

Abstract
Abnormal mesolimbic control of cortical cholinergic activity has been hypothesized to contribute to the cognitive symptoms of schizophrenia. Stimulation of NMDA receptors in nucleus accumbens (NAC) increases acetylcholine (ACh) release in prefrontal cortex (PFC), an activation thought to contribute to attentional processing. Thus, the effects of intra-NAC perfusion of NMDA (250–400 μM) on ACh release in PFC were determined in rats receiving lesions of the ventral hippocampus (VH) as neonates (nVHLX), a neurodevelopmental model of schizophrenia, or as adults (aVHLX). NMDA elevated ACh release (100–150% of baseline) in adults sham-lesioned as neonates or in aVHLX rats. Adult nVHLX were unresponsive to NAC NMDA receptor stimulation. The inability of nVHLX to respond to NMDA emerged over development as a separate experiment demonstrated that evoked ACh release was normal prior to puberty (100–150% increase) yet, in these same nVHLX animals, absent after puberty. Amphetamine-evoked ACh release was assessed in nVHLX animals to exclude potential limitations in release capacity. Amphetamine produced greater increases in ACh release than in shams, indicating that nVHLX does not impair the capacity of cholinergic neurons to release ACh. Finally, the ability of 13 days of pretreatment with clozapine (1.25 mg/kg/day) to reinstate NMDA-evoked cortical ACh efflux was determined. Clozapine treatment normalized NMDA-evoked ACh release in nVHLX animals. These experiments reveal that mesolimbic regulation of cortical ACh release is disrupted in post-pubertal nVHLX rats and normalized by low-dose treatment of clozapine; supporting the usefulness of nVHLX animals for research on the neuronal mechanisms underlying the cognitive symptoms of schizophrenia.
Keywords: acetylcholine, hippocampus, prefrontal cortex, nucleus accumbens, basal forebrain, schizophrenia, clozapine
Persistent cognitive impairments have been increasingly considered a major hindrance for patients with schizophrenia to return to a productive life. The NIMH-sponsored initiatives MATRICS and CNTRICS (Kerns, et al 2008; Barch, et al 2009) assisted in bringing this issue back to the forefront, focused the attention of researchers on specific cognitive domains and tasks and, importantly, emphasized the importance of developing more animal models of these impairments that are broadly valid and useful (Hagan and Jones 2005; Nuechterlein, et al 2004).
Since the original descriptions by Kraepelin, impairments in attention have been considered a central and perhaps even fundamental component of the cognitive symptoms of schizophrenia (for review see Nuechterlein and Dawson 1984; Braff and Light 2004). As cortical cholinergic neurotransmission has been extensively demonstrated to be necessary for the mediation of attentional performance (for review see Sarter, et al 2005a), we hypothesized that a dysregulated cortical cholinergic input system is an integral component of the forebrain circuitry responsible for mediating the persistent cognitive impairments of schizophrenia (for review see Sarter, et al 2005b). Although evidence indicating abnormal cholinergic neurotransmission in schizophrenia has been accumulating (Hyde and Crook 2001; Raedler, et al 2003), it has remained difficult to determine the presumably dynamic characteristics of such dysregulation in humans based on post-mortem analyses, thus presently limiting this research mostly to studies using animal models (Kozak, et al 2007; Sarter, et al 2009).
Research in intact animals has revealed a critical role for a distributed, interconnected neural system consisting mainly of the PFC, NAC, and the area in the basal forebrain from which the forebrain cholinergic system originates (BFCS), in the regulation of cortical ACh release (Brooks, et al 2007; Zmarowski, et al 2005) and the mediation of attention (Sarter, et al 2005a, b; Himmelheber, et al 2000; Kozak, et al 2006). Specifically, we found that activation of NAC NMDA receptors stimulates ACh release in PFC (Brooks, et al 2007; Zmarowski, et al 2005). Moreover, this evoked release was evident in prefrontal but not in posterior parietal cortex (Zmarowski, et al 2007). Effective attentional processing, including the motivated maintenance or recovery of attentional performance following performance-taxing events (such as distractors), involves the mesolimbic recruitment of the BFCS (Sarter, et al 2006). This is of relevance for understanding the neuronal mechanisms mediating the attentional impairments in schizophrenics because during periods of remission, such impairments are robustly evoked by performance-taxing demands (reviewed in Sarter, et al 2009). Thus, the present experiment assessed this interplay between the NAC and PFC ACh release in an animal model of schizophrenia.
In the present study, we chose to focus on the model of damage to the developing ventral hippocampus (VH; Lipska, et al 1993; O’Donnell, et al 2002) because a) projections from VH have been shown to influence the operations of the PFC (Bhardwaj, et al 2003; Lipska, et al 2003; Tseng, et al 2006) and b) hippocampal efferents play an important role in gating the responsiveness of NAC neurons to prefrontal inputs (O’Donnell and Grace 1995; Goto and O’Donnell 2002). Furthermore, neonatal lesions of the VH (nVHLX) result in abnormal dopamine-glutamate/GABA interactions in the PFC (Tseng, et al 2007; Tseng, et al 2008). Although the determination of the attentional capacities of nVHLX animals requires more research, the available evidence indicates that such impairments are present (Marquis, et al 2008). Thus, nVHLX animals model the abnormal prefrontal-mesolimbic interactions that are generally hypothesized to represent a core component of the disease. We hypothesized that these abnormal interactions also include the BFCS and prefrontal cholinergic inputs. The evidence described below confirms that the mesolimbic regulation of PFC ACh release is fundamentally disrupted in nVHLX animals, further supporting the usefulness of this model for studying the neuronal mechanisms responsible for the cognitive symptoms of schizophrenia.
