

Abstract
The sodium periodate mediated oxidative cleavage of the C-C bond of twelve epoxides is reported with yields of the corresponding carbonyl compounds in up to 91%. This is a two-step reaction that proceeds through a rate-limiting epoxide opening to a vicinal diol that is cleaved in situ to the corresponding carbonyl compound. This method serves as a chemoselective alternative to ozonolysis.
The oxidative cleavage of olefins has been extensively studied in organic synthesis.1 However, the cleavage of C-C bonds via epoxides is a less explored route. A major advantage of accessing carbonyl compounds through epoxides rather than olefins is that selective epoxidation can be performed on compounds with multiple double bonds.2 This creates another possibility for site-specific oxidative cleavage of olefins (Scheme 1). A similar strategy was utilized in the synthesis of the multi-functionalized molecule (−)-terpestacin.3
Scheme 1.
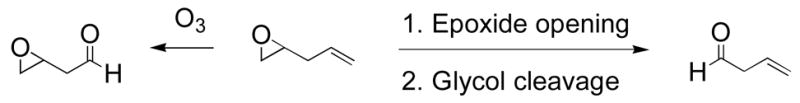
Chemoselective C-C bond cleavage.
The most common method for oxidative cleavage is ozonolysis, a process that can be dangerous on larger scales. Alternative one-pot processes include the use of RuCl3•H2O5 or OsO46 for vicinal dihydroxylation in combination with oxidants such as NaIO4, oxone, and NaOCl to facilitate conversion of the glycol to the carbonyl compound. The ability of periodate to cleave vicinal diols led us to study the cleavage of the C-C bond of epoxides, which we assumed would proceed though a diol intermediate.7 It has been reported that treatment of the vicinal diol derived from limonene with sodium periodate does facilitate C-C bond cleavage, resulting in the ketoaldehyde.8 The two-step process involving isolation of the diol was low yielding but the one-pot treatment of the epoxide with periodate at elevated temperature does afford the oxidative cleavage product in higher yield.8a However, the generality of this method was not tested with other terpene epoxides. A few cases in the literature have reported the reaction of periodic acid (HIO4) or metaperiodic acid (H5IO6) with epoxides, resulting in the corresponding carbonyl compound.9 These reactions were only demonstrated on a small number of epoxides and employed harsh reaction conditions, often with elevated temperatures. To the best of our knowledge, a systematic study of the reaction of epoxides, both aliphatic and aromatic, with aqueous sodium periodate has not been performed. In this communication, the one-pot cleavage of the C-C bonds of several epoxides in the absence of catalyst is explored (Scheme 2).
Scheme 2.

General scheme for epoxide C-C bond cleavage with sodium periodate.
During the course of our work, Ochiai demonstrated the novel use of iodosylbenzene for the one-pot oxidative cleavage of olefins.10 In some cases, a considerable amount of epoxide was detected and implicated as a reaction intermediate. A few aromatic epoxides were then tested in the reaction with iodosylbenzene to yield substituted benzaldehydes.10 Similar findings were reported in a recent paper by Liu, in which a Mn-porphyrin catalyst was used in combination with NaIO4 to facilitate the oxidative cleavage of olefins.11 Although no epoxide was detected, the reaction was thought to proceed through an epoxide intermediate, with the epoxidation being the rate limiting step. The authors were able to cleave the C-C bond of cyclohexene oxide and styrene oxide using their method, however, terminal alkenes and 1,1-disubstituted alkenes, such as 1-octene and β-pinene respectively, were not good substrates for this reaction.11 The corresponding epoxides were not tested in the Mn-porphyrin mediated C-C cleaving reaction. While the cleavage of styrene oxide can be envisioned to proceed through the acyclic diol, it was puzzling how a trans-diol from cyclohexene oxide could be cleaved by sodium periodate. These reports prompted us to disclose our results, where epoxides are converted to the corresponding carbonyl compounds using aqueous sodium periodate without the need for the use of transition metal or acid catalyst.
Initial studies were performed with (+)-β-pinene oxide 1 with the goal of forming nopinone 2 (Scheme 3). Unfortunately, a significant amount of rearrangement product 3 was observed and nopinone 2 could only be obtained in up to 32% yield.12 This occurred using a variety of solvents and solvent mixtures including water, methanol, and THF. A similar ring rearrangement occurred in the reaction with α-pinene oxide as well. We then turned our interest toward simpler aliphatic and aromatic epoxides. Water was chosen as one of the co-solvents in order to partially dissolve the periodate and to provide a slightly acidic environment to promote epoxide opening. We also found that both THF and CH3CN solvents worked well for this reaction and all subsequent reactions were performed using either THF/H2O or CH3CN/H2O.
Scheme 3.

