

Abstract
In this study, we report a phospholipase C (PLC)-mediated mechanism for the redistribution of interendothelial adherens junctions in response to melanoma cell contacts with the endothelium. We demonstrated that contact of melanoma cells to human umbilical vein endothelial cells (HUVEC) triggered rapid endothelial [Ca2+]i response through PLC-IP3 pathway. In addition, alternation of endothelial adherens junctions following contact of melanoma cells was evidenced by the changes in immunological staining patterns of vascular endothelial (VE)-cadherin. A PLC inhibitor, U73122 was shown to significantly diminish [Ca2+]i response and reduce the occurrence of melanoma cell–induced VE-cadherin reorganization. Moreover, inhibition of PLC attenuated melanoma cell transendothelial migration. However, melanoma cell-associated VE-cadherin breakdown was not sensitive to Ly294002, an inhibitor of phosphatidylinositol-3-kinase (PI3K), whereas inhibition of PI3K resulted in a reduction of melanoma cell transmigration. Taken together, our findings implicate that by inducing the PLC-Ca2+ signaling pathway, melanoma cells disrupt EC junctions to breach the endothelium and promote transvascular homing of tumor cells.
Keywords: Endothelial adherens junction, intracellular calcium, signaling, tumor cell migration, vascular endothelial-cadherin
2. INTRODUCTION
Attachment of tumor cells to the endothelium (EC) is critical for cancer metastases. The stepwise interaction of tumor cells with EC includes tumor cell adhesion to the endothelial surface, tumor cell-mediated breaching of endothelial junction, and tumor cell penetration into the subendothelial space to form secondary tumors (1,2). A number of adhesion molecules including integrins and cell adhesion molecules are involved in stabilizing tumor cell arrest on the EC surface. Increased expression of beta1 as well as beta3 integrins has been shown in malignant melanoma cells compared to normal melanocytes, suggesting a potential involvement of cell adhesion molecules in the metastatic cascade (3,4). Endothelial receptors including vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) have been shown to promote cell adhesion on EC surface (5,6). However, how these initial cell adhesion events between tumor and endothelial cells signal downstream pathways that directly regulate the integrity of endothelial barrier require further elucidation.
Intracellular calcium ([Ca2+]i) may play an important role in initiating a sequence of signaling events in response to environmental cues, including cell adhesion. Exposure of EC to hydrogen peroxide triggers a transient rise in endothelial [Ca2+]i, which in turn, modulates the integrity of endothelium via a calcium-dependent mechanism (7). The binding of thrombin to its receptor causes a rise in [Ca2+]i and activates Ca2+-sensitive protein kinase C (PKC), resulting in cytoskeletal rearrangement (8). Furthermore, transmigrating leukocytes recruit [Ca2+]i-dependent signaling pathways in EC to induce junction disassembly, thereby permitting transvascular migration in response to inflammatory signals (9–12). Therefore, it is possible that malignant tumor cells utilize similar initial signaling events to compromise endothelial junction integrity, thus promoting a permissive microenvironment for tumor metastasis.
Interendothelial adherens junctions account for the majority of barrier function of normal endothelium, and are responsible for regulating the passage of proteins and circulating cells (13). Adherens junctions are characterized by the localization of vascular endothelial (VE)-cadherin, a 130-kDa transmembrane protein that contains an extracellular domain which binds to neighboring endothelial VE-cadherin. VE-cadherin cytoplasmic tail interacts directly with important structural, cytoskeletal and signaling proteins including alpha-actinin, alpha/beta-catenin, plakoglobin, and actin (14). VE-cadherin actively participates in most of the stages of transmigration of inflammatory cells. The loss of homophilic binding of VE-cadherins weakens the adhesive interaction between neighboring EC, thereby permitting transvascular cell penetration (15). Homophilic binding activity of VE-cadherin is, in part, regulated by tyrosine-phosphorylation of its cytoplasmic tail (16) suggesting a direct role for protein tyrosine kinases in regulating interendothelial junctions, and subsequent transvascular cell migration.
Tumor cell-initiated signaling events that lead to changes in EC integrity have not been extensively characterized. In this study, we monitored the [Ca2+]i in human umbilical vein endothelial cells (HUVEC) following contact with human melanoma cells. We showed that transient rise in endothelial [Ca2+]i was elicited specifically by melanoma cells, and this response recruited the classical [Ca2+]i release mechanism via pertussis toxin (PT)-sensitive G proteins in EC. In addition, we demonstrated that the loss of VE-cadherin integrity in EC appeared at the interendothelial border in response to melanoma cell contacts. The regulation of melanoma cell-mediated junctions was independent of tyrosine phosphorylation of VE-cadherin. Most importantly, melanoma cells induced junction disassembly in the manner strongly related to phospholipase C (PLC) activation, since inhibition of PLC significantly reduced the disruptions of VE-cadherin by melanoma cells. Phosphatidylinositol-3-kinase (PI3K), however, was not responsible for the melanoma cell-associated VE-cadherin redistribution since we did not observe any abrogation of junction breakdown. In addition, melanoma transendothelial migration was diminished in the absence of PLC or PI3K activity. These findings suggest an involvement of PLC in endothelial signaling pathways recruited by melanoma cells in breaching the vasculature.
3. MATERIALS AND METHODS
3.1. Cell culture and preparation
Human umbilical vein endothelial cells (HUVEC; American Type Culture Collection, Manassas, VA) were maintained in Ham’s F12-K media (Biofluids Inc., Gaithersberg, MD) supplemented with 10 % fetal bovine serum (FBS; Biofluids Inc.), 10 μg/ml endothelial cell growth supplement (Sigma Chemical Co., St. Louis, MO), 100 μg/ml heparin (Sigma Chemical Co.), 100 units/ml penicillin-streptomycin (Biofluids Inc.) at 37 °C under 5 % CO2, and used in passages 3~9. Cells were transferred and grown to a confluence on 25-mm round glass coverslips coated with 30 μg/ml fibronectin (Sigma Chemical Co.). A2058 human melanoma cells (passages 15~20) were obtained from NCI (NIH, Bethesda, MD) and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Biofluids Inc.) supplemented with 10 % FBS in a standard cell culture condition (5 % CO2/37 °C). Neonatal melanocytes (Cambrex-Clonetics, E. Rutherford, NJ) were maintained in manufacturer’s EGM-2-MV medium. Melanoma cells or melanocytes were detached by a brief trypsinization and suspended in culture media and rocked for 1 hr at 37 °C prior to an assay.
