

Abstract
Many cancers, including breast cancer, harbor loss of function mutations in the catalytic domain of the PTEN phosphatase or have reduced PTEN expression through loss of heterozygosity (LOH) and/or epigenetic silencing mechanisms. However, specific phenotypic effects of PTEN inactivation in human cancer cells remain poorly defined without a direct causal connection between the loss of PTEN function and the development or progression of cancer. To evaluate the biological and clinical relevance of reduced or deleted PTEN expression, a novel in vitro model system was generated using human somatic cell knock-out technologies. Targeted homologous recombination allowed for a single and double allelic deletion which resulted in reduced and deleted PTEN expression, respectively. We determined that heterozygous loss of PTEN in the non-tumorigenic human mammary epithelial cell line, MCF-10A, was sufficient for activation of the PI3K/Akt and MAPK pathways, while the homozygous absence of PTEN expression led to a further increased activation of both pathways. The deletion of PTEN was able to confer growth factor-independent proliferation which was confirmed by the resistance of the PTEN−/− MCF-10A cells to small molecule inhibitors of the EGF receptor. However, neither heterozygous nor homozygous loss of PTEN expression was sufficient to promote anchorage-independent growth, but the loss of PTEN did confer apoptotic resistance to cell rounding and matrix detatchment. Finally, MCF-10A cells with the reduction or loss of PTEN showed increased susceptibility to the chemotherapeutic drug doxorubicin, but not paclitaxel.
Keywords: PTEN, breast cancer, anoikis, tumor dormancy, doxorubicin
Introduction
PTEN (phosphatase and tensin homologue deleted on chromosome 10), is a tumor suppressor gene that dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3), the product of the lipid kinase, phosphatidylinositol 3-kinase (PI3K). PTEN antagonizes activated PI3K to maintain normal cell growth or arrest, survival or apoptosis. PTEN and PI3K exist in a tight regulatory loop, and a reduction or deletion of PTEN or the acquisition of an activating mutation in PI3K leads to abnormal activation of the PI3K pathway.
PTEN is the second most frequently mutated gene in human cancers following TP53. Immunohistochemistry studies of tumors from breast, pancreatic, and ovarian cancers have demonstrated a loss of PTEN protein in 30–50% of samples (1–5). In breast cancer, this loss correlates highly with lymph node metastasis (5). Additionally, germline mutations in the PTEN gene are associated with multi-neoplastic, autosomally dominant human syndromes such as Cowden’s disease and Bannayan-Zonana syndrome. Each syndrome features a predisposition to the formation of different malignancies, including breast cancer in 20–50% of the affected females (6, 7). The high frequency of reduction or loss of PTEN in breast cancer suggests its potential role in initiation and/or progression of human breast cancer.
Previously, the effects of PTEN loss have primarily been measured in tumor cell lines, which harbor numerous other transforming and oncogenic mutations. This has made it difficult to determine which phenotypes are directly conferred by the loss of PTEN and to define the stages of tumorigenesis that are specifically altered in cells with PTEN loss. In order to elucidate the connection between PTEN loss and the initiation and/or progression of human breast carcinomas, somatic cell gene targeting technology was used to more closely mimic the physiological reduction or loss of PTEN in epithelial cells. This in vitro model system involved targeted homologous recombination to disrupt each allele of PTEN in the non-tumorigenic mammary epithelial cell (MEC) line, MCF-10A. Using these isogenic somatic cell PTEN knock-out lines, we determined that PTEN loss not only induces the activation of the PI3K pathway, but also the MAP kinase pathway. This increase in pathway activation led to growth factor-independent proliferation that was suppressed with either PI3K or MAPK inhibitors. PTEN loss in MCF-10A cells also increased anchorage-independent survival, but was insufficent to confer anchorage-independent growth. Also, of potential clinical importance, PTEN loss confers susceptibility to the chemotheraputic drug, doxorubicin, but not paclitaxel, two agents commonly used for breast cancer therapy.
Material and Methods
Cell lines and cell culture
MCF-10A cells were purchased from ATCC (Manassas, VA) and maintained as previously described (8) supplemented with 0.1µg/ml cholera toxin. Minimal assay media was composed of DMEM/F12 without phenol red, 1% charcoal stripped dextran treated fetal bovine serum (Hyclone, Logan, UT), 100 units/ml penicillin-streptomycin without exogenous growth factors. The MCF-10A.Bcl2 cells were created by stable transfection with the pBP/Bcl2 expression vector (9) and maintained in MCF-10A growth media supplemented with 2.5µg/ml puromycin. Cells were maintained in a 37°C incubator with 5% CO2.
PTEN+/− and PTEN−/− cell line generation
Heterozygote clones were created as previously described (10). A second construct was generated to delete exon 2 of PTEN on the second allele. Briefly, sequences with exact homology to intronic regions flanking exon 2 were cloned into the pAAV-MCS (Stratagene) via the pSept vector (11, 12). The adeno-associated virus was generated using the AAV Helper-free system from Stratagene following the manufacturer’s instructions. Individual G418 resistant clones were tested via PCR for the presence of a homozygote PTEN knockout. Positive clones were treated with a cre recombinase virus to excise the IRES-neoR gene. Cells were maintained in MCF-10A growth media as described above.
Proliferation assays
Cell were seeded in quadruplicate at 2.0×103 per well in 96-well plates in minimal assay media. The next day, the appropriate media with or without drug was added. Cell viability was quantified using MTT (Sigma, St. Louis, MO). After MTT treatment, the media was removed, the converted dye was solubilized in 0.01 mol/L glycine in DMSO, and the absorbance (450nm). For growth in the absence of drugs, a set of cells were exposed to MTT on day 0 to accurately assess starting cell number. Growth was calculated as a percent above cell number on day 0. For the Erlotinib and Gefitinib (LC Laboratories, Woburn, MA) studies, untreated wells of each cell line were used as the control of calculation percent viability.
Western blot analysis
Cell lysates were prepared in RIPA lysis buffer [0.5mol/L Tris-HCl, pH7.4, 1.5mol/L NaCl, 2.5% deoxycholic acid, 10% NP-40, 10mmol/L EDTA] supplemented with protease inhibitor cocktail EDTA-free (Roche, Mannheim, Germany) and phosphatase inhibitor cocktail II (Calbiochem, LaJolla, CA). Western blotting was performed using NuPage gels (Invitrogen, Carlsbad, CA). Primary antibodies for PTEN, p-Akt(S473), Akt, p-ERK1/2, ERK1/2 and PARP (Cell Signaling, Danvers, MA) and GAPDH (Abcam, Cambridge, MA) were used at manufacturers’ recommended dilutions.
Flow Cytometry
For sub-G1 analysis, cells were ethanol fixed and treated with RNase A (1mg/ml) and propidium iodide (PI) (20µg/ml). Cells were analyzed by a Becton Dickenson LSR-II at the Flow Cytometry Core Laboratory, CVD Immunology Group at the University of Maryland, Baltimore.
