signals; Figure 2a–b, right panels). These results highlight that more than one cytogenetic mechanism is involved in the origin of these abnormalities. By using a similar approach, the presence of an i(8q) was confirmed in two cases (Supplementary Figure S2).
Figure 2.

Chromosomal imbalances on chromosome 6. In the graphical output, negative values (left) indicate losses and positive values (right) indicate gains of tumor DNA. A) The simultaneous presence of 6p gain/6q loss was commonly identified. These chromosomal imbalances could involve either a region or the whole chromosomal arm. B) FISH validations of the above described cases. At the left, a sample with a whole 6p arm gain including the centromere; at the center, a FISH pattern compatible with the presence of an isochromosome 6p; and at the right, a 6p interstitial gain.
Delineation of 6q MDRs and analysis of potential target genes
In patients with 6q deletion, a single MDR was not identified. Instead, four MDRs (MDR-1 to MDR-4) were delineated and they were present in at least 14 of 17 patients each (Table 1, Figure 3a). The most common were MDR-2 (1.4 Mb) and MDR-3 (3.4 Mb), being present in 16 patients each (Figure 3a). These regions include AIM1, PRDM1 (MDR-2) and TNFAIP3 (MDR-3), three candidate tumor suppressor genes expressed in at least 50% of WM patients (Table 1). A special interest was put on PRDM1 and TNFAIP3, which have been previously reported as being inactivated in other B cell neoplasias (21–23).
Figure 3.

TNFAIP3 abnormalities. A) Delineation of four minimal deleted regions (MDRs) on 6q based on aCGH data (dashed lines). PRDM1 and TNFAIP3 were localized in MDR2 and MDR3, respectively. B) Partial DNA sequences from a normal sample (top) and one with TNFAIP3 frame-shift deletion (red arrow; T155fsX215). The absence of the wild type allele indicates the homozygous status of the mutation. The position of TNFAIP3 mutation at protein level is based on NP_006281.1, which represent the accepted full length TNFAIP3 polypeptide. C) The TNFAIP3 transcript expression level was significantly lower in patients with one copy of the gene (1N) when compared with the group with two copies of TNFAIP3 (2N).
Given the historic interest on 6q deletions in WM we also used a custom aCGH microarray to reach the maximum available resolution in the region between 6q15 and 6q telomere. Even analyzing 6q with the custom array on eleven patients (see Supplementary Table S2) we did not identify any biallelic deletion involving PRDM1, TNFAIP3 or other tumor suppressor genes.
Next, we sequenced the entire coding region of PRDM1 on nine patients with monoallelic deletion and no inactivating mutations were found. On the other hand, we detected a frame-shift deletion (T155fsX215) in one of 24 patients where TNFAIP3 was sequenced (Figure 3b). DNA obtained from whole genome amplification procedure was used in this sample, but the mutation was confirmed by independent PCR and DNA sequencing reactions done from genomic non-amplified DNA from the same patient. The mutation was found in one case with two copies of TNFAIP3, but the exclusive identification of the mutated allele by DNA sequencing suggests the existence of some uniparental disomy (UPD) mechanism involved in the loss of the wild-type allele (Figure 3b). The truncated protein lacks the 575 C-terminal amino acids, including the de-ubiquitinating region and multiple zinc-finger regions. In the remaining patients with monoallelic TNFAIP3 deletions, no mutations affecting the other allele were identified. However, by comparing the median normalized GEP data based on the TNFAIP3 status, a significantly lower expression level was observed in the group of patients with TNFAIP3 monoallelic deletion than patients with two copies of the gene (P = 0.0005; Figure 3c).
Recurrent interstitial deletions on 13q14 containing MIRN15A and MIRN16-1
Biallelic deletions were rare events in this cohort, with only two abnormalities being identified. One of the biallelic deletions comprises a 1.65 Mb region on 13q14. Small monoallelic deletions were also found on the same chromosome region in other three patients, thus delineating a 1.1 Mb MDR present in four of 42 cases (Table 1; Supplementary Figure S1). The MDR was similar to the described in chronic lymphocytic leukemia (CLL) and also includes the microRNA genes MIRN15A and MIRN16-1, which were already described as playing a key role in CLL pathogenesis (24). Besides the microRNAs, DLEU7 was localized inside the MDR.