METHODS AND MATERIALS
Animals
Male Wistar rats (Charles River, Wilmington, MA, USA) were used. Neonatal rats were bred in our colony and were weaned on postnatal day (PND) 21. Animals were maintained on a 12:12 h light/dark cycle (lights on 0600 h) in a temperature- and humidity-controlled AAALAC-approved animal facility. Animals were individually housed in plastic cages lined with corn cob bedding (Harlan Teklad, Madison, WI, USA) with ad libitum access to food and water. All procedures were approved by The Ohio State University Institutional Animal Care and Use Committee in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Neonatal Ventral Hippocampal Lesions (nVHLX)
Neonatal surgery was performed when pups weighed 15–20 g (~PND 7–9). Pups were anaesthetized by hypothermia until they were immobile. The ventral hippocampal lesion was made by penetrating the skull with an infusion syringe (10 μL syringe, 26G needle) at coordinates AP −3.0 mm, ML ±3.5 mm, DV −5.0 mm relative to Bregma. Ibotenic acid (Sigma-Aldrich, St. Louis, MO; 10 μM) was then infused into the VH at a rate of 0.15 μL/min for a total of 0.3 μL. After the infusion was complete, the needle was left in place for an additional 3 min to allow diffusion of the excitotoxin and prevent backflow up the needle tract. This procedure was then repeated in the contralateral hemisphere. For sham-surgery, 0.3 μL of artificial cerebrospinal fluid (aCSF, see below) was infused at the same rate. After surgery, the pups were warmed on a heating pad and returned as a group to the litter’s nest area. Pups and dams were left undisturbed until weaning on PND 21.
Adult Ventral Hippocampal Lesions (aVHLX)
Adult male Wistar rats (PND 64–75 at the time of lesion) were used. Animals were anesthetized with inhalant isofluorane (2%, 0.6 L/min, O2) and 0.3 μL ibotenic acid (10 μM) was bilaterally infused (2 locations/hemisphere) into the VH at a rate of 0.15 μL/min at the following coordinates (in mm from Bregma): AP −4.4, ML ± 5.0, DV −6.0 and AP −4.4, ML ± 5.0, DV −8.0. After each infusion, the needle remained in place for 3 min before being removed. Following surgery, the animals were returned to their home cages and left undisturbed for 28 days.
Guide Cannula Implantation
Animals to be tested pre-puberty were implanted with microdialysis guide cannula on approximately PND 30. Animals were anesthetized using isoflurane. Rats were implanted unilaterally with stainless steel guide cannulae (SciPro Inc., Sanborn, NY, USA) in the mPFC and the NAC. The mPFC guide cannula (0.38 mm o.d., 15 mm shaft) was implanted at a 20° rostral angle at AP: +4.2, ML: ±0.6, DV: −0.6 mm relative to Bregma. The NAC cannula (0.38 mm o.d., 20 mm shaft) was implanted at a 0° (i.e. vertical) angle at AP: +1.5, ML ±0.8, DV −5.6 mm relative to Bregma. Cannulae were fixed to the skull using skull screws and dental cement. In order to prevent clogging of the cannulae shafts, stylets ending flush with the guide cannulae were inserted. Antibiotic ointment (Neosporin) was applied to the area around the incision, and the animals were returned to their home cages.
Animals to be tested post-puberty were cannulated between PND 56 and 70. Surgical procedures and cannula angles were the same as those described for pre-puberty cannulation, with the following coordinates: PFC, AP +4.2, ML ±0.6, DV −0.6 mm; NAC, AP +1.3, ML ±1.0, DV −5.8 mm relative to Bregma.
General Microdialysis Procedures
The effects of NAC infusions, amphetamine, or clozapine, and their respective vehicles, were all tested as within-subjects variables and in with counterbalanced order of drug and vehicle. Animals had at least one day between two successive microdialysis sessions. During a standard microdialysis session, animals were allowed to habituate to the test chamber (microdialysis bowl: diameter = 39 cm, height = 32cm, Carnegie Medicine, Stockholm, Sweden) for 15 min, after which concentric dialysis probes (mPFC: 0.2 mm o.d., 3.0 mm membrane tip; NAC: 0.2 mm o.d., 2.0 mm membrane tip; SciPro Inc.) were inserted into the guide cannulae. Animals were then perfused with aCSF [NaCl (126.5 mM), NaHCO3 (27.5 mM), KCl (2.4 mM), Na2SO4 (0.5 mM), KH2PO4 (0.5 mM), CaCl2 (1.1 mM), MgCl2 (0.8 mM), dextrose (1 mM)], at a 1.25 – 2.0 μL/min flow rate. The perfusion medium did not include an acetylcholinesterase inhibitor. Following probe insertion, a 3-hr washout period was observed to allow for stable levels of basal ACh release. After this washout period, four baseline collections were taken in 15-min intervals. Following baseline collections, drugs were perfused into the NAC over the course of one hour, during which four collections were taken. After drug perfusion, aCSF was once again perfused through the NAC probe and three additional collections were taken to determine if ACh returned to basal levels. Immediately following session completion, probes were removed from the cannulae and stylets were replaced in the cannulae. Animals were then returned to their home cages. Between dialysis sessions, each dialysis probe was stored in an antibacterial agent (ProClin 150 Reagent, BAS, Inc., W. Lafeyett, IN, USA) to ensure sterility.