Survival assays
Cell were seeded in quadruplicate at 1.5×104 per well in 96-well plates in minimal assay media. The next day, the appropriate concentrations of doxorubicin (Calbiochem, LaJolla, CA) or paclitaxel (Invitrogen, Carlsbad, CA) were added to the cells. On day 5, cell viability was quantified using MTT. Untreated wells of each cell line were used as the control of calculation percent viability.
Results
PTEN heterozygous and homozygous loss promotes activation of both the PI3 kinase and MAP kinase pathways in mammary epithelial cells
Reduction of PTEN expression or complete loss is observed in approximately 40% of human breast cancers (2, 5, 13). To determine the oncogenic phenotype of reduced or absent PTEN expression in human breast epithelial cells, we generated isogenic human MECs using MCF-10A cells. MCF-10A breast epithelial cells were chosen for targeted PTEN knock-out because these cells are human, mostly diploid, non-tumorigenic cell line and karyotype analysis of late passage cells demonstrate genetic stablility (data not shown). The use of non-tumorigenic cell lines allow us to assess any oncogenic effects directly resulting from loss of PTEN. Several independently derived heterozygous knock-out clones (PTEN+/−) were identified containing one active PTEN allele. Subsequent targeting of the second allele was accomplished to yield homozygous PTEN (PTEN−/−) knock-out clones.
Somatic cell gene knock-out was accomplished via homologous recombination between the genomic locus and the targeting vector to delete exon II of PTEN and replace it with a promoterless, IRES-neoR gene flanked by LoxP sites (Fig. 1A). For each round of targeting, positive clones were verified by PCR. Subsequent removal of the IRES-neoR cassette was accomplished by treatment with cre recombinase. At least three single, independent isogenic PTEN+/− and PTEN−/− clones from separate infections were isolated and used to account for any clonal variations.
Figure 1. Single and biallelic deletion of the PTEN gene.
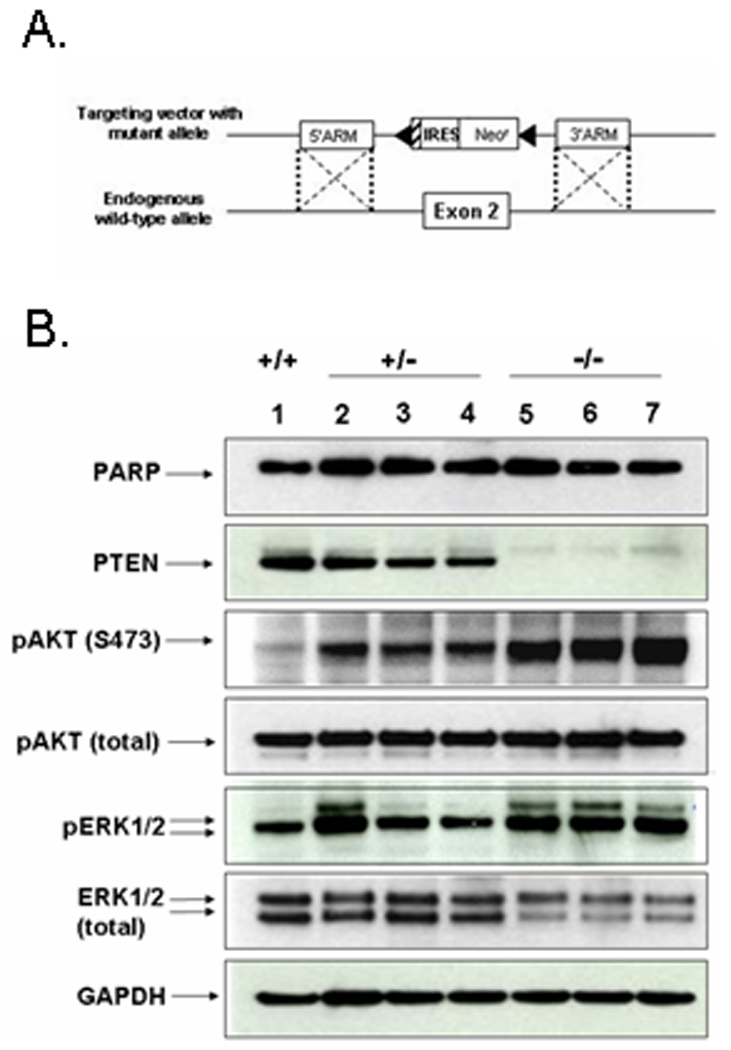
(A.) To remove each copy of PTEN, our targeting construct contained a neomycin resistance cassette flanked by LoxP sites and homologous sequences to the intronic regions 5’ and 3’ of exon 2. (B.) MCF-10A cells (lane 1), PTEN+/− (lanes 2–4), and PTEN−/− clones (lanes 5–7) were harvested at sub-confluence during exponential growth. PTEN expression is reduced in the PTEN+/− clones and absent in the PTEN−/− clones. With a reduction and deletion of PTEN, activated AKT (pAKT) is increased (upper bands) while total AKT levels remain equivalent. Activated ERK (pERK1/2) levels are also increased in the clones. GAPDH was used as a loading control.
The MCF-10A PTEN+/− clones showed a decrease in PTEN levels (Fig. 1B, lanes 2–4), and the PTEN−/− clones completely lacked PTEN expression (Fig. 1B, lanes 5–7). The absence of PTEN mRNA in the PTEN−/− clones was verified by real-time PCR (data not shown). All PTEN−/− clones maintained increased pAKT levels over their PTEN+/− and parental counterparts. Interestingly, activated ERK (pERK1/2) levels were also increased in the PTEN−/− clones over the MCF-10A parental cells. While the PTEN+/− clones showed an increase in activated ERK levels from the parental cells, the increase was less dramatic or consistent among the PTEN+/− clones (Fig. 1B). However, in conjunction with an increase in pERK1/2 in the PTEN−/− clones, decreased total ERK levels were consistently observed.
PTEN loss confers growth factor-independent proliferation
To determine whether the activation of the PI3K and MAPK pathways altered proliferation rates, MCF-10A, PTEN+/−, and PTEN−/− clones were media and analyzed over a period of nine days. Interestingly, at early passage, the PTEN+/− and PTEN−/− clones with reduced or deleted PTEN grew significantly slower than parental MCF-10A cells (Fig. 2A, P<0.05). While at later passage the MCF-10A cells maintain a similar growth rate to that of their earlier passage counterparts, the proliferation rates of the PTEN+/− and PTEN−/− clones increase over time (Fig 2A). The parental MCF-10A, PTEN+/−, and PTEN−/− clones do not undergo any significant cell death over the first seven days due to the absence of a sub-G1 population (Fig. S1). However, once the cells achieve contact inhibition by day 7, all cells begin to die, shown by a drop in viability and the presence of a sub-G1 peak. The increase proliferation rate is likely due to a variety of mechanisms downstream of AKT activation, such as increased cyclin D1 expression, inhibition of Forkhead transcription factors, or reduction of p27Kip1, all of which positively regulate G1/S cell cycle progression (reviewed in (14)).