TRAF3 inactivation is a recurrent event in WM and its inactivation is associated with constitutive activation of the non-canonical NF-kB pathway
The remaining biallelic deletion was identified on 14q32.32. By aCGH we found one biallelic deletion including TRAF3, AMN, and CDC42BPB in one patient (MC1353; Figure 4a). Additionally, a focal monoallelic deletion was identified in a second patient in the same region (MC1341; Figure 4a). These findings were validated by cIgM-FISH, showing the presence of the abnormality in more than 90% of IgM positive cells in both cases (Figure 4b). In the sample with the monoallelic deletion we identified a missense mutation (D483N) in the remaining allele (Figure 4c). This substitution affects an amino acid highly conserved throughout vertebrate evolution, thus suggesting its functional relevance. Next, in order to determine the frequency of TRAF3 abnormalities in our entire WM cohort, we screened by cIgM-FISH 15 additional patients without aCGH data. We found a TRAF3 monoallelic deletion in one additional patient (Figure 4b), and, again, the abnormality was found in the majority of the IgM positive cells (88%). Moreover, we identified a nonsense mutation (K286X) on the remaining allele (Figure 4c). This truncated form of TRAF3 lacks the MATH domain, which is critical for the interaction of TRAF3 with NIK and TNF receptors (25, 26). In summary, three of 57 (5.3%) patients studied have TRAF3 biallelic deletions or inactivating mutations and UPD. In one of the three patients, the TRAF3 abnormalities were found at diagnosis, suggesting that its presence is not a consequence of treatment.
Figure 4.

TRAF3 abnormalities. A) Chromosome (left panel) and gene view (right panel) showing mono and biallelic deletions on 14q32.32, including TRAF3 (highlight in red, inside the dashed box). B) cIgM-FISH validations. A TRAF3 monoallelic deletion was identified in a third patient (MC1383) with no aCGH data. The NFKBIA probe was used as a surrogate CEP14 probe. C) Partial DNA sequences of both cases with TRAF3 monoallelic deletion (MC1341 and MC1383), confirming the presence of a mutation in the remaining allele (c.1800G>A and c.1209A>T, leading to D483N and K286X substitutions, respectively). The positions of TRAF3 mutations at the cDNA and protein level are based on NM_145725.1 and NP_663777.1, which represent the accepted full length TRAF3 transcript and polypeptide, respectively.
We previously showed in MM that TRAF3 inactivation is correlated with an increased NF-kB transcriptional signature, measured as a NF-kB index (12). When analyzed our WM cohort, four of 22 WM patients clearly split as an outlier subgroup with the highest NF-kB indexes (Figure 5a). Two of these four patients do not produce active TRAF3 (MC1341 and MC1353), consistent with the association between TRAF3 inactivation and increased NF-kB transcriptional activity. Additionally, other of the remaining patients from the outlier group with the highest NF-kB indexes expressed TRAF3 at the second lowest level (normalized expression value of 0.62 versus median normalized expression value of 1.41 in the entire cohort; see Supplementary Table S6), suggesting its potential inactivation. However, no DNA or RNA samples were available and neither aCGH nor sequencing could be performed to confirm the TRAF3 status in this patient. Finally, to confirm that TRAF3 inactivation was associated with activation of the non-canonical NF-kB pathway, we examined the NFKB2 sub-cellular localization. As predicted, nuclei of patients with TRAF3 inactivation contained NFKB2 while NFKB2 was cytoplasmic in patients without inactivation (Figure 5b).
Figure 5.