Specific Microdialysis Procedures for Individual Experiments
Experiment 1a: Animals Lesioned as Neonates and Tested as Adults
Animals sustaining ventral hippocampal lesions as neonates (nVHLX) and their sham controls were tested as adults in two 7-hour dialysis sessions as described in General Microdialysis, consisting of intra-NAC perfusion of aCSF or NMDA (400 μM) in counterbalanced order (see figure legends for number of animals per group). This concentration of NMDA was the higher concentration tested in Zmarowksi et al., 2007 and although not producing greater cortical ACh release than the lower concentration (250 μM), was selected for Experiment 1a in order to minimize a potential false-negative finding. As this higher concentration did not evoke PFC ACh release in nVHX animals, subsequent experiments were conducted with the lower concentration, in part in order to minimize non-selective effects (see Zmarowski et al., 2005, 2007).
Experiment 1b: Animals Lesioned as Adults and Tested as Adults
Animals sustaining ventral hippocampal lesions as adults (aVHLX) and their sham controls were tested as adults in two 7-hour sessions as described in General Microdialysis, consisting of intra-NAC perfusion of aCSF or NMDA (250 μM) in counterbalanced order.
Experiment 2: Animals Lesioned as Neonates and Tested Before and After Puberty
nVHLX rats and their sham controls were tested in five, 7 hour sessions, as described in General Microdialysis. Each animal received two dialysis sessions before puberty (PND 30 – 38) and three sessions after puberty (PND 56 – 64). The pre-puberty sessions included aCSF or NMDA (250 μM) perfused into the NAC in counterbalanced order. The post-puberty sessions included aCSF, NMDA (250 μM), or systemic amphetamine (AMPH, 2 mg/kg, i.p.) presented in counterbalanced order.
Experiment 3: Treatment with Clozapine
At PND 58, nVHLX and their sham controls were randomly divided into two drug pretreatment groups. Animals received once daily injections (i.p.) of either clozapine (1.25 mg/kg, pH = 7.4, Sigma-Aldridge, St. Louis, MO) or saline. The selection of clozapine dose guided by several observations. First, studies in schizophrenic patients demonstrated moderately beneficial cognitive effects following low-dose treatment with first- and second-generation antipsychotics (see Mishara and Goldberg, 2004 for a meta-analysis). Second, we have demonstrated that low-dose treatment of clozapine (2.5 mg/kg) in rats, defined in terms of 50% D2 receptor occupation (Kapur et al., 2003), enhances the attentional performance of rats in a different model of schizophrenia (Martinez and Sarter, 2008; Rueter et al., 2004). Third, as we and others (Ichikawa et al., 2002; Li et al., 2005) observed that doses higher than 1.25 mg/kg increased basal cortical ACh release (an effect that would have confounded this experiment), we further reduced the dose to 1.25 mg/kg. Injections continued for 13 days with guide cannulae implantation surgery occurring on injection day 7. Microdialysis testing occurred on day 11 and 13 of drug treatment injection regimen with injections given immediately prior to dialysis probe insertion. Dialysis sessions for this experimental group were identical to those described in Experiment 1 with each subject receiving, in counterbalanced order, aCSF or NMDA (250 μM) during drug perfusion collections.
ACh Quantification
ACh samples were stored (−80° C) until analyzed using high performance liquid chromatography (HPLC) with electrochemical detection. Using an autosampler (ESA Inc., Chelmsford, MA), 15 μL of each sample was injected through a UniJet microbore analytical column (1 × 50 mm, BAS Inc.) using a sodium phosphate mobile phase (35 mM Na2HPO4, 483.55 μM EDTA, 0.005% microbicide reagent ProClin, pH = 8.5; flow rate of 0.15 ml/min) to separate ACh and choline. The sensitivity of the ACh assay was enhanced by using a pre-column immobilized enzyme reactor (IMER; BAS, Inc.), containing choline oxidase and catalase, to oxidize extracellular choline. Following separation of ACh, a post-column enzyme reactor (BAS, Inc.), was used to convert ACh into the reporting molecule H2O2 (Potter et al., 1983). H2O2 was quantified with a peroxidase-wired glassy carbon electrode (Model #5041 analytical cell, ESA Inc.) with an applied potential of −200 mV (Huang et al., 1995).
Histological Procedures
At the end of each experiment, animals were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.), intracardially perfused first with heparinized saline and then with formalin. The brains were extracted, prepared with a 30% sucrose solution, frozen, sliced with a cryostat, mounted on microscope slides, and stained with cresyl violet for verification of cannulae placements and extent of lesion.
Statistical Analyses
In each experiment, baseline (collections 1–4) values of extracellular ACh (fmol/15 μL) were compared using one-way repeated measures analysis of variance (ANOVA) for differences as a function of dialysis session or treatment group. In the absence of significant differences, subsequent drug effects were expressed as a percentage of the mean baseline for the treatment group. For each experiment, an overall ANOVA was conducted on ACh values using a two-way, within-subjects ANOVA with drug GROUP and TIME as within-subjects measures. In Experiment 2, an overall three-way ANOVA was initially conducted on within-subject factors of GROUP, TIME, and AGE followed by separate two-way ANOVAs at each age to reveal the source of certain interactions. In all ANOVAs, the Huynh-Feldt correction was utilized to reduce Type I errors associated with repeated measures ANOVAs (Vasey and Thayer, 1987). When appropriate, a minimum number of post-hoc comparisons were conducted using t-tests (alpha = 0.05 for all comparisons). All statistical tests were performed using SPSS for Windows (V15.0; SPSS Inc. Chicago, IL).
RESULTS
Ibotenic acid-induced lesions of ventral hippocampus
Figure 1 shows photomicrographs depicting representative coronal sections of the ventral hippocampal region in the brains of animals from each of the four lesion conditions; adult sham surgery (a), lesioned as adults (b), neonatal sham surgery (c), and neonatal lesions (d). At both ages, infusions of ibotenic acid led to comparable lesions within the ventral CA3 and CA1 portions of the hippocampus as well as the ventral dentate gyrus and subiculum. Infusions of vehicle in shams did not result in appreciable damage to the VH. Only rats with these anatomical profiles were included in the results.