Attempted cleavage of (+)-β-pinene oxide.
Six aliphatic epoxides, including terminal epoxides, were screened in the cleavage reaction (Table 1).13 Facile conversion of cyclohexene oxide, limonene oxide, and 3-carene oxide was achieved in moderate to high yields with just two equivalents of sodium periodate (entries 4, 5, and 6). The lower yield of adipaldehyde is likely due to the high volatility of the product. Less reactive substrates, such as epoxyoctane and 1,2-epoxy-9-decene (entries 1 and 3), required at least four equivalents of periodate to facilitate full consumption of starting material. The reaction carried out in the THF/H2O solvent system gave impure aldehyde, contaminated with a significant amount of unidentified byproducts. However, aqueous acetonitrile eliminated this byproduct and gave essentially analytically pure aldehyde product (entry 1). It is possible that the lipophilic tails of these epoxides are forming micelles, preventing the polar epoxide heads from reacting with aqueous periodate. The reactions of 1,2-epoxy-9-decene and limonene oxide (entries 3 and 5) demonstrate the chemoselectivity of this reaction, which is complementary to conventional ozonolysis.
Table 1.
Oxidative cleavage of aliphatic epoxides
 | |||||
|---|---|---|---|---|---|
| Entry yieldb | Substrate | Product | Solvent | NaIO4a (equiv) | |
| 1 |
|
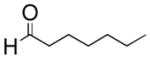
|
CH3CN/H2O | 5 | 73% |
| 2 |

|

|
CH3CN/H2O | 5 | 58%c |
| 3 |

|

|
CH3CN/H2O | 4 | 78% |
| 4 |

|

|
THF/H2O | 2 | 67% |
| 5 |
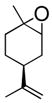
|
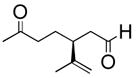
|
THF/H2O | 2 | 91% |
| 6 |

|

|
THF/H2O | 2 | 88% |

|

|
||||
| 7 | X = H | CH3CN/H2O | 2 | 84% | |
| 8 | X = p-Cl | CH3CN/H2O | 2 | 34% | |
| 9 | X = p-F | THF/H2O | 2 | 62% | |
| 10 | X = o-Br | CH3CN/H2O | 2 | 71%d | |
| 11 |