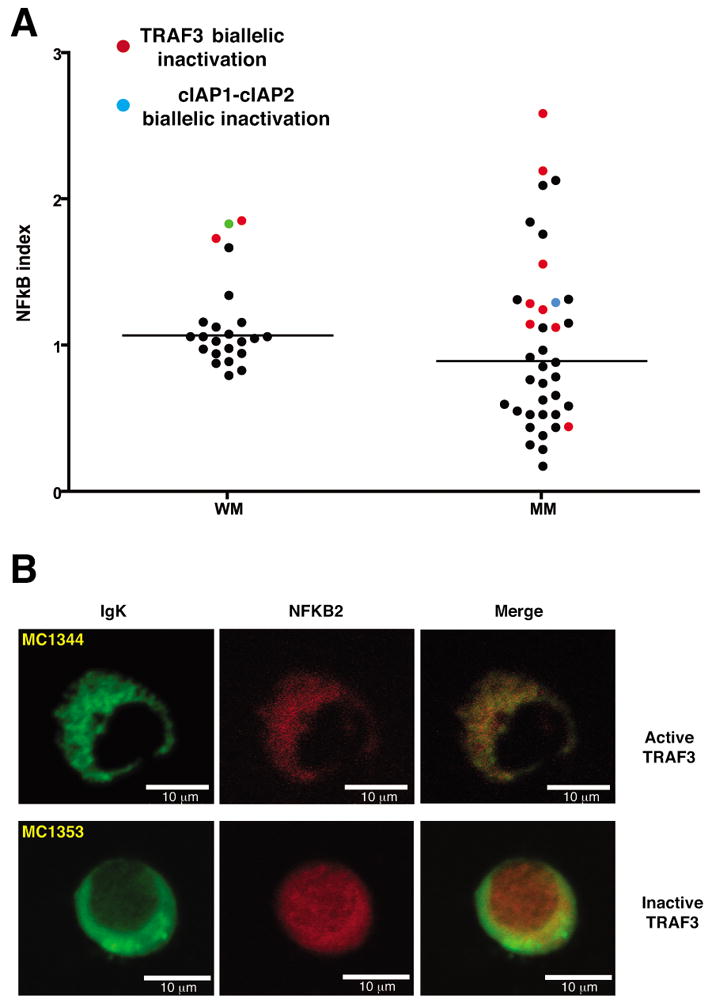
TRAF3 inactivation is associated with activation of the non-canonical NF-kB pathway. A) NF-kB index in WM. Our MM cohort is included for comparison. The patients with TRAF3 and cIAP1-cIAP2 inactivation are highlighted in red and blue, respectively. An association between patients with TRAF3 inactivation and high NF-kB index is clearly observed in WM. The case with the second highest index value showed a very low TRAF3 expression (green dot; see Supplementary Table S6), suggesting its inactivation. However, no DNA or cDNA was available for performing aCGH or DNA sequencing. The bars correspond to the median values. B) Immunofluorescence staining confirmed the association between TRAF3 inactivation and nuclear localization of NFKB2, indicating p100 to p52 processing (patient MC1353). A patient with close-to-median NF-kB index and active TRAF3 was used as a negative control (MC1344).
Abnormalities in other NF-kB regulators
Next, in an attempt to identify additional genetic abnormalities affecting NF-kB pathways, we screened the GEP and aCGH data for spiked expression and abnormalities of other regulator genes. By GEP, we did not identify differential expression of any of the main target genes involved in the regulation of NF-kB pathways. By aCGH, we identified large monoallelic deletions comprising cIAP1/cIAP2 (~35 Mb) and CYLD (~15 Mb) in one patient sample each (Supplementary Table S7). These two genes are negative regulators of the NF-kB signaling and their inactivation was previously shown in MM patients and HMCLs (12). Next, we screened our entire cohort of 57 patients by cIgM-FISH, but no additional imbalances in cIAP1/cIAP2 or CYLD were identified. Finally, all patients were screened for NIK translocations/amplifications, but no abnormalities were identified. A summary of the observed abnormalities affecting genes involved in the NF-kB signaling pathways is shown in Supplementary Table S7.
Finally, we found a monoallelic deletion affecting exons 1 to 4 of PAX5, which was confirmed by PCR and sequencing across the breakpoint (data not showed). This gene encodes a B-cell lineage specific activator protein involved at early stages of B-cell differentiation (27). PAX5 is involved in t(9;14)(p13;q32) translocations recurring in small lymphocytic lymphomas of the plasmacytoid subtype, and in derived large-cell lymphomas (28). We analyzed our entire cohort by cIgM-FISH and aCGH, but no additional imbalances affecting this gene were identified. We sequenced the entire coding and promoter region of PAX5 in 16 patients but no mutations were identified.