Figure 1.

Representative coronal sections of the ventral hippocampal region of animals that received sham-lesions as adults (a) or neonates (c), or infusions of ibotenic acid as adults (b) or neonates (d; scales: 200 μm; all 4 sections are from Bregma AP levels −4.5 – −4.8 mm). At both ages, the lesions removed the ventral CA3 and CA1 portions of the hippocampus and extended posteriorly to include the ventral dentate gyrus and subiculum. Damage to the adjacent piriform (Pir) and enthorinal cortex remained generally very limited. Additional landmark structure indicated on the sections from sham-lesioned brains are the dorsal lateral geniculate nucleus (DLG), the ventral part of the medial geniculate nucleus (MGV) as well as the substantia nigra (SN).
Experiment 1a: NMDA-evoked ACh release in nVHLX tested as adults
The effects of NAC perfusion of NMDA on prefrontal ACh release in rats with sham or ibotenic acid (nVHLX) as neonates and tested as adults are illustrated in Figure 2. Mean (± S.E.M.) absolute basal ACh release across the four treatment groups was 5.07 ± 0.27 fmol/15 μL. As basal ACh release did not differ among the groups (F3,42 = 0.94, P = 0.43), subsequent data were expressed as a percent change from each group’s baseline. An overall analysis revealed that ACh efflux varied as a function of lesion group, drug infused, and time (collection interval) (F10,130 = 2.60, P = 0.006). Subsequent analyses revealed that the response to drug infusions differed significantly between sham and lesioned rats (F1,13 = 17.37, P = 0.001). In response to NMDA infusions, cortical ACh release in sham-lesioned rats markedly increased and remained elevated throughout the period of NMDA infusion and for one additional collection interval thereafter (F10,80 = 3.91, P < 0.001). Stimulated levels returned to baseline levels during the 10th collection (t8 = −1.10, P = 0.30). In striking contrast, in nVHLX rats, infusions of NMDA did not increase PFC ACh release, as indicated by the absence of a significant difference between the effects of perfusion of NMDA and aCSF (F10,50 = 1.94, P = 0.120).
Figure 2.

Mean (± S.E.M.) ACh efflux in the mPFC of sham-lesioned controls (n = 9) and nVHLX rats (n = 6) receiving, in pseudo-randomized order, perfusions of vehicle (aCSF) or NMDA (400 μM) into the NAC shell during two separate dialysis sessions as adults. Following baseline collections (1–4), aCSF or NMDA was perfused for 1 hr (5–8). Following drug perfusion, aCSF was re-perfused for 45 min (9–11) until the conclusion of the dialysis session. In sham animals, perfusion of NMDA produced a robust increase in cortical ACh efflux above that observed during aCSF perfusion. In nVHLX animals, however, perfusion of NMDA failed to increase cortical ACh efflux above baseline levels.
Experiment 1b: NMDA-evoked ACh release in aVHLX rats
This experiment determined whether the ventral hippocampal lesion-induced attenuation of the NAC NMDA-induced increases in PFC ACh release depended upon the age at which the VH was lesioned. The effects of intra-NAC perfusion of NMDA or the aCSF control on ACh efflux in PFC, in rats receiving hippocampal lesions as adults (aVHX), are depicted in Figure 3. Mean (± S.E.M.) absolute basal ACh efflux across the four treatment groups was 5.78 ± 0.33 fmol/15 μL. As there were no significant differences among the groups in basal efflux (F3,36 = 0.94, P = 0.43), subsequent data were expressed as a percent change from each group’s baseline. In contrast to rats lesioned as neonates, NMDA-evoked ACh release in aVHLX animals was identical to that seen in sham-lesioned controls (F1,12 = 0.01, P = 0.94). Both lesioned and control rats exhibited comparable elevations of ACh following NMDA (F1,12 = 0.10, P = 0.76).
Figure 3.
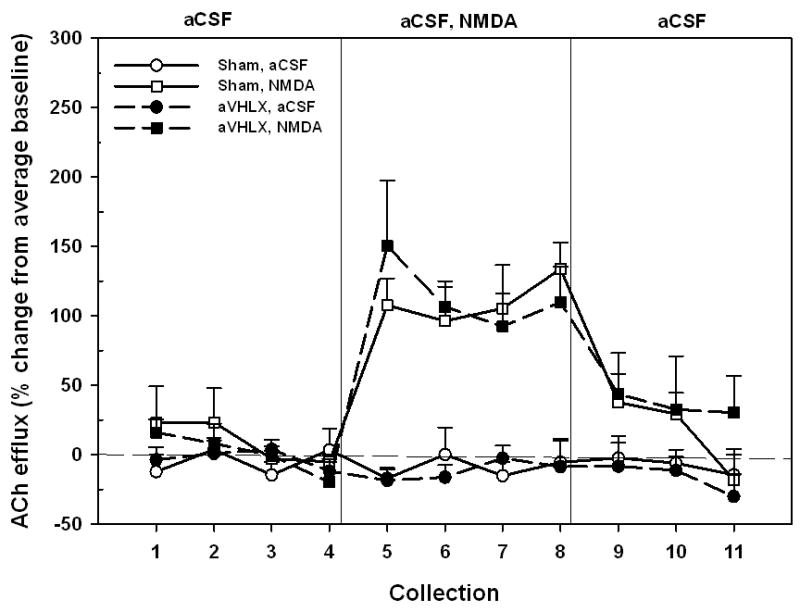
Mean (± S.E.M.) ACh efflux in the mPFC of sham-lesioned controls (n = 6) and aVHLX rats (n = 6) receiving, in pseudo-randomized order, perfusions of vehicle (aCSF) or NMDA (250 μM) into the NAC shell during two separate dialysis sessions. Following baseline collections (1–4), aCSF or NMDA was perfused for 1 hr (5–8). Following drug perfusion, aCSF was re-perfused for 45 min (9–11) until the conclusion of the dialysis session. Perfusion of NMDA resulted in a sustained increase in cortical ACh efflux, above that observed during aCSF perfusion, in both sham and aVHLX animals. There was no effect of lesion on the NMDA-stimulated release.