For the negative control of Ca2+ assay, tumor cell conditioned medium (TCM) was obtained from the supernatant of suspended tumor cells which had been gently rocked for 1 hr in assay buffer in a standard culture condition (5 % CO2/37 °C). Co-culture medium used for negative control of immunohistochemistry assay was prepared by incubating endothelial cells with melanoma in 1:3 concentration ratio for 4 hr.
3.2. Pharmacological inhibitors
In some experiments, HUVEC were treated with specific inhibitors including thapsigargin (TG; Molecular Probes, Inc., Eugene, OR), U73122 (Sigma Chemical Co.), pertussis toxin (PT; List Biological Laboratories, Inc., Campbell, CA), and Ly294002 (Calbiochem, San Diego, CA). Cells were incubated in culture media containing 1.0 μM TG for 10 min, or 2~10 μM U73122 for 5 min followed by a 30 min incubation in fresh media prior to a Ca2+ assay. For PT pretreatment, endothelial cells were exposed to 0.5 μg/ml PT in culture media for 2 hr at 37 °C prior to Fura-2-AM loading. Ly294002 was applied to cells at 20 μM for 2 hr.
3.3. Ca2+ ratiometric measurements
Prior to Ca2+ assays, HUVEC plates were incubated with fresh culture media for 30 min followed by incubation with 10 μM Fura-2-AM (Molecular Probes, Inc.) for an additional 30 min. Coverslips were then mounted to the assay chamber containing assay buffer (137 mM NaCl, 4.9 mM KCl, 1.2 mM MgSO4, 1.2 mM NaH2PO4, 20 mM HEPES, 15 mM D-glucose, 1.5 mM CaCl2, 0.1 % w/v fraction V bovine serum albumin, BSA [pH 7.4]) and allowed to equilibrate for 30 min prior to [Ca2+]i measurements. Melanoma cells were washed twice with assay buffer following the regeneration, and added to HUVEC monolayer at a concentration of 3 × 106 cells/chamber at the specified time point during the [Ca+2]i measurement.
The procedure for a digital Ca2+ ratiometric assay is detailed elsewhere (17). A computer software package (Axon Imaging Workbench 2.2; Axon Instruments, Inc., Foster City, CA) was used to control the excitation light (340 and 380nm band pass filters; Chroma Technology Co., Brattleboro, VT), sample and record the emitted fluorescence (510 nm) images from the Fura-2-AM loaded cells in the field of view once every 6 sec. The background fluorescence was subtracted from each image. Ratio images of the cells at rest were collected initially for the first 100 s to establish a [Ca2+]i baseline. Following the establishment of a baseline [Ca2+]i, melanoma cell solution was perfused into the chamber and raw images were collected for an additional 8~10 min.
A calibration curve was constructed by acquiring 340/380 values (background subtracted) of 10 μM Fura-2 pentapotassium salt solution using the Calcium Calibration Kit #1 (Molecular Probes, Inc.) and used in an off-line calibration of the ratio data. All experiments were carried out at room temperature.
As a negative control for the specificity of signals initiated by receptor-ligand interaction, 15 μm diameter polystyrene microspheres (Polyscience, Inc., Warrington, PA) at 3 × 106 beads/chamber were used in Ca2+ assays.
[Ca2+]i response was defined as the absolute difference between the peak [Ca2+]i over the baseline value. Baseline [Ca2+]i was determined by taking an average of the [Ca2+]i values of cells at rest, prior to addition of melanoma cells. This baseline [Ca2+]i typically varied within a standard deviation of 15 % of the mean value. Therefore, [Ca2+]i amplitudes consisting of over 20 % of the baseline levels were defined as positive responses over the background noise. The percentage of responding cells was determined by taking a ratio of cells with over 20 % peak [Ca2+]i to total cells assayed.
3.4. Cell fixation and immunohistochemistry
Cells on a glass cover slip were fixed using 5 % formaldehyde in PBS for 15 min at room temperature and washed 3 times in PBS. Fixed cells were treated for 20 min with 0.3 % Triton X-100, 5 % calf serum (CS), and 2.5 % goat serum (GS) in PBS to permeabilize the cell membrane. Permeabilized cells were exposed to 1 μg of primary antibody (anti-VE-cadherin; mouse monoclonal IgG1; Santa Cruz Biotechnology, Santa Cruz, CA) in PBS with 0.3 % Triton X-100/5 % CS/2.5 % GS per coverslip, and incubated overnight at 4 °C. Secondary antibody (AlexaFluor-488 conjugated goat anti-mouse IgG; Molecular Probes, Inc.) was applied to the coverslips at concentration of 1 μg/ml in PBS with 5 % CS/2.5 % GS, and incubated at 4 °C for 1 hr. The immunofluorescence stained plates were washed 3 times in PBS and observed using Olympus IX51 fluorescence microscope (Olympus Inc., Tokyo, Japan) at excitation/emission wavelengths of 488 nm/520 nm, respectively. Negative control consisting of secondary antibody alone was applied to cells followed by an incubation in PBS/0.3 % Triton X-100/5 % CS/2.5 % GS to determine any non-specific secondary antibody staining.