Figure 2. PTEN loss confers growth in minimal media.
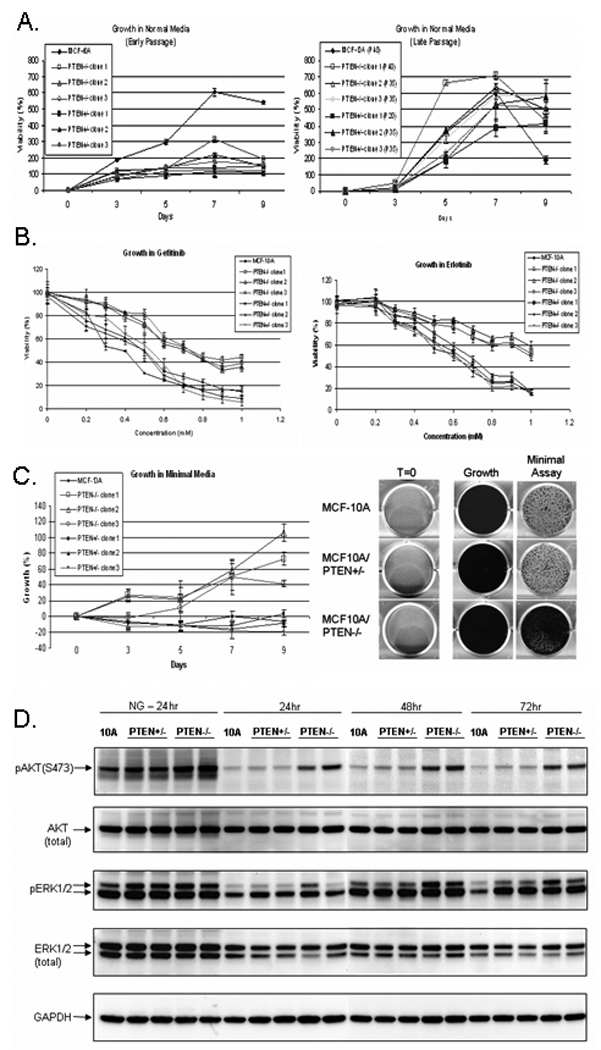
(A.) Early and late passage growth of MCF-10A cells, PTEN+/− clones, and PTEN−/− clones in normal MCF-10A growth media. Points, means of 2 independent experiments performed in quadruplicate; bars, SD. (B.) Cell growth of MCF-10A cells, PTEN+/− clones, and PTEN−/− clones after exposure to increasing doses of the EGFR antagonists Gefitinib and Erlotinib. Points, means of 3 independent experiments performed in quadruplicate; bars, SD. (C.) Cell growth in minimal media. Points, means of 2 independent experiments performed in quadruplicate; bars, SD. On day 6, MCF-10A cells, PTEN+/−, and PTEN−/− cells were fixed and stained with a solution of 10% PBS-buffered formalin and 0.25% crystal violet. Representative wells from the MCF-10A, PTEN+/−, and PTEN−/− cells are shown. (D.) MCF-10A, two PTEN+/−, and two PTEN−/− clones were plated in either normal growth media (NG) or minimal assay media and harvested at the indicated times by direct addition of RIPA lysis buffer.
A well-known characteristic of MCF-10A MECs is their EGF requirement for cellular proliferation. Growth factor-independent proliferation is a common hallmark in cancer cells containing oncogenic phenotypes and aberrantly activated signaling (15). Since the PTEN+/− and PTEN−/− clones have an increase in activated PI3K and MAPK pathways, we examined whether the increased activation of these pathways was sufficient to confer EGF-independent growth by treating the cells with increasing concentrations of the clinically administered EGFR small molecule inhibitors, Gefitinib and Erlotinib (Fig 2B). Compared to the parental and PTEN+/− clones, the PTEN−/− clones were significantly more resistant to growth inhibition via the EGFR inhibitors indicating a decreased requirement of EGF for proliferation. To confirm this observation, the MCF-10A cells, PTEN+/−, and PTEN−/− clones were maintained in minimal assay media devoid of exogenous growth factors for nine days. As previously observed using compounds to disrupt EGF signaling, the MCF-10A cells and PTEN+/− clones showed reduced growth (Fig. 2C). However, the PTEN−/− cells survived and continued to slowly proliferate, although considerably slower than in media supplemented with EGF. At later passage, PTEN−/− cell proliferation in the absence of mitogens became more robust (Fig. S2).
Immunoblots confirmed the increased activity of each pathway in the presence and absence of mitogens (Fig. 2D). After 24hrs in growth media, the levels of pAKT are highest in the PTEN−/− clones, and slightly more elevated in the PTEN+/− clones compared to the MCF-10A parental cells. Assessment of the same set of cells grown in media without mitogens demonstrated reduced levels of pAKT. Although there is a minimal level of detectable pAKT in the parental and PTEN+/− clones, the PTEN−/− clones continue to maintain elevated pAKT levels at 24, 48, and 72hrs.
Phosphorylated ERK levels were also slightly elevated in all PTEN+/− and PTEN−/− clones compared to the MCF-10A parental cells when grown in normal culture media (Fig. 2D). The levels of activated ERK were similar between the PTEN+/− and PTEN−/− clones. This result differed from the earlier immunoblot results (Fig. 1B) likely due to different harvest times after replating. Cells in Figure 1B were harvested during exponential growth three days after replating, while cells in Figure 2D were harvested only 24 hours after replating when they are not yet in exponential growth. However, after three days in media devoid of growth factors, the PTEN+/− cells have increased pERK levels over the MCF-10A parental cells, and the PTEN−/− cells have pERK levels higher than the PTEN+/− clones, similar to the pattern of ERK phosphorylation seen after three days in culture media (Fig. 1B). This pattern of increasing levels of activated ERK, from the MCF-10A parental cells to their PTEN−/− isogenic counterparts, indicates that even under growth factor reduced conditions, loss of PTEN allows for activation of the ERK signaling pathway.
To determine if the improved cell growth in PTEN−/− cells resulted from differences in cell cycling or reduced apoptosis, we performed flow cytometry measurements of DNA content throughout the growth in minimal media (Fig. 3). These results showed that both MCF-10A and PTEN+/− cells undergo apoptotic DNA cleavage beginning on day 3 that increases to 87% and 81%, respectively, by day 9 while PTEN−/− cells show no signs of apoptosis. The increased viability of the PTEN−/− cells is primarily from resistance to apoptosis, rather than a difference in cell cycling. The elevation of apoptosis in MCF-10A and PTEN+/− cells during exposure to minimal media is clearly sufficient to offset any cell growth and keep the cell population from increasing.