|

|
CH3CN/H2O | 2 | 41%e |
| 12 |

|

|
CH3CN/H2O | 2 | 64% |
A significant drop in pH occurred with the addition of NaIO4 to the THF/H2O mixture (pH 6 to pH 4).
Isolated yield.
Starting material was recovered in 39% yield.
Based on recovered starting material.
Only PhCHO was observed.
Periodate cleavage is known to prefer cis-diols as substrates, whereas acid-induced epoxide opening is known to create a trans-diol intermediate.14 While this may not be of consequence to linear epoxides, it is somewhat unexpected to observe such facile conversion of the cyclic examples. This leads us to believe that isomerization of the diol may be occurring under the acidic reaction conditions.15 It is also worth noting that in the case of epoxydodecane, a significant amount of unreacted starting material was recovered (Table 1, entry 2). This implies that epoxide opening is the rate-limiting step of the reaction. The trisubstituted epoxides generally gave products in higher yield compared to monosubstituted epoxides, reflecting the ease of epoxide hydrolysis in the former.
Initial attempts to perform this reaction on styrene oxide in THF/H2O resulted in a mixture of benzaldehyde and phenacetaldehyde. The production of the latter is due to a rearrangement that occurs before ring opening.16 Carrying out the reaction in CH3CN/H2O exclusively provided benzaldehyde in 84% isolated yield (entry 7). It should be pointed out that the reaction of styrene with iodosylbenzene gave phenacetaldehyde in 85% yield along with 5% benzaldehyde, whereas styrene oxide gave only benzaldehyde in 89% yield, which is comparable to the results realized in our reaction.10
Extension to halogenated aromatic epoxides gave fair to moderate yields for p-fluoro, p-chloro, and o-bromo-styrene oxide (Table 1, entries 8–10). We believe these lower yields may be due to the incomplete hydrolysis of the epoxide. The recovery of starting material in the o-bromo derivative leads us to believe that the epoxide-opening is the rate-limiting step. Disubstituted aromatic epoxides such as the TBS-protected cinnamyl epoxide and trans-stilbene oxide (entries 11 and 12) gave comparable results.
The oxidative cleavage of the C-C bond of twelve different epoxides has been demonstrated in yields up to 91%. We believe this is a two-step reaction that proceeds through a rate-limiting epoxide ring-opening, leading to a vicinal diol intermediate that is highly reactive toward periodate oxidation. Apparently, the trans-diols formed from cyclic epoxides isomerize under the acidic reaction conditions before undergoing C-C bond cleavage. More highly substituted epoxides appear to be more reactive, which may be due to the ease of initial epoxide opening. This aqueous one-pot procedure provides access to a variety of different carbonyl compounds, which are important intermediates in organic synthesis. This method, in combination with known selective epoxidation methods, should provide an appealing alternative to ozonolysis.
Acknowledgments
DDD and MAT thank the National Institute on Drug Abuse (DA07215; Professor Alexandros Makriyannis P.I.) for generous support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Kuhn FE, Fischer RW, Herrmann WA, Weskamp T. In: Transition Metals for Organic Synthesis. Beller M, Bolm C, editors. Vol. 2. Wiley-VCH; Weinheim: 2004. p. 427. [Google Scholar]
- 2.Barlan AU, Zhang W, Yamamoto H. Tetrahedron. 2007;63:6075–6087. doi: 10.1016/j.tet.2007.03.071.Bäckvall JE, Ericsson AM, Juntunen SK, Najera C, Yus M. J Org Chem. 1993;58:5221–5225.Hydroxyl-directed epoxidation is also possible: Sharpless KB, Michaelson RC. J Am Chem Soc. 1973;95:6136–6137.
- 3.Trost BM, Dong G, Vance JA. J Am Chem Soc. 2007;129:4540–4541. doi: 10.1021/ja070571s. [DOI] [PubMed] [Google Scholar]
- 4.Criegee R. Angew Chem, Int Ed. 1975;14:745–752. [Google Scholar]
- 5.(a) Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J Org Chem. 1981;46:3936–3938. [Google Scholar]; (b) Yang D, Zhang C. J Org Chem. 2001;66:4814–4818. doi: 10.1021/jo010122p. [DOI] [PubMed] [Google Scholar]
- 6.(a) Pappo R, Allen DS, Jr, Lemieux RU, Johnson WS. J Org Chem. 1956;21:478–479. [Google Scholar]; (b) Whitehead DC, Travis BR, Borhan B. Tetrahedron Lett. 2006;47:3797–3800. [Google Scholar]
- 7.Fatiadi AJ. Synthesis. 1974;4:229–272. [Google Scholar]
- 8.(a) de Lima Castro F, Kover RX, Kover WB, Jones J., Jr J Braz Chem Soc. 1999;10:112–116. [Google Scholar]; (b) Suemune H, Kawahara T, Sakai K. Chem Pharm Bull. 1986;34:550–557. doi: 10.1248/cpb.34.3440. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kim J, Matsuyama S, Suzuki T. J Labelled Compd Radiopharm. 2004;47:921–934. [Google Scholar]; (b) Sankaranarayanan S, Chattopadhyay S. Tetrahedron: Asymm. 1998;9:2627–2633. [Google Scholar]; (c) Nagarkatti JP, Ashley KR. Tetrahedron Lett. 1973;14:4599–4600. [Google Scholar]
- 10.Miyamoto K, Tada N, Ochiai M. J Am Chem Soc. 2007;129:2772–2773. doi: 10.1021/ja070179e. [DOI] [PubMed] [Google Scholar]
- 11.Liu S-T, Reddy KV, Lai R-Y. Tetrahedron. 2007;63:1821–1825. [Google Scholar]
- 12.Pellegata R, Ventura P, Villa M, Palmisano G, Lesma G. Synth Commun. 1985;15:165–170. [Google Scholar]
- 13.Representative procedure for the C-C cleavage of epoxides. To a round-bottom flask equipped with a magnetic stir-bar, finely powdered sodium periodate (20–50 mmol, 99% pure) was stirred with the appropriate solvent mixture (40 mL) for five minutes. The epoxide (10 mmol) was then added and the reaction mixture was stirred at room temperature. Upon reaction completion, as monitored by TLC, the white precipitate that had formed was filtered away and washed with 30 mL Et2O, creating two distinct layers. The aqueous layer was extracted with two 30 mL portions of Et2O, washed with water and brine, dried (MgSO4), filtered, and concentrated in vacuo. Although most examples provided pure carbonyl compounds the crude reaction mixtures could be purified via column chromatography on silica gel eluting with EtOAc/Hexanes.
- 14.(a) Waters WA. Mechanisms of Oxidation of Organic Compounds. Wiley; New York: 1964. p. 72. [Google Scholar]; (b) Buist GJ, Bunton CA, Hipperson WCP. J Chem Soc B. 1971:2128–2142. [Google Scholar]
- 15.Aqueous sodium periodate has a pH of 4.
- 16.Chen J, Che C-M. Angew Chem, Int Ed. 2004;43:4950–4954. doi: 10.1002/anie.200460545. [DOI] [PubMed] [Google Scholar]