3.5. Immunoprecipitation and Western blots
Fully confluent monolayer of HUVEC were maintained in culture at 2.5 × 106 cells per 10 cm tissue culture plate. 1 ml of conditioned medium was withdrawn from the HUVEC culture plate immediately prior to assays, and used to resuspend melanoma cells at 1.0 × 107 cells/ml and added back to the confluent HUVEC plate. At designated times following the introduction of melanoma cells, HUVEC were scraped and resuspended into PBS at 4 °C. Whole cell extracts were prepared by resuspending 2 × 107 cells in 500 μl of lysis buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA [pH 8.0], 2 mM Na3VO3, 10 mM NaF, 10 mM Na4P2O7, 1 % NP-40, 1 mM PMSF, 2 ng/ml pepstatin A). Lysates were incubated on ice for 30 min followed by a centrifugation at 16,000 g for 5 min at 4 °C. Pellets were discarded and the supernatant was precleared with protein G-Agarose (Santa Cruz Biotechnology). Precleared lysates were incubated with anti-VE-cadherin preabsorbed with protein G-Agarose over night at 4 °C under a continuous mixing. Pellets were collected after 4 washes with lysis buffer and mixed with 2× SDS running buffer (0.2 % bromophenol blue, 4 % SDS, 100 mM Tris [pH 6.8], 200 mM DTT, 20 % glycerol) in 1:1 ratio. Samples were boiled for 3 min and 20 μl was loaded onto a 6 % SDS-PAGE gel and proteins were transferred to a 0.2 μm nitrocellulose filter (Schleicher and Schuell, Keene, NH) by electroblotting. Primary antibodies included anti-VE-cadherin and anti-phospho-tyrosine (mouse monoclonal IgG1; Cell Signaling Technology, Beverly, MA). Secondary antibody was peroxidase-conjugated goat anti-mouse IgG (Sigma Chemical Co.). Proteins were detected using the Enhanced Chemiluminescence Detection System (Amersham Pharmacia Biotech, Arlington Heights, IL). Vascular endothelial growth factor (VEGF; Sigma Chemical Co.) was used as an inducer for tyrosine phosphorylation of VE-cadherin.
3.6. Transendothelial migration
The transendothelial migration assay was carried out in a modified 48-well chemotactic Boyden chamber (18). In brief, 3 × 105 of HUVEC were seeded on fibronectin-coated polycarbonate filter with 8 μm pore size (Neuro Probe, Inc., Cabin John, MD) and incubated for 36~48 hr in culture medium. Prior to an assay, chemoattractant solution, type IV collagen (BD Bioscience, Bedford, MA), was added into the bottom wells of the chamber at a concentration of 100 μg/ml in assay medium (Ham’s F12-K and 0.1% BSA), and the filter with confluent HUVEC was placed over the filled wells. A top plate was screwed in and 2 × 104 of melanoma cells in suspension were pipetted into each well. The chamber assembly was subsequently incubated for 4 hr at 5% CO2/37 °C. For some experiments, monolayers were pretreated with 10 μM U73122 for 5 min or 20 μM Ly294002 for 2 hr, and then the drug-containing medium was replaced and the filter was washed with fresh assay medium 3 times prior to melanoma cell addition. At the end of the assay, the chamber was disassembled and the filter was stained with Protocol Brand Hema3 solution (Fisher Scientific, Pittsburgh, PA). Cells on the top of the filter were removed by scraping. Melanoma cell transendothelial migration was evaluated by counting the number of cells under 10 × magnification. 7 fields of a filter were quantified and averaged. At least 3 filters were analyzed. For negative control, HUVEC monolayer without melanoma cell addition was evaluated in the presence of type IV collagen, and migrated cells were counted after 4 hr. No significant EC migration was observed.
4. RESULTS
4.1. Melanoma cell contact induces endothelial [Ca2+]i
In order to determine whether melanoma cells induce changes in endothelial [Ca2+]i, [Ca2+]i in HUVEC following melanoma contact was examined. HUVEC responded to melanoma cell contact with a single [Ca2+]i peak that gradually returned to baseline over the duration of an assay (Figure 1). The population of HUVEC responding to melanoma contact was 80 ± 4 %, and melanoma cell contact induced an increase of 121 ± 22 % (HUVEC; n = 11) in peak [Ca2+]i magnitudes over resting baseline [Ca2+]i. Neither polystyrene beads nor melanocytes induced significant [Ca2+]i response in HUVEC to suggest a requirement for specific cell-cell interactions between melanoma and endothelial cells (Figure 1). Tumor cell conditioned medium (TCM; n = 3) obtained from the supernatant of suspended melanoma cells did not trigger [Ca2+]i response (data not shown) indicating that direct contact between melanoma and EC was required for a Ca2+ response. To further elucidate the role of heterotrimeric G- proteins in eliciting [Ca2+]i, HUVEC were cultured in a presence of PT prior to melanoma cell contact. Application of PT to endothelial cells resulted in a partial attenuation of calcium response, inducing 51 ± 19 % in [Ca2+]i magnitude and 59 ± 9 % in the population of cell responding (Figure 2). Therefore, melanoma binding to endothelium induces [Ca2+]i partially via G protein-coupled receptor.
Figure 1.
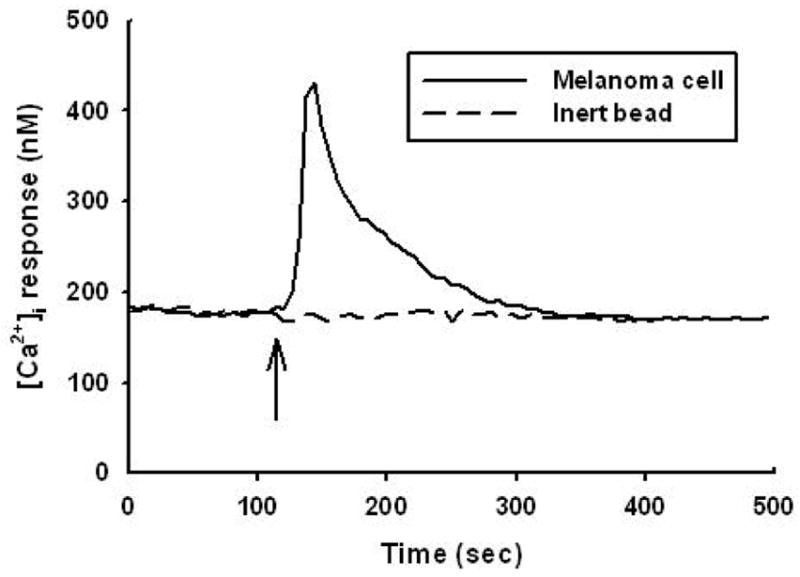
Endothelial cells release [Ca2+]i in response to contact with melanoma cells. Melanoma cells elicited a threefold increase of [Ca2+]i over baseline in HUVEC. In contrast, inert beads did not trigger significant [Ca2+]i response. Arrows indicate the time point at which tumor cells or inert beads were added. The result is representative of 11 independent experiments.