Figure 3. MCF10A and PTEN+/− cells undergo apoptosis, while the PTEN−/− cells survive in minimal media.
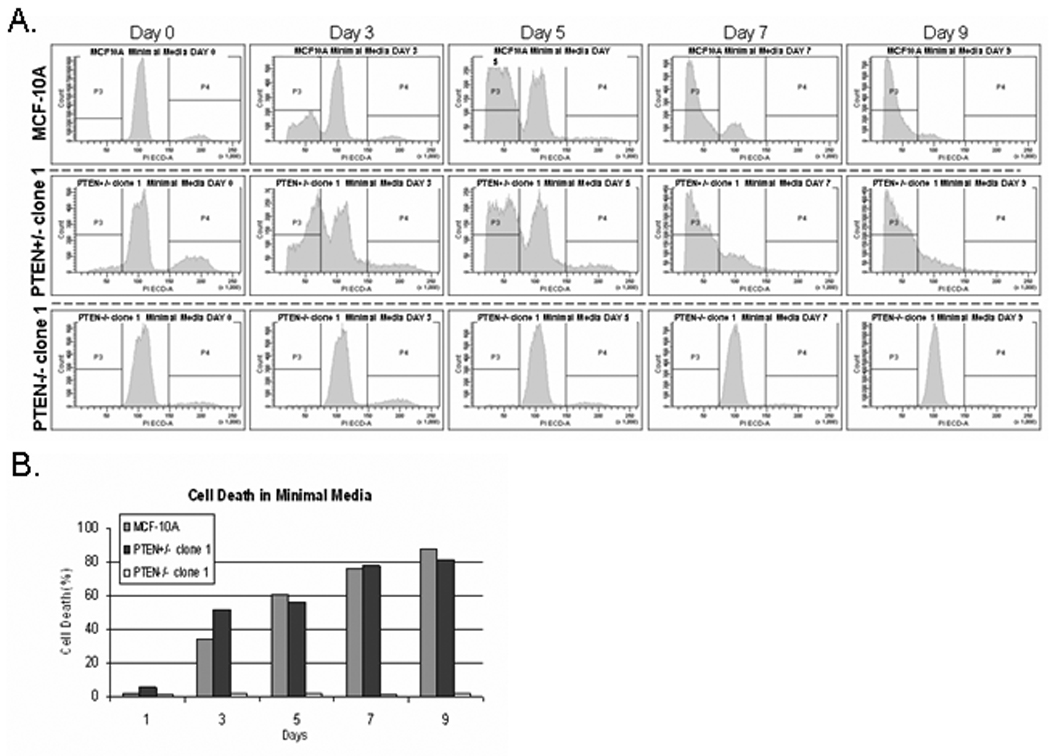
(A.) Flow cytometry analysis of PI stained MCF10A and PTEN+/− cells reveals massive cell death beginning at day 3 and continuing through day 9 until only a sub-G1 population is left. However, the PTEN−/− cells continue to survive and slowly cycle due to the presence of a minimal G2 peak present until day 9 when the cells become growth arrested at confluence. (B.) Graphical representation of the cell death minimal media.
Growth factor independent proliferation due to PTEN loss can be inhibited by pharmacological blockade of phosphoinositidyl-3-kinase and MAPK pathways
To confirm the requirement of active PI3K and MAPK pathways for continued cell proliferation in the absence of growth factors, the PTEN−/− cells were treated with inhibitors of each pathway and growth factor-independent proliferation was assessed. Only the PTEN−/− cells were used in this experiment because the parental and heterozygote clones do not grow under these conditions (Fig. 2C). Increasing concentrations of the PI3K inhibitor, LY294002, in minimal assay media was added to the cells, and after five days, a dose-dependent inhibition of growth was observed. The addition of 10µM LY294002 led to nearly complete inhibition of proliferation of all PTEN−/− clones (Fig. 4A). Similarly, the PTEN−/− clones were grown in the presence of the MEK1/2 inhibitor, U0126 (Fig. 4B). There was also a dose-dependent growth inhibition of the PTEN−/− cells following exposure to the MEK inhibitor. In the presence of 1µM of U0126, growth factor-independent proliferation was inhibited by over 50% and almost completely inhibited with 2.5µM U0126.
Figure 4. Inhibition of the PI3K and MAP kinase pathways suppresses proliferation in minimal media of the PTEN−/− mammary epithelial cells.
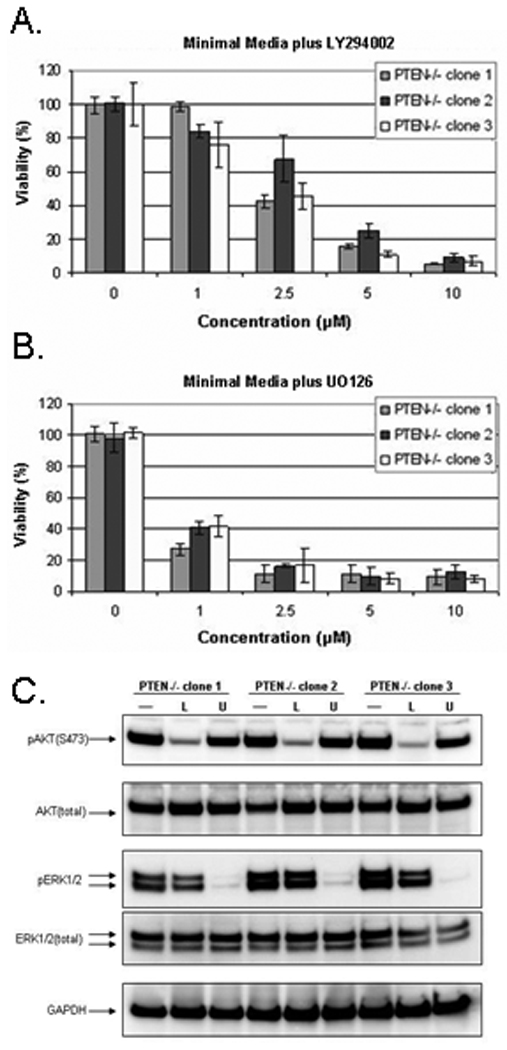
(A.) Percent growth of three PTEN−/− clones in minimal assay media with increasing concentration of LY294002. (B.) Percent growth of three PTEN−/− clones in minimal assay media with increasing concentration of U0126. (C.) The three PTEN−/− clones were plated in minimal assay media and allowed to recover overnight. The next day, fresh minimal assay media was added +/− 10µM LY294002 (L) or 10µM U0126 (U) for 1 hour. Western blot analysis demonstrating pAKT reduction in the cells treated with the PI3K inhibitor, LY294002, and pERK reduction in the cells treated with the MEK inhibitor, U0126.