Experiment 2: Emergence of Cholinergic Dysregulation Post-Puberty
This experiment determined whether the attenuated ability of accumbens NMDA receptor activity to stimulate prefrontal ACh release in nVHX rats emerged gradually over the course of development. The same groups of nVHLX rats, and their sham controls, were tested for sensitivity to intra-accumbens NMDA infusions both pre- (days 30–38) and post-puberty (days 56–64) and the results are summarized in Figure 4. Furthermore, this experiment utilized a more demanding but also more stringent within-subject experimental design to reproduce the collective evidence obtained from Experiment 1. Both experimental approaches are necessary in order to exclude potential confounds that may be based on interactions between the lesions and repeated microdialysis sessions that are separated by an average of 26 days (Experiment 2) or on interactions between differential periods between the time of the lesion and cannulation surgery and the first insertion of a probe (Experiment 1).
Figure 4.

Mean (± S.E.M.) ACh efflux in the mPFC of sham-lesioned controls (n = 6) and nVHLX rats (n = 6) tested at pre-pubertal (panel a; Days 30–38) and again at post-pubertal ages (panel b; Days 56–64) for the effects of intra-NAC perfusions of aCSF or NMDA (250 μM). When rats were tested pre-puberty, both sham and nVHLX exhibited an NMDA-stimulated release of ACh (collections 5–8). However, when these same rats were retested post-puberty, the responsivity of nVHLX rats to NMDA was lost and only sham-lesioned rats exhibited a drug-induced stimulation of ACh.
Basal ACh levels in dialysates obtained pre- versus post-puberty did not differ among sham-lesioned and lesioned animals (F12,120 = 1.86, P = 0.07; 5.11 ± 0.19 fmol/15 μL). For clarity of presentation, the data for animals tested pre-puberty (Fig. 4, top) are presented separately from those obtained from these same animals tested post-puberty (Fig. 4, bottom). An omnibus ANOVA revealed significant differences in ACh release as a function of lesion condition, NMDA/aCSF infusion, collection interval and time of testing (F10,100 = 2.67, P = 0.01). More targeted comparisons indicated that intra-NAC perfusion of NMDA stimulated prefrontal ACh release in both sham-lesioned and lesioned rats when tested pre-puberty (top panel; F10,100 = 4.31, P = 0.002). However, there was no overall difference between the two lesion conditions (F1,10 = 0.32, P = 0.58). In contrast, the data depicted in the bottom panel resembled our previous results (Fig. 2) in nVHLX rats tested as adults. When tested post-puberty, the response to NMDA perfusion differed markedly between sham controls and rats lesioned as neonates (F1,10 = 66.56, P < 0.001). Finally, this developmental change in the ability of nVHLX rats to respond to NMDA is highlighted by differences in a direct comparison between lesioned animals tested pre-puberty and these same rats tested post-puberty (F10,50 = 2.88, P = 0.01). The collective evidence from Experiments 1 and 2 conclusively indicates that the inability of intra-NAC NMDA to stimulate prefrontal ACh release in nVHLX animals emerges only after puberty.
Effects of systemic amphetamine
In light of the complete lack of response to NMDA in the nVHLX groups tested as adults (Fig. 2) or post-puberty (Fig. 4, bottom), an additional microdialysis session was conducted in the post-pubescent rats in order to determine whether the cortical cholinergic transmission of these nVHLX rats was sensitive to other stimulatory conditions and, in particular, to one that has been demonstrated not to require NAC activity (Arnold, et al 2000). The ability of systemically-administered amphetamine to stimulate prefrontal ACh release (Fig. 5) was a function of drug, lesion condition, and collection interval (F10,100 = 3.47, P = 0.009). Both sham controls and nVHLX rats responded to amphetamine with large and protracted increases in cortical ACh efflux, and, in contrast with the insensitivity to NMDA seen in lesioned rats (Figs. 1, 3, 4), nVHLX rats exhibited a greater response to amphetamine than did controls (F1,10 = 13.98, P = 0.004). These results reject the possibility that as a result of the neonatal lesion, the adult cortical cholinergic input system fundamentally lacks the capacity to respond to stimulation.
Figure 5.