Figure 2.
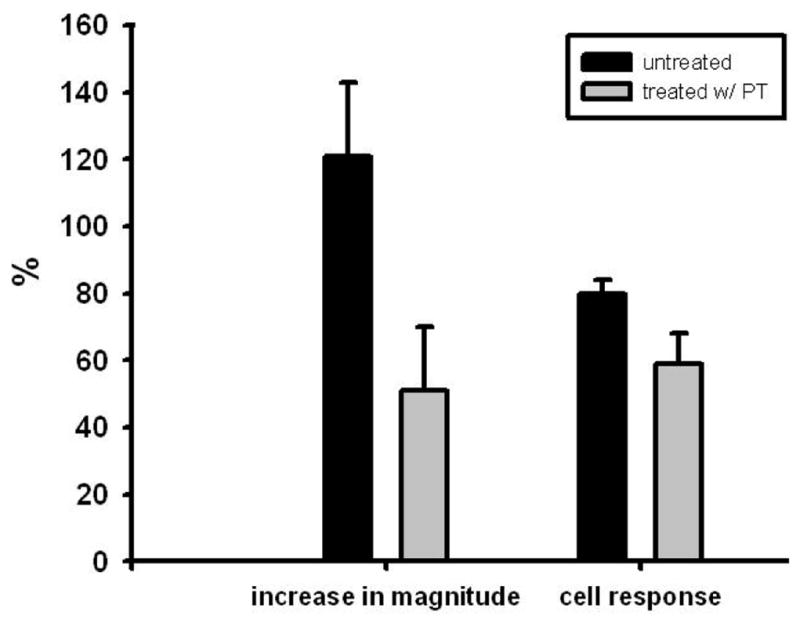
Pertussis toxin inhibits endothelial [Ca2+]i responses. HUVEC were preincubated with 0.5 μg/ml PT in culture medium for 2 hr. Melanoma cell contact induced an increase 121 ± 22 % (n = 11) in peak [Ca2+]i magnitude in untreated endothelial cells, whereas the magnitude reduced to 51 ± 19 % (n = 12) in PT-pretreated cells. The population of responding cells decreased to 59 ± 9 % (n = 12) compared with 80 ± 4 % (n = 11) in untreated endothelial cells. Results are presented with mean ± SEM.
4.2. Endothelial [Ca2+]i activation in response to melanoma cell contact occurs through PLC-IP3 pathway
We were interested in gaining insight into signaling events downstream of Ca2+, therefore we employed thapsigargin (TG), a plant-derived sesquiterpene-lactone, which is an irreversible inhibitor of endoplasmic reticulum (ER) Ca2+-ATPase to target intracellular Ca2+ stores. TG blocks Ca2+ uptake into inositol-1, 4, 5-triphosphate (IP3)-sensitive as well as insensitive intracellular Ca2+ stores (19). Due to the store-operated calcium release following TG application, the initial baseline [Ca2+]i was elevated from 237 ± 25 nM (control; n = 11) to 715 ± 10 nM (n = 3) prior to melanoma cell contact. Addition of melanoma cells did not elicit further endothelial [Ca2+]i activity over the elevated baseline (Figure 3). These results indicate that endothelial [Ca2+]i response to melanoma cell contacts originates from TG-sensitive intracellular stores, such as the ER.
Figure 3.

TG-treatment abolished melanoma cell-induced endothelial [Ca2+]i responses. HUVEC were pretreated with TG for 10 min followed by 30 min regeneration in fresh assay buffer prior to experiments. Elevated [Ca2+]i baseline is due to the store-operated calcium release following TG application. Contact with melanoma cells did not elicit additional [Ca2+]i activity. Arrow indicates the time point at which tumor cells were added to the assay chamber. Similar results were obtained in 3 independent experiments.
To further dissect intracellular mechanisms regulating the release of [Ca2+]i in EC following contact with melanoma cells, U73122, an amino-steroid inhibitor of PLC (20), was used. U73122 was applied to HUVEC at 2, 5, 7, and 10 μM for duration of 5 min, prior to melanoma cell contact. A dose-dependent attenuation in the percentage of HUVEC responding as well as the magnitude of peak [Ca2+]i response was observed with a complete abrogation of [Ca2+]i response occurring at 10 μM (Figure 4). Together with the observations from using TG, these data show melanoma binding to endothelium activates PLC/IP3 signaling and the release of Ca2+ from ER stores.
Figure 4.

PLC inhibitor U73122 inhibits endothelial [Ca2+]i responses following contact melanoma cells. HUVEC were pre-incubated with 2, 5, 7 and 10 μM U73122 for 5 min prior to an assay. A complete elimination of melanoma cell-induced [Ca2+]i responses was observed at the concentration of 10 μM. Results are presented with mean ± SEM; n = 3~5.
4.3. Melanoma cell contact induces adherens junction breakdown in EC
Tumor metastasis requires disruption of intercellular junctions, however whether the melanoma binding to EC can trigger these changes has not been extensively examined. We specifically addressed whether melanoma cells were capable of promoting the reorganization of VE-cadherin. HUVEC monolayers were fixed and stained with mAb against VE-cadherin after culturing in the absence or presence of A2058 human melanoma cells and distribution of adherens junctions were analyzed by fluorescence microscopy. Control EC monolayer, which did not contain melanoma cells, stained strongly at cell periphery for VE-cadherin (Figure 5A). Co-culturing melanoma and EC for 10 min did not lead to significant changes in the intercellular junctions (Figure 5B), whereas after 45 min of coculturing, junctions were disrupted at the site of melanoma contact (Figure 5C), suggesting that melanoma cells initiate reorganization of adherens junctions. It is unlikely that the gaps which were observed were due to shadows created by the melanoma cells since the disrupted junctions did not overlap with cells and coculturing melanoma cells and EC for 10 min did not reorganize adherens junctions. In addition, cell-cell contact was required for VE-cadherin reorganization since medium obtained from melanoma-EC cocultures for 4 hr was not sufficient to induce junction redistribution (data not shown). These observations, suggest that melanoma binding to EC initiates signaling events that target VE-cadherin and compromise the integrity of the endothelium.