To verify inhibition of the PI3K and MAPK pathways by LY294002 and U0126 respectively, immunoblots were performed. In the absence of the PI3K or MEK1/2 inhibitors, the PTEN−/− clones displayed high levels of pAKT and pERK (Fig. 4A). After LY294002 treatment, all PTEN−/− clones showed a significant drop in pAKT levels. Following U0126 treatment, the levels of pERK dropped to almost undetectable levels.
Anchorage-independent survival and growth
Since PTEN loss highly correlates with increased breast cancer lymph-node metastasis (16–18), it was next determined if PTEN loss alone would lead to the transformation of non-tumorigenic breast epithelial cells. Anchorage-independent growth in soft agar is a property of transformed cells that best correlates with in vivo tumorigenicity (19). The MCF-10A non-tumorigenic parental cells and PTEN−/− clones were plated in soft agar and incubated for 21 days. MCF7 breast cancer line was used as a positive control for colony growth and only incubated for 14 days due to the formation of multiple, large colonies. As expected, the MCF-10A cells did not form colonies. Likewise, the PTEN−/− cells were unable to form colonies in soft agar and unable to form tumors in SCID mice (n=5) after 24 weeks (data not shown). However, colony formation in soft agar and in vivo tumor growth rely on anchorage-independent proliferation, but is not a sufficient test for increased resistance to anoikis, or apoptosis after matrix detachment. Since previous data has shown that activation of the PI3K pathway contributes to cell survival after detachment (20), the MCF-10A, PTEN+/−, and PTEN−/− cells were next tested for apoptotic resistance during cell rounding and detachment. Included in these studies, as a control for resistance to cell rounding and anoikis, were MCF-10A cells overexpressing the anti-apoptotic gene Bcl-2 (8). PARP cleavage, an indicator of apoptosis, was examined after the cells were treated with Latrunculin-A (LA) to induce cell rounding (Fig. 5A). LA is a specific inhibitor of actin polymerization which has been used to induce rapid rounding of MCF-10A cells while allowing the cells to maintain attachment to the tissue culture dishes (8). The MCF-10A, PTEN+/−, and PTEN−/− cells treated with vehicle control in minimal assay media demonstrated similar, low levels of PARP cleavage. The MCF-10A.Bcl2 cells, with verified resistance to apoptosis, had undetectable PARP cleavage. Upon addition of LA, the MCF-10A parent line and PTEN+/− clones undergo significant PARP cleavage while the PTEN−/− cells maintain high levels of full-length PARP, similar to that of the MCF-10A.Bcl2 cells. To examine whether PTEN−/− cells exhibited a general resistance to apoptosis, all cells were treated with the death receptor ligand, TRAIL. Binding of TRAIL to transmembrane death receptors stimulates apoptosis via the extrinsic pathway, which is independent of Akt and the mitochondria. Within 2hrs of TRAIL treatment, all MCF-10A cells and variants began to undergo apoptosis confirmed by the increase of cleaved PARP levels. The apoptotic resistance of PTEN−/− cells therefore seems restricted to the intrinsic apoptosis pathway, since it cannot prevent apoptosis that occurs downstream of mitochondrial apoptosis signaling. Additionally, to determine anoikis resistance, the cells were plated over low-attachment plates. Without tissue culture treated plastic, the cells remained in suspension. After detachment for 24hrs, the MCF-10A parental cells and PTEN+/− clones undergo massive cell death demonstrated by high levels of cleaved PARP and the presence of a sub-G1 peak (Fig. 5B and C). The PTEN−/− cells maintained high levels of full-length PARP and a lower percentage of cells in sub-G1 indicating their resistance to anoikis. pERK levels are only very slightly elevated in the PTEN+/− clones, these cells maintained similar levels of PARP cleavage to the parental cells. Although, the first PTEN−/− clone revealed an increase in pERK levels compared to the other PTEN−/− clone, no differences in the levels of cleaved PARP were observed between clones. Therefore while MEK activation was required for continued cell growth (Fig. 4), the levels of ERK activation were relatively independent from apoptosis during detachment. It is more likely that AKT activation was responsible for the resistance to anoikis, since high levels of pAKT were maintained in the suspended PTEN−/− cells.
Figure 5. PTEN loss promotes resistance to apoptosis upon cell rounding and anoikis.
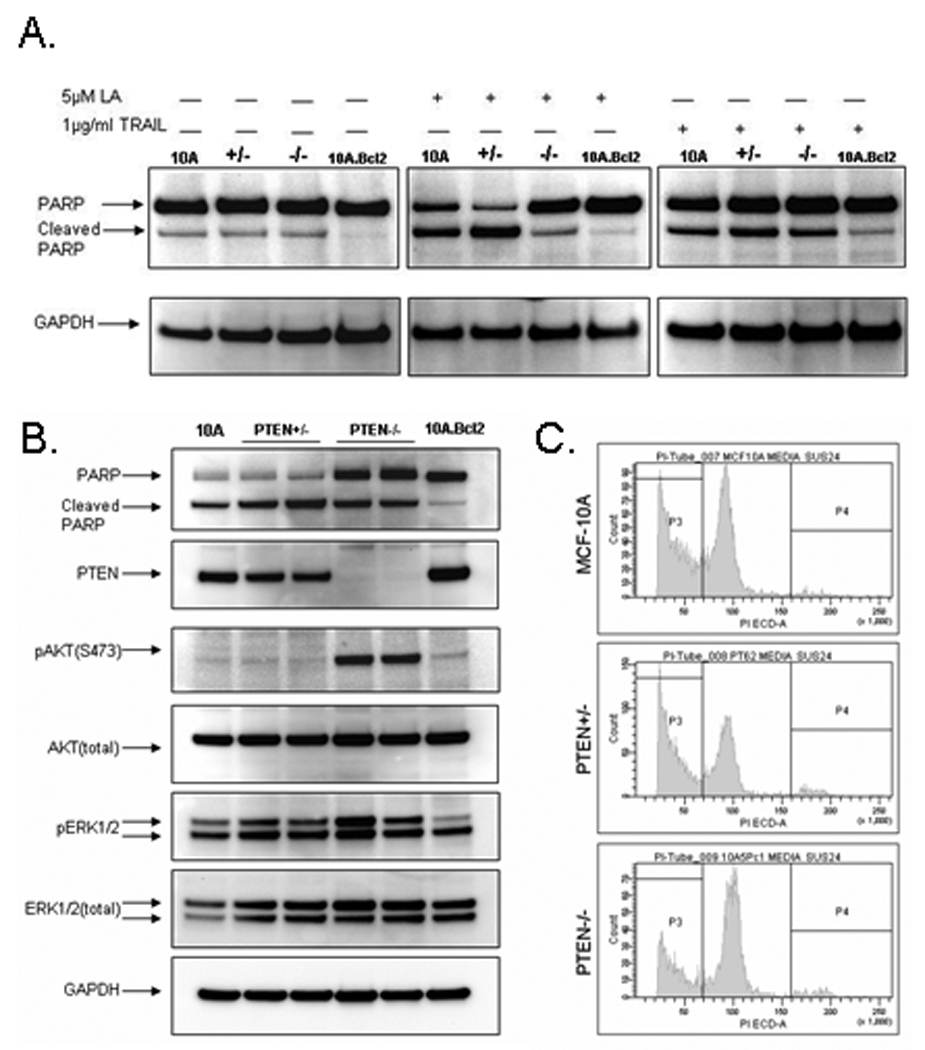
(A.) MCF-10A, a PTEN+/− clone, a PTEN−/− clone, and the MCF-10A.Bcl2 cells were plated in minimal assay media. The next day the media was changed to fresh minimal media +/− 5µM Latrunculin-A (LA) to induce cell rounding for 24hr or 1µg/ml TRAIL for 2hr. and harvested at the indicated times by direct addition of RIPA lysis buffer. (B.) MCF-10A, two representative PTEN+/− clones, two representative PTEN−/− clones, and the MCF-10A.Bcl2 cells were incubated in suspension in normal growth media for 24hr. (C.) Flow cytometry analysis of MCF-10A, PTEN+/− clone 1, and PTEN−/− clone 1 incubated in suspension in DMEM/F12 for 24hrs. A larger percentage of MCF-10A (42.8%) and the PTEN+/− cells (49.4%) undergo apoptosis than the PTEN−/− cells (29.6%).