Discussion
Although Waldenström’s macroglobulinemia is considered a distinct clinicopathological entity, the absence of morphological, immunophenotypic or chromosomal disease-specific markers makes difficult its differential diagnosis from other B-cell neoplasias, as MM, marginal zone lymphomas (MZL) and CLL. We present here the first high-resolution genomic study in WM aiming to elucidate the genetic basis of the disease.
Overall, the WM karyotype is stable, even using this high-resolution approach, with few recurrent abnormalities identified. Considering the amount and type of recurrent chromosomal abnormalities observed is clear that WM placed closer to MZL and CLL than MM, as was previously suggested by using gene expression signatures (20). Thus, WM shares with MZL the simultaneous presence of trisomies 3 and 18 (29). Furthermore, we found 7q and 11q deletions in our WM cohort, comprising the same chromosome regions previously identified in MZL and B-CLL (30, 31).
Chromosome 13 deletions are recurrent events in CLL and MM besides WM, but different chromosomal regions are affected by the deletion, ranging from monosomies in MM to small 13q14.3 interstitial deletion in CLL patients (32, 33). WM patients show the same type of interstitial deletion pattern found in CLL patients, including MIRN15A and MIRN16-1 in the MDR (33). It was proposed that these MIRs have tumor suppressor function since their expression is inversely correlated to the expression of the anti-apoptotic BCL2 protein in CLL. Both MIRs negatively regulate BCL2 at a posttranscriptional level and BCL2 repression induces apoptosis in a leukemic cell line model (34). These findings together with a recent report in SMZL (35), lead to the assumption that this abnormality is not restrict to B-CLL and its dysregulation is common to several indolent B-cell malignancies.
On the other hand, other recurrent chromosomal abnormalities, as whole or partial gains of chromosome 4 and 8q arm seem to be specific of WM, helping to distinguish it from other indolent B-cell malignancies. Gain of 6p arm was always concomitant with 6q deletion, thus suggesting its origin as being secondary to 6q deletion. A similar mechanism seems to be operating in chromosome 8, as simultaneous 8p losses and 8q gains occur in a recurrent fashion, thus highlighting the presence of abnormalities involving both chromosomal arms as being common events in WM.
At gene level, remarkable findings affecting genes involved in the regulation of the NF-kB signaling pathways were identified. In fact, we found biallelic deletions or inactivating mutations and UPD affecting two negative regulators of NF-kB pathways, TRAF3 and TNFAIP3.
TRAF3 encodes a putative ubiquitin-ligase that plays a key role as a negative regulator of the non-canonical NF-kB pathway (36). Loss of TRAF3 results in the accumulation of the serine/threonine-protein kinase NIK leading to the constitutive activation of the non-canonical NF-kB pathway (37). Recent studies have characterized TRAF3 as a tumor suppressor gene (12, 38). In TRAF3−/− HMCLs, the reintroduction of TRAF3 was associated with an inhibition of NFKB2 processing (12). Furthermore, the conditional knock out of TRAF3 in B-cells is associated with extended B-cell survival and B-cell hyperplasia (38). Here, we identified biallelic TRAF3 inactivation in at least 5.3% (3/57) WM samples. Our findings suggest that the constitutive activation of the non-canonical pathway, as a consequence of TRAF3 inactivation, might be a recurrent event in WM pathogenesis. Considering that we analyzed only ~70% of patients by aCGH and sequencing, we assume that the prevalence of TRAF3 abnormalities might be even higher. These findings and our previous work showing TRAF3 as being the tumor suppressor gene more commonly inactivated in MM (12) confirm that TRAF3 inactivation is not specific of MM, being a common event in B-cell malignancies.
In addition, an inactivating mutation was found in TNFAIP3. TNFAIP3 encodes A20, a TNF/CD40/Toll like receptor-inducible zinc finger protein that acts as a negative regulator of the NF-kB signaling pathway by negatively modulating the function of upstream IKK-activating signaling proteins such as TRAF6, RIP, and, possibly, IKKγ (39, 40). Its inactivation leads to constitutive activation of the inhibitor of IkB kinase (IKK). TNFAIP3 knock out mice develop severe inflammation and cachexia, failing to terminate TNF-induced NF-kB responses (41). Recently, TNFAIP3 polymorphisms have been associated with autoimmune diseases as rheumatoid arthritis and systemic lupus erythematosus (42–44). In addition, TNFAIP3 biallelic deletions have been identified in diffuse large B-cell lymphomas and ocular adnexal marginal B cell lymphomas, thus supporting that TNFAIP3 inactivation might be relevant in the pathogenesis of B-cell tumors (22, 23).