Figure 5.

Melanoma cells induce junction opening and disassembly of VE-cadherin homophilic binding. Endothelial cells were fixed and stained for VE-cadherin using VE-cadherin mAb. Panel (A) shows 100× fluorescence images of continuous VE-cadherin staining patterns without melanoma cell contact. Contact of melanoma to EC for 10 min did not induce junction opening in panel (B). Contrastively, disassembled junctions caused by melanoma cell contacts at 45 min are shown in panel (C). Asterisks indicate the sites where gaps formed. The results are representative of 3 experiments. For negative control, secondary antibody alone did not contribute to any background signal of nonspecific binding (data not shown).
4.4. Phospholipase C but not PI3K mediates melanoma cell-dependent disruption of adherens junctions in EC
We demonstrated that endothelial [Ca2+]i was elicited through PLC/IP3 pathway in response to melanoma cell contact. Furthermore, melanoma cell contact with endothelial cells induced dissociation of VE-cadherin (Figure 6A). Therefore, we wished to determine whether the loss of VE-cadherin integrity required PLC/IP3 signaling. As shown in Figure 6B, PLC inhibitor U73122 used at 10 μM markedly reduced the dissociation of VE-cadherin that was induced by the addition of melanoma cells. Although the majority of junctions remained intact when PLC was inhibited, a modest number of gaps were apparent at the cell margins where melanoma cells contacted EC. This observation implies that melanoma cells induce, in addition to PLC, other signaling cascades that are required for penetrating the EC barrier. PI3K inhibitor Ly294002 did not reduce VE-cadherin dissociation initiated by melanoma cells (Figure 6C). Therefore, melanoma cells induce reorganization of VE-cadherin through PLC independent of PI3K.
Figure 6.

PLC activity participates in melanoma cell-mediated junction regulation. (A) VE-cadherin staining patterns (100×) in response to melanoma cell contacts at 45 min. (B) HUVEC were pretreated with 10 μM U73122 for 5 min prior to melanoma cell addition. A majority of junctions remained continuous at 45 min in the 100× fluorescence image while only a small population of endothelial cells responded to melanoma cell contacts by opening of junctions. (C) Pretreatment with 20 μM Ly294002 did not reduce VE-cadherin disassembly (100×) at 45 min. Asterisks indicate the sites where gaps formed. The results are representative of 3 experiments.
4.5. Melanoma contact does not alter tyrosine-phosphorylation of VE-cadherin in EC
Tyrosine phosphorylation of adherens junction components has been shown to influence junction reorganization (16). Therefore, we examined whether melanoma cells upon binding EC induce tyrosine phosphorylation of VE-cadherin. VE-cadherin was immunoprecipitated and resolved by SDS-PAGE electrophoresis, and then subsequently probed with anti-phosphotyrosine or anti-VE-cadherin antibodies. Consistent with previous reports (21) there was virtually no detectable phosphorylation of VE-cadherin from tightly confluent endothelial monolayer (Figure 7). When melanoma cells were added to a confluent HUVEC monolayer, no changes in phosphorylation of VE-cadherin were observed, even after 45 min of direct coculturing of melanoma and EC (Figure 7). Therefore, melanoma cells do not induce changes in VE-cadherin phosphorylation suggesting that VE-cadherin reorganization does not require tyrosine phosphorylation.
Figure 7.
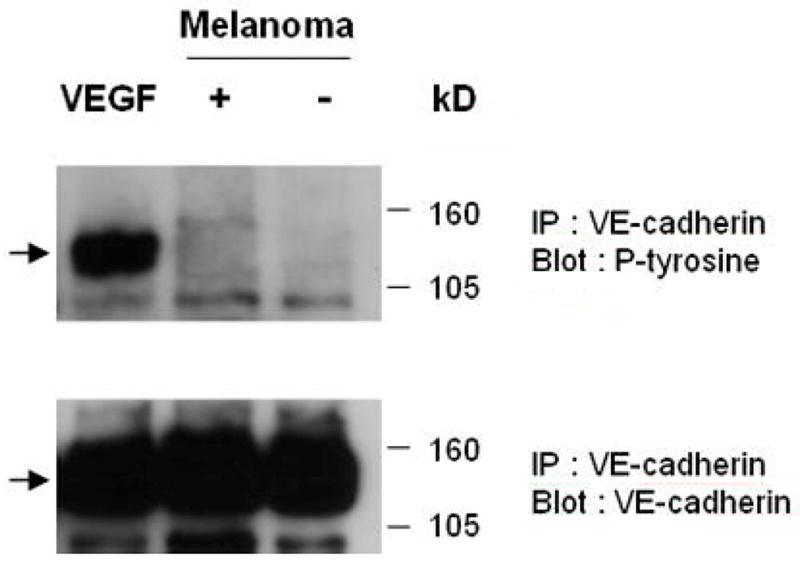
Tyrosine phosphorylation of VE-cadherin was not altered upon melanoma cell contact. HUVEC lysates were immunoprecipitated with VE-cadherin Ab after 45 min with and without melanoma cell contacts. Precipitated proteins were analyzed by immunoblotting with an Ab against phosphotyrosine. The same blot was stripped and then reprobed with Ab to VE-cadherin. VE-cadheirn was not found to be tyrosine phosphorylated in both cases (+ and −). VEGF, an inducer of VE-cadherin tyrosine phosphorylation, used at the concentration of 50 ng/ml altered the phosphotyrosine after 30 min. Results are representative of 7 experiments.