PTEN loss sensitizes cells to the chemotherapeutic drug doxorubicin but not paclitaxel
Studies suggest that loss of PTEN expression correlate with poor prognosis, as well as resistance to chemotherapies (21, 22). To determine whether PTEN loss mediates chemotherapeutic resistance and increases cell survival, the PTEN isogenic MCF-10A cells were exposed to increasing concentrations of doxorubicin and paclitaxel. Primary normal breast epithelial cells are alive and metabolizing, but not actively proliferating. To mimic healthy, growth arrested epithelial cells, MCF-10A cells were plated at high density (1.5×104) per well in a 96-well plate in minimal assay media. After 24hrs, the cells attached as approximately a 90% confluent monolayer. Although PTEN−/− cells have the ability to grow in minimal media, they remain contact inhibited and growth arrest at confluence, therefore plating densities used matched those of the MCF-10A parental cells. Drugs were added after growth arrest to determine cell survival. Interestingly, the PTEN+/− and PTEN−/− cells responded differently to the drugs. Doxorubicin similarly reduced the percentage of surviving cells in both the PTEN+/− and PTEN−/− cells (Fig. 6A). Even at the low concentration of 5nM, doxorubicin reduced the PTEN+/− and PTEN−/− clones by 10 and 24%, respectively. No difference in susceptibility was observed between the isogenic cells following exposure to paclitaxel (Fig. 6B).
Figure 6. PTEN loss sensitizes mammary epithelial cells to the commonly used breast cancer chemotherapeutic doxorubicin but not to paclitaxel.
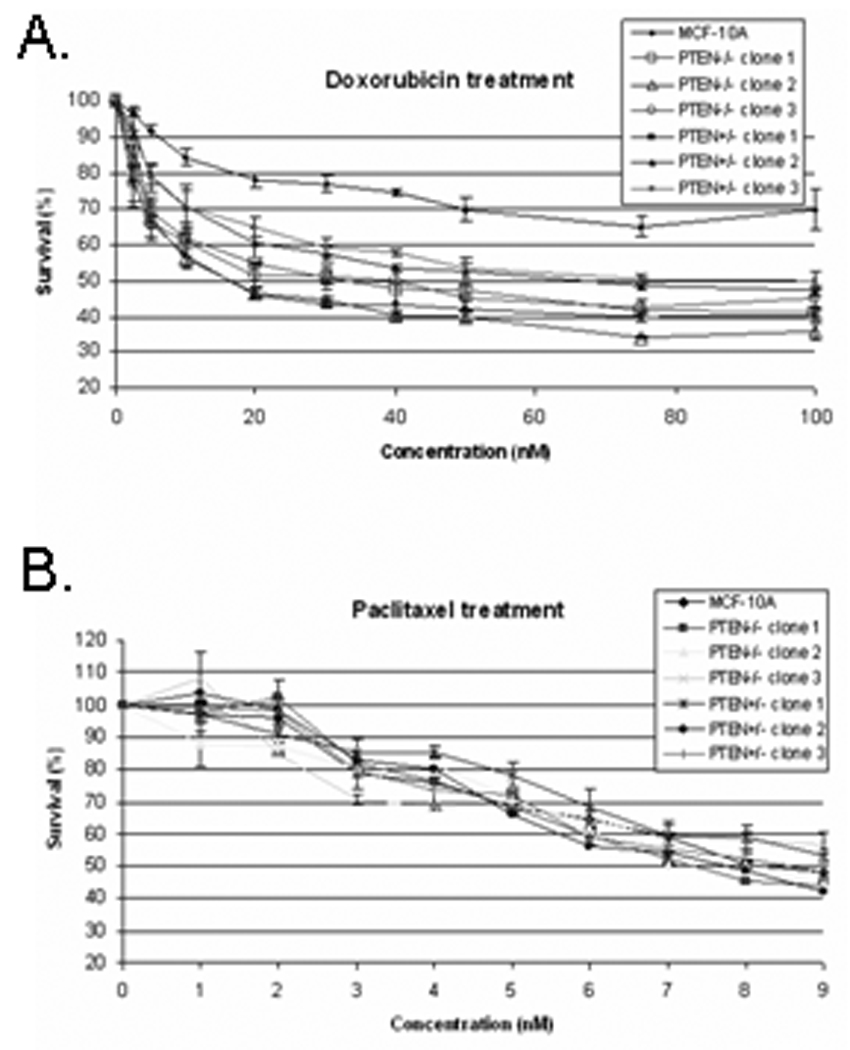
Cell growth of MCF-10A cells, PTEN+/−, and PTEN−/− clones after exposure to increasing doses of doxorubicin (A.) and paclitaxel (B.). Points, means of 3 independent experiments performed in quadruplicate; bars, SD.