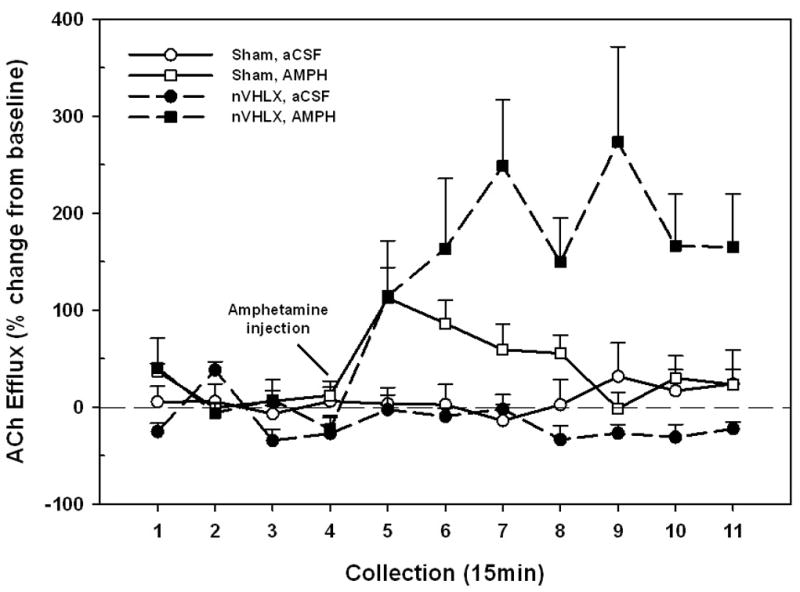
Mean (± S.E.M.) ACh efflux in the mPFC of shams (n = 6) or nVHLX (n = 6) rats receiving systemic injections of amphetamine (AMPH; 2 mg/kg, i.p.) as adults. Following baseline collections (1–4), AMPH was injected. In contrast to the effects of NMDA (Figs. 2 and 4b), administration of amphetamine resulted in robust increases in cortical ACh efflux in both sham and nVHLX groups, with lesioned animals exhibiting a potentiated release relative to that seen in control rats.
Experiment 3: Reversal by Semi-Chronic Treatment with Clozapine
The effects of semi-chronic treatment with saline (top panel) or clozapine (bottom panel) on the ability of intra-NAC NMDA to stimulate cortical cholinergic transmission in nVHLX animals are summarized in Figure 6. Mean (± S.E.M.) basal ACh release did not differ among sham-lesioned and lesioned rats following treatment with saline (7.45 ± 0.13 fmol/15 μL; F1,14 = 2.09, P = 0.17) or clozapine (5.45 ± 0.14 fmol/15 μL; F1,14 = 0.001; P = 0.98), although the overall differences in basal levels between the two pre-treatment conditions did differ (t30 = 7.54, P < 0.001). While the elevated baselines following saline pre-treatment were a bit unusual, they were judged not be of biological significance as the responses to NMDA infusions (see below) were very similar to those seen in non pre-treated rats (Experiments 1–2). Thus, subsequent analyses of ACh release were conducted and expressed as a percent change from these baselines. Semi-chronic pretreatment with saline did not affect the absence of NMDA-evoked ACh release in nVHLX rats nor the increase in ACh release seen in sham-lesioned controls. ACh release in saline-treated animals (Fig. 6, top) closely resembles the data obtained in Experiment 1a (Fig. 2). An overall analysis revealed a highly significant interaction between the effects of NMDA/aCSF, lesion condition, and collection interval (F10, 140 = 9.28, P < 0.001). Post hoc comparisons confirmed that intra-NAC perfusion of NMDA produced the expected increase in ACh efflux, relative to aCSF, in sham-lesioned controls (F1,7 = 231.21; P < 0.001) but not in nVHLX rats (F1,7 = 3.06; P = 0.82).
Figure 6.

Mean (± S.E.M.) ACh efflux in the mPFC of sham (n = 8) or nVHLX (n = 8) rats following pretreatment (daily injections for 13 days) with either saline (0.9%) or clozapine (1.25 mg/kg, i.p.) prior to microdialysis testing as adults. Each animal received, in pseudo-randomized order, perfusions of vehicle (aCSF) and NMDA (250 μM) into the NAC shell during two dialysis sessions. The period for baseline infusions of aCSF and NMDA was the same as those reported in previous experiments (Figs. 2–4). In the control condition of saline-pretreatment (Panel a), the results of Experiment 1A (Fig. 2) were replicated. Namely, NMDA perfusion increased cortical ACh efflux in sham control animals whereas this effect was absent in nVHLX animals. Following pre-treatment with clozapine (Panel b), nVHLX animals exhibited a sham-control-like stimulation of ACh release following intra-NAC perfusion of NMDA.
In contrast, semi-chronic administration of clozapine resulted in a marked NMDA-evoked increase in PFC ACh release in nVHLX animals. The effect of clozapine was evident, statistically, by the absence of interactions between the effects of drug, lesion condition and collection (F10, 140 = 0.74, P = 0.68). Specifically, following clozapine pre-treatment, the NMDA-evoked increases in ACh release in nVHLX rats (F1,7 = 21.33; P = 0.02) were indistinguishable from that seen in sham lesioned rats (F1,14 = 0.32, P = 0.58).
DISCUSSION
These experiments revealed several novel findings regarding the impaired regulation of the cortical cholinergic system in rats sustaining lesions of the VH as neonates. First, the ability of NMDA receptor activation in the NAC to stimulate cortical ACh release, as seen in sham-lesioned controls, was completely lost in rats lesioned as neonates. Second, this lesion-induced deficit was age-dependent in that rats comparably lesioned as adults exhibited control-like elevations in cortical ACh release following intra-NAC perfusions of NMDA. Furthermore, the deficit in nVHLX rats emerged over the course of development as it appeared after puberty but was not present prior to puberty. Finally, semi-chronic treatment with a low-dose of clozapine (for justification of dose see Methods) in adulthood restored the ability of NMDA to evoke cortical ACh release in nVHLX rats. The discussion that follows addresses several issues related to the interpretation of these findings; including a) the ability of intra-NAC NMDA perfusions to stimulate cortical cholinergic transmission, b) the consequences of ventral hippocampal lesions during early development on the subsequent organization of the hippocampal-accumbens-basal forebrain-prefrontal cortical distributed system, and c) the implications for the validity and usefulness of neonatal ventral hippocampal lesions as an animal model of schizophrenia.