4.6. Activations of PLC and PI3K are necessary for efficient transendothelial migration of melanoma cells
The above experiment suggests that melanoma cells can induce biochemical changes in EC that correlate with changes in the VE-cadherin organization. However, we were interested in determining the functional significance of PLC and Ca2+ induction and whether these biochemical events could directly enhance the ability of melanoma cells to transverse the EC. Melanoma cells were added to the upper chamber of a Boyden migration assay system in which a confluent monolayer of HUVEC was seeded on the upper surface of the fibronectin-coated filter. In the lower chamber collagen IV, which is a chemoattractant for melanoma cells, was present. When added to HUVEC, melanoma cells started to migrate though the monolayer towards type IV collagen in the bottom wells. In the presence of the collagen, approximately 550–650 melanoma cells/mm2 migrated to the backside of the fibronectin-coated filter without endothelial monolayer after 4 hr. As shown in Figure 8, 280 ± 15 melanoma cells/mm2 transmigrated through untreated HUVEC, suggesting that EC were acting as a barrier to melanoma migration. To further assess the role of PLC or PI3K during melanoma cell transendothelial migration, HUVEC were pretreated with 10 μM U73122 or 20 μM Ly294002 prior to a transmigration assay. The migrated melanoma cell counts were normalized to the control in which melanoma cell migrated through untreated HUVEC. Pretreatment with U73122 or Ly294002 inhibited melanoma cell transmigration by 37 ± 3 % (176 ± 11 cells/mm2; p < 0.001) or 33 ± 6 % (187 ± 17 cells/mm2; p < 0.001), respectively. The observations indicate that PLC and PI3K signals are required for efficient melanoma cell transmigration across endothelium.
Figure 8.
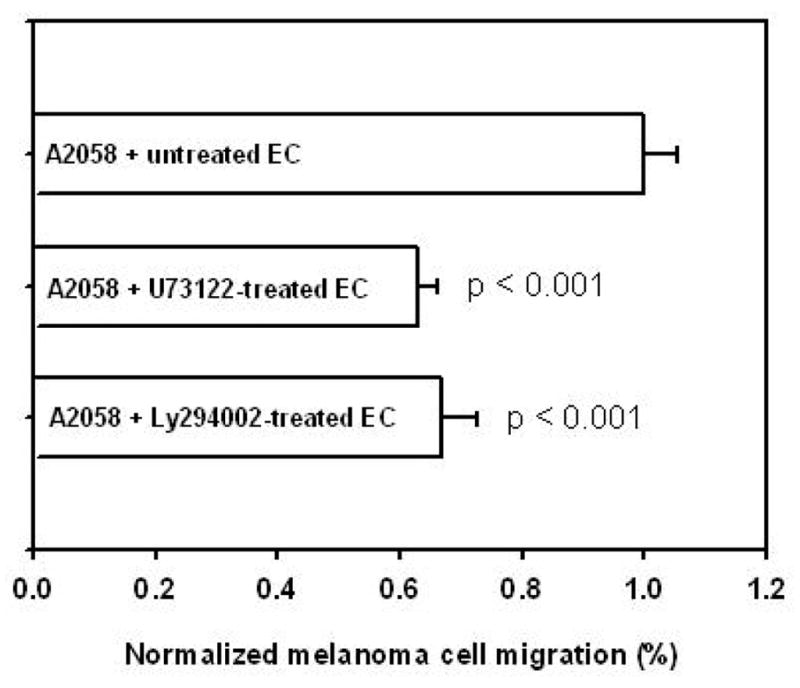
PLC and PI3K signals are required for melanoma cell transendothelial migration. Melanoma cell transendothelial migration was assessed using modified Boyden chamber. HUVEC were treated with 10 μM U73122 or 20 μM Ly294002 prior to melanoma cell addition on endothelium. Melanoma cells transmigrated through treated endothelial cells with type IV collagen in the bottom wells of the chamber during 4 hr assay. Values were calculated as percentages of control in which melanoma migrated through untreated endothelium (A2058 + untreated EC). Pretreatment with U73122 or Ly294002 inhibited transmigration by 37 ± 3 % (A2058 + U73122–treated EC; p < 0.001) or 33 ± 6 % (A2058 + Ly294002–treated EC; p < 0.001), respectively. Results are presented with mean ± SEM; n = 3~5.
5. DISCUSSION
In this study, we have demonstrated that melanoma cell contact with HUVEC recruited classic [Ca2+]i release in a PT-sensitive manner. Melanoma cell contact with the endothelium caused a reorganization of adherens junctions. In addition, we have shown that melanoma cell-induced dissociation of VE-cadherin junctions depends on phospholipase C activity. Activation of PLC was also required for efficient transendothelial migration of melanoma cells.
Tumor cell-induced endothelial [Ca2+]i activity and disassembly of VE-cadherin complex have been previously reported (22,23). However, the exact role of Ca2+ signaling in controlling the downstream junction regulation remained unclear. Melanoma cells resemble leukocytes in inducing endothelial [Ca2+]i,, which was specifically generated by cell-cell interaction and not by cell-cell contact associated mechanical forces. We were able to target PLC activation as an essential intermediate within this signaling cascade initiating endothelial [Ca2+]i response. Results from the deactivation of Ca2+-ATPases by TG further suggest that a specific pool of intracellular Ca2+ sensitive to PLC/IP3-dependent pathway, is released in response to melanoma-endothelial contacts. Additionally, application of PT to HUVEC prevented nearly half of melanoma cell-initiated increase in [Ca2+]i magnitude. Activation of PLC-Ca2+ that acted through a G protein–linked receptor was utilized by melanoma cells which were in contact with HUVEC. Others have also reported this partial attenuation of [Ca2+]i response in bovine aortic endothelial cell following PT treatments (24). However, these observations, in fact, suggest other pathways are operational within a subpopulation of endothelial cells in response to Ca2+ agonist stimulation. As one potential explanation for the heterogeneity, tyrosine kinase-linked receptor has been reported to play a role in initiating endothelial Ca2+ signals following the tumor cell contact (23,25). These findings point toward a complex network of signaling pathways being recruited by tumor cell interaction with the endothelium.