Discussion
The loss of PTEN expression or the acquisition of activating PI3K mutations (PIK3CA) occurs in approximately 50–75% of breast cancers illustrating the importance of the PI3K pathway in breast cancer. Notably, loss of PTEN expression and PIK3CA mutation are mutually exclusive events (23), likely because PTEN and PI3K exist in a tight, regulatory loop, strictly controlling phosphatidylinositol levels. The loss of PTEN or acquisition of an activating mutation in PIK3CA are reciprocal alterations, either of which would result in increased levels of PIP3 and remove the selective pressure to convert PIP3 to its PIP2 counterpart. Our findings support the role of PTEN loss in breast cancer based on the ability of PTEN−/− cells to proliferate in the absence of growth factors and their resistance to anoikis. However, PTEN loss is insufficient to promote active tumorigenesis of the MCF-10A cells suggesting a need for other oncogenic events (24). This result is contradictory to recent data in which the overexpression of two clinically relevant PI3K mutations (H1047R and E545K) conferred anchorage-independent growth of MCF-10A cells in soft agar (25). However in these studies, the expression of the PIK3CA mutant cDNA is under control of a CMV promoter that may increase PIK3CA expression to levels not observed in primary tumors or derived cell lines. In support of this hypothesis, knock-in of the same PIK3CA activating mutations, H1074R and E545K, did not cause anchorage-independent growth of MCF-10A cells (26). Similar to MCF-10A PTEN−/− cells, knockin mutant PIK3CA cells were not tumorigenic, did not form colonies in soft agar, and did not alter acinar growth in 3-D Matrigel culture.
Although metastasis is the cause of 90% of human cancer deaths (27), the metastatic process presents numerous challenges to tumor cells, including apoptosis that results from detachment (anoikis) or cell shape change (amorphosis) (28). Resistance to apoptosis allows tumor cells to survive these challenges (29), but does not promote immediate tumor outgrowth at the secondary site, yielding a period of tumor dormancy (30). There is currently tremendous clinical interest in such dormant tumor cells, since their presence in the bloodstream strongly predicts poor patient outcome in breast cancer (31, 32). The importance of defining the mechanisms that promote tumor dormancy is also emphasized by the observation that breast tumor patients who are diagnosed early with no detectable regional metastases have over a 30% chance of recurrence when followed for 10–15 years (33, 34). Our results indicate that PTEN loss induces a dormant tumor cell phenotype by promoting resistance to apoptosis without inducing complete anchorage-independent growth. Recent evidence shows that MECs, which have not fully transformed to anchorage-independent growth, are still fully capable of metastasizing to the lung in a dormant state and then recurring once growth-initiating oncogenes are activated (35, 36). Systems based on fibroblasts or exogenous overexpression of PI3KCA display active tumor growth, while our system based on homologous knockout of PTEN in MCF-10A MECs more effectively models the dormant phenotype of carcinoma cells. However, such dormant tumor cells are typically difficult to treat with traditional chemotherapies, since they persist without active cell division. Defining which types of chemotherapy are able to effectively target tumor cells in such a dormant state will be critical to treating metastatic recurrence.
A variety of chemotherapeutic agents converge on a common final pathway leading to apoptotic cell death. Certain studies have shown that activation of the PI3K pathway enhances the survival of cancer cells in response to such agents and contribute to chemotherapy resistance. However, these previous studies employed an overexpressed, constituently active AKT1 (37, 38) which may not recapitulate the physiologically active AKT levels due to PTEN loss or PIK3CA mutations. While other studies overexpressed an oncogene such as constitutively-active Ras (39) or HER2 (40) in the MCF7 breast adenocarcinoma line which already contains a PIK3CA mutation (E545K) (41). Here, to more closely recapitulate physiologic levels active AKT, we used the MCF-10A isogenic PTEN knock-out clones to determine chemotherapeutic response to doxorubicin and paclitaxel. Surprisingly, the PTEN−/− clones were more susceptible to doxorubicin than their parental PTEN expressing counterparts. However, no difference in survival was observed between the isogenic clones when treated with paclitaxel. The susceptibility of the PTEN+/− and PTEN−/− cells to doxorubicin and not paclitaxel may be explained by the different mechanisms of action of each drug. Paclitaxel is a microtubule (MT) stabilizing compound which interferes with the normal breakdown of this cytoskeletal component. This drug immediately and adversely affects cell function as MT inherent dynamic instability is necessary for their function to transport other cellular components. Doxorubicin is known to intercalate within the DNA and inhibition of topoisomerase II progression, eliciting DNA damage. Upon DNA damage, normal cells growth arrest to either repair the damage or undergo apoptosis if the damage is substantial. However, constitutively active PI3K and pathway components have been shown to override DNA damage-induced cell arrest (42–44). Haploinsufficency and deletion of PTEN may allow for cell cycle progression and death due to massive DNA damage. Further work to elucidate the mechanisms by which PTEN expression loss may contribute to chemotherapy susceptibility is warranted.
There is a significant clinical relevance for the creation and characterization of this PTEN isogenic model system. Using this model, we have shown that PTEN expression loss in MECs results in constitutive AKT activation and induces multiple phenotypic alterations characteristic of breast tumor cells, including growth factor–independent proliferation and protection from anoikis. PTEN expression loss also confers increased susceptibility to doxorubicin but not paclitaxel. Together, these data support the notion that the cancer-associated PTEN expression loss may significantly contribute to breast cancer cell survival and tumor dormancy.
Supplementary Material
Acknowledgements
This work was supported by the Susan G. Komen Breast Cancer Foundation Fellowship, PDF104506 (MIV), T32-DK067872 from the NIH, National Institute of Diabetes And Digestive And Kidney Diseases (MBW), R01-CA115699 from the National Cancer Institute (TW), R01-CA124704 from the National Cancer Institute (SSM), the Breast Cancer Research Foundation (BHP), R01-CA107331 and R01-GM58888 from the National Cancer Institute and General Medicine at NIH (DJW). We thank Agnes Cheung for developing the MCF-10A.Bcl2 cell line and Regina Harley for flow cytometry experiments.