Here, we identified a homozygous inactivating mutation at TNFAIP3 in one of 24 patients. Additionally, monoallelic deletions including TNFAIP3 were identified in 38% of patients. In this sub-set of patients, no mutations were identified in the remaining allele, but the TNFAIP3 transcript expression levels were significantly lower than in the group of patients with two copies of the gene. These findings are suggestive of TNFAIP3 haploinsufficiency in WM. Indeed, a recent report using TNFAIP3 +/− knock out mice have suggested that TNFAIP3 haploinsufficiency is associated with increased expression of NF-kB target genes (45). However, additional studies are needed to prove the effect of TNFAIP3 monoallelic deletions in the activation of the NF-kB target genes in WM patients.
To date, few recurrent cytogenetics abnormalities have been reported in WM and the molecular consequences of these aberrations are largely unknown (4, 5). To our knowledge, this is the first study showing the inactivation of tumor suppressor genes in WM and identifying a potential target gene on the 6q arm. Overall, we showed TRAF3 and TNFAIP3 inactivation in 5.3% and 4.2%, respectively. In addition, 38% of patients have monoallelic TNFAIP3 deletions. Moreover, these genes are negative regulators of NF-kB signaling pathways, highlighting the importance of these pathways in sustaining growth in WM. Additional mutational screening of other regulators is ongoing to determine the extent of constitutive NF-kB activation in this malignancy.
Finally, our findings might have therapeutic implications. Bortezomib, a first in class proteasome inhibitor, has demonstrated anti-tumor activity in refractory and relapsed MM (46) and more recently in untreated and relapsed WM (47–49). The inhibition of the NF-kB pathways is thought to be a principal mechanism of action of bortezomib. The identification of mutations affecting regulators of the NF-kB pathways highlights their roles in WM pathogenesis and identifies a subset of patients who might be benefit from proteasome-inhibitors based treatments.
Supplementary Material
Acknowledgments
This work was funded by the International Waldenstrom's Macroglobulinemia Foundation. We thank Chris Gooden, Michael Bittner, Wee Joo Chng, Michael Sebag and Sandra Montgomery for helpful assistance. E.B. is supported by IWMF 2S grant from the IWMF. J.J.K. is supported by a MMRF Fellowship and the Gene and Mary-Lou Kurtz fellowship in myeloma. P.L.B. is supported by R01-AG020686 and SPORE P50-CA100707-01. R.F. is supported by R01-CA83724-01, SPORE P50-CA100707-01 and P01-CA62242 from the National Cancer Institute, and the Donaldson Charitable Trust Fund. R.F. is a Clinical Investigator of the Damon Runyon Cancer Research Fund.
Financial Support: E.B. is supported by IWMF 2S grant from the International Waldenström’ s Macroglobulinemia Foundation. J.J.K. is supported by a MMRF Fellowship and the Gene and Mary-Lou Kurtz fellowship in myeloma. P.L.B. is supported by R01-AG020686 and SPORE P50-CA100707-01. R.F. is supported by CA 972 74, R01-CA83724-01, SPORE P50-CA100707-01 and P01-CA62242 from the National Cancer Institute, and the Donaldson Charitable Trust Fund. R.F. is a Clinical Investigator of the Damon Runyon Cancer Research Fund.
Footnotes
Institution where work was done: Mayo Clinic Scottsdale
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Authorship contributors E.B., J.J.K., M.C., P.L.B., R.F. design the study; E.B., J.J.K., S.V.W., R.V., R.F.J.C., M.B. performed the research; X.L., V.H.J.Z., T.P.T., K.H. collected data; P.G., M.G., S.H., S.V.R., A.K.S., A.D., I.G. contributed with patient samples; E.B., R.F. wrote the paper; all authors reviewed and gave final approval of the manuscript.