VE-cadherin has been suggested to play a major role in regulating tumor cell transendothelial migration (22,26). Disruption of EC monolayer around the edges of the cell membranes at interendothelial junctions caused by melanoma cells is similar to the gap formations observed following histamine or tumor necrosis factor (TNF)-alpha stimulation (27,28). However, melanoma-induced gaps were highly localized and limited to the sites of direct melanoma–EC contacts. Polystyrene beads or co-culture medium obtained by melanoma–EC coincubation did not induce VE-cadherin rearrangement. Therefore, specific cell–cell interactions between melanoma and endothelial cells are likely required to initiate the gap formation. Candidate adhesion molecules that could mediate these interactions include endothelial E-, P-selectin and VCAM-1, all of which have been reported to induce Ca2+ and cytoskeleton reorganization upon engagement during PMN-EC contact (29). Therefore, we propose that adhesion molecules such as selectin/sialyl Lewis X and VCAM-1/VLA-4 potentially mediate melanoma cell-induced junction reorganization. Future studies will be needed to address the roles of these molecules in signaling junction rearrangement.
Since melanoma cell attachment to the endothelium was shown to release endothelial Ca2+ through PLC, the effect of PLC inhibitor, U73122, has been investigated. A high dose of PLC inhibitor that was effective to fully attenuate [Ca2+]i response significantly reduced the occurrence of melanoma–induced VE-cadherin disruption. PLC–Ca2+ is a critical pathway for VE-cadherin junction regulation initiated by melanoma cell; however, an incomplete abrogation of gap formations implies that other pathways also play a role in signaling VE-cadherin disassembly. For example, p38 mitogen-activated protein kinase (MAPK) has been indicated as an important upstream component associated with VE-cadherin regulation (data not shown). Others have also reported that p38 MAPK is required for the hyper-permeability of endothelium following TNF-alpha stimulation (28). Mechanical shear-induced activation of endothelial cells may involve VE-cadherin and p38 MAPK in addition to VEGF-receptor (30). These reports suggest involvement of other signaling events, which explain why inhibition of PLC does not completely abrogate the melanoma cell-induced adherens junction breakdown. Furthermore, Kielbassa-Schnepp et al. showed that endothelial Ca2+ is considered to be a critical upstream second messenger during leukocytic diapedesis, especially in transvascular migration of monocytes through microvascular endothelial cells (10). In their study, removal of endothelial Ca2+ response significantly reduces the number of transmigrating monocytes while not affecting the granulocyte extravasation or the overall cell adhesion to the endothelium. Together with our results, these studies suggest that the [Ca2+]i is a critical component of the junction regulation for subsequent transvascular cell migration. In addition, PI3K has been implicated to be a regulator of tight junction integrity (31); however, our data suggest PI3K is not necessary for adherens junction reorganization, although PI3K activity is required for efficient transmigration of the melanoma cells across the EC barrier. These results would suggest that for the reorganization of adherens junctions that PI3K is not upstream of PLC and that disruption of VE-cadherin is not sufficient for melanoma cell migration. Furthermore, the combinatorial effects of multiple signaling pathways, including PLC and PI3K, are necessary for disrupting the endothelium and allowing transmigration of metastatic melanoma cells.
Levels of phosphorylation of adherens junctions are considered to be a determinant of intercellular adhesiveness (21). In particular, VE-cadherin undergoes tyrosine phosphorylation in a loose monolayer, but not in tightly confluent cells. However, we did not see melanoma contact induce significant change in phosphotyrosine of VE-cadherin; therefore, mechanisms other than a direct attenuation of VE-cadherin binding activity may be recruited by melanoma cells as they transverse the endothelial barrier.
It has been reported that PKC regulates endothelial barrier function via altering actin binding protein and myosin light chain kinase (MLCK) activity. MLCK generates actin-myosin interaction facilitating cytoskeleton reorganization. PKC has also been shown to play a direct role in the loss of VE-cadherin junctions and rearrangement of focal adhesion (8,32). Therefore, PKC may be a candidate of downstream effector in [Ca2+]i pathway to mediate junction redistribution through Ca2+/calmodulin-associated manner.
Taken together, we report a PLC-dependent mechanism for the disassembly of VE-cadherin junctions upon melanoma cell contacts with the endothelium. The findings provide important insights into the complexity of cellular signaling cascades that are initiated and ultimately culminate at the transendothelial invasion and metastasis of tumor cells.
Acknowledgments
Authors thank Dr. David A. Antonetti (Penn State University, Cellular and Molecular Physiology Department) for helpful discussions. Technical assistance of Polung Yang (Penn State University, Veterinary Science Department) is appreciated. This work was supported by NIH-CA97306, NSF-BES138474 and the Health Research Formula Funding Program (SAP 41000-20604).