Accumbens NMDA receptors and cortical cholinergic transmission
Intra-accumbens perfusions of NMDA reliably evoked prefrontal ACh release in several groups of rats, including those that were sham-lesioned as neonates or adults, lesioned as adults, or lesioned as neonates and tested pre-puberty. The ability of NMDA perfusions in NAC to stimulate cholinergic transmission in PFC has previously been demonstrated in intact rats (Brooks, et al 2007; Zmarowski, et al 2005,2007), and the present evidence indicates that NMDA-evoked increases in release were generally unaffected in all groups except animals lesioned as neonates and tested post-puberty.
The neuronal circuitry mediating the effects of NAC NMDA receptor stimulation to prefrontal cholinergic transmission is not completely understood. Glutamatergic inputs to the shell region of the NAC arise from multiple sources, including ventral hippocampus (subiculum; Groenewegen, et al 1987), amygdala (Yim and Mogenson, 1982), and prefrontal cortex (O’Donnell and Grace, 1995; Gruber and O’Donnell, 2009). In the NAC, NMDA receptors are located, at high density, on medium spiny (MSN) projection neurons in NAC (Martin and Siggins, 2002). These projection neurons may modulate cortical ACh release by directly regulating basal forebrain activity. A high proportion of basal forebrain neurons, many of which are cholinergic (Henny and Jones, 2008), are innervated by GABAergic projections, most likely from the NAC (Ingham, et al 1988; Zaborsky and Cullinan, 1992). Moreover, accumbens projections may contact basal forebrain GABAergic corticopetal neurons (Henny and Jones, 2008) and these inputs, through their contacts with cortical GABAergic interneurons (Henny and Jones, 2008; Zaborsky, et al 1999), have the potential to regulate cortical ACh release through local mechanisms. Finally, the ability of NAC efferents to modulate cortical cholinergic transmission may involve more indirect mechanisms such as projections back to VTA (Usuda, et al 1998) and then to basal forebrain (Gaykema and Zaborsky, 1996), or projections from VTA directly to prefrontal cortex (Lewis and O’Donnell, 2000) for local regulation of ACh release. Additional research will be required to identify the contributions of these various circuits.
Several observations suggest that the loss of NMDA-evoked ACh release in nVHLX animals tested post-puberty was not secondary to NMDA-induced damage to the NAC projection neurons or the basal forebrain cortical cholinergic system. First, three of the four treatment groups in this study (shams at both age groups and aVHLX rats) remained responsive to intra-NAC perfusions. Second, even nVHLX rats exhibited a control-like response to NMDA when tested pre-puberty. Third, nVHLX rats that had not responded to NMDA as adults exhibited a normal response following semi-chronic treatment with clozapine. Finally, nVHLX rats that were unresponsive to NMDA displayed elevated cortical ACh release following systemic amphetamine.
Mechanistic considerations
The effects of ibotenic-acid induced lesions of the VH on cortical cholinergic transmission depended markedly on the age at which the lesion was sustained and the age at the time of testing. This lesion, at this sensitive period of development, disrupts the maturation of the NAC-BF-PFC circuit that is revealed by the effects of stimulation of NAC NMDA receptors post-puberty. In adult rats, the activity of ventral hippocampal projections, in interaction with dopaminergic inputs from VTA, regulate the accumbens’ responsivity to prefrontal excitatory inputs (Grace, 2000; O’Donnell and Grace, 1995). In addition, VH projections constitute a major source of excitatory input to PFC (Lewis and O’Donnell, 2000). The collective evidence on the age-dependent effects of lesions to the VH indicates anatomical, molecular, and neurochemical alterations consistent with the theory that neonatal, but not adult, lesions induce a reorganization of this circuit, and that the maladaptive consequences of this plasticity functionally express themselves around the time of puberty. For example, the length and branching of basilar dendrites, as well as their density, was reduced in PFC and NAC of nVHX, but not aVHX, rats (Flores et al., 2005). These anatomical changes are accompanied by an increased expression of NR1 mRNA in PFC that is seen post-but not pre-puberty (El-Rawas et al., 2009). Electrophysiological recordings of NAC-modulation of prefrontal activity also reveal maturationally-dependent impairments in nVHX rats. VTA-evoked depolarizations are accompanied by a high frequency of NAC (Brady and O’Donnell, 2004) or PFC (O’Donnell, et al 2002) spike firing in post-pubescent rats (but not pre-pubsecent rats) lesioned as neonates but a marked suppression of spike firing in controls and rats lesioned as adults. The enhanced activation of PFC neurons may be secondary to a puberty-dependent loss of D2 modulation of prefrontal interneurons (Tseng et al., 2008) and might be expected to contribute to ‘noisy’ cortical information processing. The heightened responsivity of NAC neurons following the lesion was normalized by subchronic administration of haloperidol (Goto and O’Donnell, 2002) or lesions of the PFC (Goto and O’Donnell, 2004).
A previous study identified deficits in the regulation of cortical cholinergic transmission following nVH lesions that, as in the present study and those referenced above, became evident only during or after puberty (LaPlante, et al 2004). Specifically, systemic administration of a D1 agonist stimulated prefrontal ACh release to a greater degree in lesioned than in control rats. Moreover, local perfusion of a D1 agonist did not affect ACh release in the PFC of controls but stimulated release in rats lesioned as neonates.
Taken together, numerous plastic changes within the NAC-BF-PFC circuit, ranging from structural modification of dendritic trees to variations in the expression of receptors, result from nVH lesions in interaction with brain maturation. In conceptual terms, such lesions serve to model such maturational abnormalities in this circuit, and the effects of NMDA infusions into the NAC reveal these abnormalities.