References
- 1.Fonseca R, Hayman S. Waldenström macroglobulinaemia. Br J Haematol. 2007;138:700–20. doi: 10.1111/j.1365-2141.2007.06724.x. [DOI] [PubMed] [Google Scholar]
- 2.McMaster ML. Familial Waldenström’s macroglobulinemia. Semin Oncol. 2003;30:146–52. doi: 10.1053/sonc.2003.50063. [DOI] [PubMed] [Google Scholar]
- 3.McMaster ML, Goldin LR, Bai Y, et al. Genomewide linkage screen for Waldenström’s macroglobulinemia susceptibility loci in high-risk families. Am J Hum Genet. 2006;79:695–701. doi: 10.1086/507687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schop RF, Kuehl M, Van Wier SA, et al. Waldenström macroglobulinemia neoplastic cells lack immunoglobulin heavy chain locus translocations but have frequent 6q deletions. Blood. 2002;100:2996–3001. doi: 10.1182/blood.V100.8.2996. [DOI] [PubMed] [Google Scholar]
- 5.Terre C, Nguyen-Khac F, Barin C, et al. Trisomy 4, a new chromosomal abnormality in Waldenström’s macroglobulinemia: a study of 39 cases. Leukemia. 2006;20:1634–6. doi: 10.1038/sj.leu.2404314. [DOI] [PubMed] [Google Scholar]
- 6.Schop RF, Jalal SM, Van Wier SA, et al. Deletions of 17p13.1 and 13q14 are uncommon in Waldenström macroglobulinemia clonal cells and mostly seen at the time of disease progression. Cancer Genet Cytogenet. 2002;132:55–60. doi: 10.1016/s0165-4608(01)00526-x. [DOI] [PubMed] [Google Scholar]
- 7.Hayden MS, Gosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh S, Karin M. Missing Pieces in the NF-κB Puzzle. Cell. 2002;109:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 9.Senftleben U, Cao Y, Xiao G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–9. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 10.Basseres DS, Baldwin A. Nuclear factor-kB and inhibitor of kB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–30. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 11.Courtois G, Gilmore T. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–43. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 12.Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–44. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Annunziata CM, Davies R, Demchenko Y, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105:13520–5. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lucas PC, Yonezumi M, Inohara N, et al. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J Biol Chem. 2001;276:19012–9. doi: 10.1074/jbc.M009984200. [DOI] [PubMed] [Google Scholar]
- 16.Morgan JA, Yin Y, Borowsky AD, et al. Breakpoints of the t(11;18)(q21;q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res. 1999;59:6205–13. [PubMed] [Google Scholar]
- 17.Lipson D, Aumann Y, Ben-Dor A, Linial N, Yakhini Z. Efficient calculation of interval scores for DNA copy number data analysis. J Comput Biol. 2006;13:215–28. doi: 10.1089/cmb.2006.13.215. [DOI] [PubMed] [Google Scholar]
- 18.de Smith AJ, Tsalenko A, Sampas N, et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet. 2007;16:2783–94. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- 19.Ahmann GJ, Jalal SM, Juneau AL, et al. A novel three-color, clone-specific fluorescence in situ hybridization procedure for monoclonal gammopathies. Cancer Genet Cytogenet. 1998;101:7–11. doi: 10.1016/s0165-4608(97)00058-7. [DOI] [PubMed] [Google Scholar]
- 20.Chng WJ, Schop R, Price-Troska T, et al. Gene-expression profiling of Waldenstrom macroglobulinemia reveals a phenotype more similar to chronic lymphocytic leukemia than multiple myeloma. Blood. 2006;108:2755–63. doi: 10.1182/blood-2006-02-005488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasqualucci L, Compagno M, Houldsworth J, et al. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med. 2006;203:311–7. doi: 10.1084/jem.20052204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honma K, Tsuzuki S, Nakagawa M, et al. TNFAIP3 is the target gene of chromosome band 6q23.3-q24.1 loss in ocular adnexal marginal zone B cell lymphoma. Genes Chromosomes Cancer. 2008;47:1–7. doi: 10.1002/gcc.20499. [DOI] [PubMed] [Google Scholar]
- 23.Thelander EF, Ichimura K, Corcoran M, et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia. Leuk Lymphoma. 2008;49:477–87. doi: 10.1080/10428190701817282. [DOI] [PubMed] [Google Scholar]