References
- 1.Nicolson GL. Metastatic tumor cell interactions with endothelium, basement membrane and tissue. Curr Opin Cell Biol. 1989;1:1009–1019. doi: 10.1016/0955-0674(89)90073-2. [DOI] [PubMed] [Google Scholar]
- 2.Tang DG, Honn KV. Adhesion molecules and tumor metastasis: an update. Invas Metast. 1994;14:109–122. [PubMed] [Google Scholar]
- 3.Albelda SM, Mette SA, Elder DE, Stewart R, Damjanovich L, Herlyn M, Buck CA. Integrin distribution in malignant melanoma: association of the beta 3 subunit with tumor progression. Cancer Res. 1990;50:6757–6764. [PubMed] [Google Scholar]
- 4.Tawil NJ, Gowri V, Djoneidi M, Nip J, Carbonetto S, Brodt P. Integrin alpha3beta1 can promote adhesion and spreading of metastatic breast carcinoma cells on the lymph node stroma. Int J Cancer. 1996;66:703–710. doi: 10.1002/(SICI)1097-0215(19960529)66:5<703::AID-IJC20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 5.Bevilacqua MP. Endothelial-leukocyte adhesion molecules. Annu Rev Immunol. 1993;11:767–804. doi: 10.1146/annurev.iy.11.040193.004003. [DOI] [PubMed] [Google Scholar]
- 6.Siegel G, Malmsten M. The role of the endothelium in inflammation and tumor metastasis. Int J Microcirc Clin Exp. 1997;17:257–272. doi: 10.1159/000179238. [DOI] [PubMed] [Google Scholar]
- 7.Siflinger-Birnboim A, Lum H, Del Vecchio PJ, Malik AB. Involvement of Ca2+ in the H2O2-induced increase in endothelial permeability. Am J Physiol. 1996;270:L973–978. doi: 10.1152/ajplung.1996.270.6.L973. [DOI] [PubMed] [Google Scholar]
- 8.Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis C, Tiruppathi C. Ca2+ signaling and PKCalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J physiol. 2001;533.2:433–445. doi: 10.1111/j.1469-7793.2001.0433a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silversrein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kielbassa-Schnepp K, Strey A, Janning A, Missiaen L, Nilius B, Gerke V. Endothelial intracellular Ca2+ release following monocyte adhesion is required for the transendothelial migration of monocytes. Cell Calcium. 2001;30:29–40. doi: 10.1054/ceca.2001.0210. [DOI] [PubMed] [Google Scholar]
- 11.Pfau S, Leitenberg D, Rinder H, Smith BR, Pardi R, Bender JR. Lymphocyte adhesion-dependent calcium signaling in human endothelial cells. J Cell Biol. 1995;128:969–978. doi: 10.1083/jcb.128.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziegelstein RC, Corda S, Pili R, Passaniti A, Lefer D, Zweier JL, Fraticelli A, Capogrossi MC. Initial contact and subsequent adhesion of human neutrophils or monocytes to human aortic endothelial cells release an endothelial intracellular calcium store. Circulation. 1994;90:1899–1907. doi: 10.1161/01.cir.90.4.1899. [DOI] [PubMed] [Google Scholar]
- 13.Dejana E, Corada M, Lampugnani MG. Endothelial cell-to-cell junctions. FASEB J. 1995;9:910–918. [PubMed] [Google Scholar]
- 14.Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, Dejana E. The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin) J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, Dejana E. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volberg T, Zick Y, Dror R, Sabanay I, Gilon C, Levitzki A, Geiger B. The effect of tyrosine-specific protein phosphorylation on the assembly of adherens-type junction. EMBO J. 1992;11:1733–1742. doi: 10.1002/j.1460-2075.1992.tb05225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 18.Harvath L, Falk W, Leonard EJ. Rapid quantitation of neutrophil chemotaxis: use of a polyvinylpyrrolidone-free polycarbonate membrane in a multiwell assembly. J Immunol Methods. 1980;37:39–45. doi: 10.1016/0022-1759(80)90179-9. [DOI] [PubMed] [Google Scholar]
- 19.Kline D, Kline JT. Thapsigargin activates a calcium influx pathway in the unfertilized mouse egg and suppresses repetitive calcium transients in the fertilized egg. J Biol Chem. 1992;267:17624–17630. [PubMed] [Google Scholar]
- 20.Smallridge RC, Kiang JG, Gist ID, Fein HG, Galloway RJ. U-73122, an aminosteroid phospholipase C antagonist, noncompetitively inhibits thyrotropin-releasing hormone effects in GH3 rat pituitary cells. Endocrinology. 1992;131:1883–1888. doi: 10.1210/endo.131.4.1396332. [DOI] [PubMed] [Google Scholar]
- 21.Lampugnani MG, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110:2065–2077. doi: 10.1242/jcs.110.17.2065. [DOI] [PubMed] [Google Scholar]
- 22.Lewalle JM, Bajou K, Desreux J, Mareel M, Dejana E, Noel A, Foidart JM. Alteration of interendothelial adherens junctions following tumor cell-endothelial cell interaction in vitro. Exp Cell Res. 1997;237:347–356. doi: 10.1006/excr.1997.3799. [DOI] [PubMed] [Google Scholar]
- 23.Lewalle JM, Cataldo D, Bajou K, Lambert CA, Foidart JM. Endothelial cell intracellular Ca2+ concentration is increased upon breast tumor cell contact and mediates tumor cell transendothelial migration. Clin Exp Metastasis. 1998;16:21–29. doi: 10.1023/a:1006555800862. [DOI] [PubMed] [Google Scholar]
- 24.Jiang L, Jha V, Dhanabal M, Sukhatme VP, Alper SL. Intracellular Ca2+ signaling in endothelial cells by the angiogenesis inhibitors endostatin and angiostatin. Am J Physiol Cell Physiol. 2001;280:C1140–1150. doi: 10.1152/ajpcell.2001.280.5.C1140. [DOI] [PubMed] [Google Scholar]
- 25.Furuichi T, Michikawa T, Mikoshiba K. Intracellular Calcium Channels. In: Carafoli E, Klee C, editors. Calcium as a Cellular Regulator. Oxford University Press; New York: 1999. [Google Scholar]
- 26.Sandig M, Voura EB, Kalnins VI, Siu CH. Role of cadherins in the transendothelial migration of melanoma cell in culture. Cell Motil Cytoskel. 1997;38:351–364. doi: 10.1002/(SICI)1097-0169(1997)38:4<351::AID-CM5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 27.Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell- to-cell afherens junctions. Arterioscler Thromb Vasc Biol. 1999;19:2286–2297. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- 28.Nwariaku FE, Chang J, Zhu X, Liu Z, Duffy SL, Halaihel NH, Terada L, Turnage RH. The role of p38 MAP kinase in tumor necrosis factor-induced redistribution of vascular endothelial cadherin and increased endothelial permeability. Shock. 2002;18:82–85. doi: 10.1097/00024382-200207000-00015. [DOI] [PubMed] [Google Scholar]
- 29.Lorenzon P, Vecile E, Nardon E, Ferrero E, Harlan JM, Tedesco F, Dobrina A. Endothelial cell E- and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J Cell Biol. 1998;142:1381–1391. doi: 10.1083/jcb.142.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci USA. 2002;99:9462–9467. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheth P, Basuroy S, Li C, Naren AP, Rao RK. Role of phosphatidylinositol 3-kinase in oxidative stress-induced disruption of tight junctions. J Biol Chem. 2003;278:49239–49245. doi: 10.1074/jbc.M305654200. [DOI] [PubMed] [Google Scholar]
- 32.Garcia JGN, Davis HW, Patterson CE. Regulation of endothelial gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163:510–522. doi: 10.1002/jcp.1041630311. [DOI] [PubMed] [Google Scholar]