REFERENCES
- 1.Bruni P, Boccia A, Baldassarre G, et al. PTEN expression is reduced in a subset of sporadic thyroid carcinomas: evidence that PTEN-growth suppressing activity in thyroid cancer cells mediated by p27kip1. Oncogene. 2000;19:3146–3155. doi: 10.1038/sj.onc.1203633. [DOI] [PubMed] [Google Scholar]
- 2.Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol. 2001;14:672–676. doi: 10.1038/modpathol.3880371. [DOI] [PubMed] [Google Scholar]
- 3.Ebert MP, Fei G, Schandl L, et al. Reduced PTEN expression in the pancreas overexpressing transforming growth factor-beta 1. Br J Cancer. 2002;86:257–262. doi: 10.1038/sj.bjc.6600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurose K, Zhou XP, Araki T, et al. Frequent loss of PTEN expression is linked to elevated phosphorylated Akt levels, but not associated with p27 and cyclin D1 expression, in primary epithelial ovarian carcinomas. Am J Pathol. 2001;158:2097–2106. doi: 10.1016/S0002-9440(10)64681-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perren A, Weng LP, Boag AH, et al. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol. 1999;155:1253–1260. doi: 10.1016/S0002-9440(10)65227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 7.Marsh DJ, Zheng Z, Zedenius J, et al. Differential loss of heterozygosity in the region of the Cowden locus within 10q22-23 in follicular thyroid adenomas and carcinomas. Cancer Res. 1997;57:500–503. [PubMed] [Google Scholar]
- 8.Martin SS, Leder P. Human MCF10A mammary epithelial cells undergo apoptosis following actin depolymerization that is independent of attachment and rescued by Bcl-2. Mol Cell Biol. 2001;21:6529–6536. doi: 10.1128/MCB.21.19.6529-6536.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinkas J, Martin SS, Leder P. Bcl-2-mediated cell survival promotes metastasis of EpH4 betaMEKDD mammary epithelial cells. Mol Cancer Res. 2004;2:551–556. [PubMed] [Google Scholar]
- 10.Lee C, Kim JS, Waldman T. PTEN gene targeting reveals a radiation-induced size checkpoint in human cancer cells. Cancer Res. 2004;64:6906–6914. doi: 10.1158/0008-5472.CAN-04-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topaloglu O, Hurley PJ, Yildirim O, Civin CI, Bunz F. Improved methods for the generation of human gene knockout and knockin cell lines. Nucleic Acids Res. 2005;33:e158. doi: 10.1093/nar/gni160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rago C, Vogelstein B, Bunz F. Genetic knockouts and knockins in human somatic cells. Nat Protoc. 2007;2:2734–2746. doi: 10.1038/nprot.2007.408. [DOI] [PubMed] [Google Scholar]
- 13.Garcia JM, Silva J, Pena C, et al. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer. 2004;41:117–124. doi: 10.1002/gcc.20062. [DOI] [PubMed] [Google Scholar]
- 14.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 15.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 16.Vranic S, Bilalovic N, Lee LM, et al. PIK3CA and PTEN mutations in adenoid cystic carcinoma of the breast metastatic to kidney. Hum Pathol. 2007;38:1425–1431. doi: 10.1016/j.humpath.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 17.Janssen EA, Soiland H, Skaland I, et al. Comparing the prognostic value of PTEN and Akt expression with the Mitotic Activity Index in adjuvant chemotherapy-treated node-negative breast cancer patients aged <55 years. Cell Oncol. 2007;29:25–35. doi: 10.1155/2007/569246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitz M, Grignard G, Margue C, et al. Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int J Cancer. 2007;120:1284–1292. doi: 10.1002/ijc.22359. [DOI] [PubMed] [Google Scholar]
- 19.Shin SI, Freedman VH, Risser R, Pollack R. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc Natl Acad Sci U S A. 1975;72:4435–4439. doi: 10.1073/pnas.72.11.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. Embo J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell RA, Bhat-Nakshatri P, Patel NM, et al. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 22.Garcia JM, Silva JM, Dominguez G, et al. Allelic loss of the PTEN region (10q23) in breast carcinomas of poor pathophenotype. Breast Cancer Res Treat. 1999;57:237–243. doi: 10.1023/a:1006273516976. [DOI] [PubMed] [Google Scholar]
- 23.Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–2559. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- 24.Ciardiello F, Gottardis M, Basolo F, et al. Additive effects of c-erbB-2, c-Haras, and transforming growth factor-alpha genes on in vitro transformation of human mammary epithelial cells. Mol Carcinog. 1992;6:43–52. doi: 10.1002/mc.2940060108. [DOI] [PubMed] [Google Scholar]
- 25.Isakoff SJ, Engelman JA, Irie HY, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 26.Gustin JP, Karakas B, Weiss MB, et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci U S A. 2009;106:2835–2840. doi: 10.1073/pnas.0813351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 28.Martin SS, Vuori K. Regulation of Bcl-2 proteins during anoikis and amorphosis. Biochim Biophys Acta. 2004;1692:145–157. doi: 10.1016/j.bbamcr.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 30.Martin SS, Ridgeway AG, Pinkas J, et al. A cytoskeleton-based functional genetic screen identifies Bcl-xL as an enhancer of metastasis, but not primary tumor growth. Oncogene. 2004;23:4641–4645. doi: 10.1038/sj.onc.1207595. [DOI] [PubMed] [Google Scholar]
- 31.Riethdorf S, Wikman H, Pantel K. Review: Biological relevance of disseminated tumor cells in cancer patients. Int J Cancer. 2008;123:1991–2006. doi: 10.1002/ijc.23825. [DOI] [PubMed] [Google Scholar]
- 32.Riethmuller G, Klein CA. Early cancer cell dissemination and late metastatic relapse: clinical reflections and biological approaches to the dormancy problem in patients. Semin Cancer Biol. 2001;11:307–311. doi: 10.1006/scbi.2001.0386. [DOI] [PubMed] [Google Scholar]
- 33.Fisher B, Jeong JH, Dignam J, et al. Findings from recent National Surgical Adjuvant Breast and Bowel Project adjuvant studies in stage I breast cancer. J Natl Cancer Inst Monogr. 2001:62–66. doi: 10.1093/oxfordjournals.jncimonographs.a003463. [DOI] [PubMed] [Google Scholar]
- 34.Wallgren A, Bonetti M, Gelber RD, et al. Risk factors for locoregional recurrence among breast cancer patients: results from International Breast Cancer Study Group Trials I through VII. J Clin Oncol. 2003;21:1205–1213. doi: 10.1200/JCO.2003.03.130. [DOI] [PubMed] [Google Scholar]
- 35.Podsypanina K, Du YC, Jechlinger M, et al. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321:1841–1844. doi: 10.1126/science.1161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinberg RA. Leaving home early: reexamination of the canonical models of tumor progression. Cancer Cell. 2008;14:283–284. doi: 10.1016/j.ccr.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt M, Hovelmann S, Beckers TL. A novel form of constitutively active farnesylated Akt1 prevents mammary epithelial cells from anoikis and suppresses chemotherapy-induced apoptosis. Br J Cancer. 2002;87:924–932. doi: 10.1038/sj.bjc.6600566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.VanderWeele DJ, Zhou R, Rudin CM. Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther. 2004;3:1605–1613. [PubMed] [Google Scholar]
- 39.Jin W, Wu L, Liang K, et al. Roles of the PI-3K and MEK pathways in Ras-mediated chemoresistance in breast cancer cells. Br J Cancer. 2003;89:185–191. doi: 10.1038/sj.bjc.6601048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knuefermann C, Lu Y, Liu B, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22:3205–3212. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- 41.Bachman KE, Argani P, Samuels Y, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3:772–775. doi: 10.4161/cbt.3.8.994. [DOI] [PubMed] [Google Scholar]
- 42.Kandel ES, Skeen J, Majewski N, et al. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol. 2002;22:7831–7841. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhan Q, Antinore MJ, Wang XW, et al. Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene. 1999;18:2892–2900. doi: 10.1038/sj.onc.1202667. [DOI] [PubMed] [Google Scholar]
- 44.Shtivelman E, Sussman J, Stokoe D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr Biol. 2002;12:919–924. doi: 10.1016/s0960-9822(02)00843-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.