- 24.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–50. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 26.Ni CZ, Oganesyan G, Welsh K, et al. Key molecular contacts promote recognition of the BAFF receptor by TNF receptor-associated factor 3: implications for intracellular signaling regulation. J Immunol. 2004;173:7394–400. doi: 10.4049/jimmunol.173.12.7394. [DOI] [PubMed] [Google Scholar]
- 27.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–62. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 28.Iida S, Rao PH, Nallasivam P, et al. The t(9;14)(p13;q32) chromosomal translocation associated with lymphoplasmacytoid lymphoma involves the PAX-5 gene. Blood. 1996;88:4110–7. [PubMed] [Google Scholar]
- 29.Dierlamm J, Pittaluga S, Wlodarska I, et al. Marginal zone B-cell lymphomas of different sites share similar cytogenetic and morphologic features. Blood. 1996;87:299–307. [PubMed] [Google Scholar]
- 30.Karhu R, Knuutila S, Kallioniemi OP, et al. Frequent loss of the 11q14-24 region in chronic lymphocytic leukemia: a study by comparative genomic hybridization. Tampere CLL Group Genes Chromosomes. Cancer. 1997;19:286–90. doi: 10.1002/(sici)1098-2264(199708)19:4<286::aid-gcc12>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 31.Sole F, Woessner S, Perez-Losada A, et al. Cytogenetic studies in seventy-six cases of B-chronic lymphoproliferative disorders. Cancer Genet Cytogenet. 1997;93:160–6. doi: 10.1016/s0165-4608(96)00191-4. [DOI] [PubMed] [Google Scholar]
- 32.Fonseca R, Oken MM, Harrington D, et al. Deletions of chromosome 13 in multiple myeloma identified by interphase FISH usually denote large deletions of the q arm or monosomy. Leukemia. 2001;15:981–6. doi: 10.1038/sj.leu.2402125. [DOI] [PubMed] [Google Scholar]
- 33.Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–71. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferreira BI, García JF, Suela J, et al. Comparative genome profiling across subtypes of low-grade B-cell lymphoma identifies type-specific and common aberrations that target genes with a role in B-cell neoplasia. Haematologica. 2008;93:670–9. doi: 10.3324/haematol.12221. [DOI] [PubMed] [Google Scholar]
- 36.Hauer J, Püschner S, Ramakrishnan P, et al. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc Natl Acad Sci U S A. 2005;102:2874–9. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He JQ, Zarnegar B, Oganesyan G, et al. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med. 2006;203:2413–8. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie P, Stunz L, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253–67. doi: 10.1016/j.immuni.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–60. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 40.Wang YY, Li L, Han KJ, Zhai Z, Shu HB. A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-kappaB and ISRE and IFN-beta promoter. FEBS Lett. 2004;576:86–90. doi: 10.1016/j.febslet.2004.08.071. [DOI] [PubMed] [Google Scholar]
- 41.Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008 Aug 1; doi: 10.1038/ng.200. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008 Aug 1; doi: 10.1038/ng.202. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plenge RM, Cotsapas C, Davies L, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–82. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-kappaB target genes. Proc Natl Acad Sci U S A. 2007;104:18601–6. doi: 10.1073/pnas.0709011104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 47.Chen CI, Kouroukis C, White D, et al. Bortezomib is active in patients with untreated or relapsed Waldenström’s macroglobulinemia: a phase II study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1570–5. doi: 10.1200/JCO.2006.07.8659. [DOI] [PubMed] [Google Scholar]
- 48.Leleu X, Eeckhoute J, Jia X, et al. Targeting NF-kappaB in Waldenström macroglobulinemia. Blood. 2008;111:5068–77. doi: 10.1182/blood-2007-09-115170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Treon SP, Hunter Z, Matous J, et al. Multicenter clinical trial of bortezomib in relapsed/refractory Waldenstrom’s macroglobulinemia: results of WMCTG Trial 03–248. Clin Cancer Res. 2007;13:3320–5. doi: 10.1158/1078-0432.CCR-06-2511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.