Causal relationships between dysregulated cholinergic neurotransmission and cognitive, specifically attentional impairments
The present results indicate that an abnormally regulated cortical cholinergic input system represents a hallmark of this animal model of schizophrenia. The significance of this finding concerns the over-arching hypothesis that a dysregulated cholinergic system contributes to, or is even primarily responsible for, the attentional impairments that result from nVHLX. First, as adults, nVHLX animals were found to exhibit attentional impairments in experiments assessing attentional set-shifting (Marquis et al., 2008) and performance in the 5-choice serial reaction time task (Le Pen et al., 2003). Furthermore, the repeatedly demonstrated impairments in sensory gating (Lipska et al., 1995; Le Pen et al., 2000; Rueter et al., 2004) likewise may reflect a fundamental disruption in cognition-associated input processing caused by nVHLX.
Second, a substantial body of evidence indicates that an abnormally regulated cortical cholinergic input system causes impairments in attention. Removal of cholinergic inputs to the cortex causes persistent impairments in attentional performance, assessed by a range of tasks (e.g., McGaughy et al., 1996 e.g., McGaughy et al., 2002). Our experiments concerning an animal model of schizophrenia that results from exposure to an escalating-dose regimen of amphetamine generated additional, relevant evidence. We observed that disruption of attentional performance in these animals was associated with a failure of the prefrontal cholinergic input system to respond to task. Prefrontal ACh release remained at baseline throughout the test session and performance remained close to chance level (Kozak et al., 2007). Importantly, the “freezing” of ACh release at baseline levels was noticeable prior to task onset, allowing a causal interpretation: the failure of the cholinergic system to depart from baseline activity levels caused the impairment, as opposed to representing merely a correlate of disrupted performance. The present evidence likewise would predict that, in vHLX animals, the attentional task-induced recruitment of prefrontal-mesolimbic-basal forebrain circuits (Sarter et al., 2006) is disrupted and that performance-associated increase in prefrontal ACh release is attenuated.
Although it will be important to demonstrate in future experiments that attentional task-performing nVHLX animals exhibit attenuated levels of ACh release, causal relationships are more difficult to derive from such studies and may be based on evidence indicating treatment-induced co-variation of ACh release and performance. We know already that, in intact animals, infusions of NMDA into the NAC benefits attentional performance, specifically in interaction with performance challenges that are thought to activate top-down mechanisms, including mesolimbic-basal forebrain circuitry (St. Peters et al., 2009). In nVHXL animals, such infusions would be expected to normalize the increases in task performance-associated increases in cholinergic neurotransmission. Unfortunately, experiments necessitating the measurement of ACh release in task-performing animals and in response to bilateral intracranial infusions are extremely challenging.
Taken together, there is growing evidence indicating that nVHLX causes impairments in attention and that abnormally regulated activity of cortical cholinergic inputs causes impairments in attention. Therefore, the unresponsive cholinergic system in adult nVHLX animals is likely to be primarily responsible for the attentional impairments that were demonstrated in these animals.
Implications for the validity and usefulness of nVHX as an animal model for schizophrenia
Increases in prefrontal cholinergic activity have been suggested to contribute to the recruitment of the “anterior attention system” and therefore to the activation of top down mechanisms that act to maintain and recover attentional performance under challenging conditions (see Introduction). NMDA perfusions into the NAC are thought to mimic, in part, the recruitment of mesolimbic-BFCS interactions that are required to optimize attentional mechanisms in response to performance challenges. As impairments of the attentional abilities of schizophrenic patients are particularly severe when challenged (for review see Sarter, et al 2009), the loss of NMDA-mediated activation of the prefrontal cholinergic system in nVHLX animals is interpreted as reflecting a loss in the ability to recruit, top-down, the mechanisms that normally support attentional performance and performance recovery (see above).
Consistent with the schizophrenia phenotype, nVHLX animals exhibit a range of cognitive deficits in the attentional and other cognitive domains (see above, see also Chambers, et al., 1996). As our research focuses on the established role of cholinergic neurotransmission for attention, the present results specifically are considered to indicate a neuronal mechanism that might be responsible for the vulnerability of schizophrenic patients to challenges on attentional capacities. As discussed elsewhere (Sarter, et al 2005b; Sarter, et al 2009) such vulnerability may not merely represent one among many cognitive problems of schizophrenic patients, but may instrumentally contribute to the severity of a wide range of cognitive impairments and thus have been proposed to represent a primary target for treatment developments (Nuechterlein, et al 2009; Young, et al 2009).
Finally, the finding that low-dose treatment with clozapine reversed the lesion-induced deficit in NMDA-mediated cortical ACh release contributes to the predictive validity of this particular neurochemical assay within the nVHX model. The finding also supports further the usefulness of low-dose antipsychotic treatments to determine the predictive validity of animal models in the absence of more efficacious cognition enhancers (Martinez and Sarter, 2008). Importantly, this normalization of NAC-BF-PFC function was not seen following the acute administration of either clozapine or haloperidol (Brooks, unpublished observations). The mechanisms underlying the ability of clozapine to restore cholinergic transmission have not been resolved. One rather straight forward possibility is that the neonatal lesion results in elevated levels of D2-mediated neurotransmission in NAC. We have previously shown, in intact rats, that intra-NAC perfusions of the D2-like agonist quinpirole blocks the ability of concurrent NMDA administration to stimulate prefrontal ACh release (Brooks, et al., 2007), suggesting the excessive D2-like receptor activity in NAC can dampen the NMDA-modulation of prefrontal ACh release. However, we have also observed abnormalities in the generation of cholinergic activity based on local prefrontal circuitry (Wescott, et al., 2009), suggesting a possible role of disrupted prefrontal circuitry (Tseng, et al., 2008) in the failure of NAC stimulation to activate PFC cholinergic activity. Finally, it seems likely, of course, that the functional consequences of an abnormally organized PFC are magnified by the inability of the cholinergic input to recruit PFC circuitry.
In summary, the results reported here demonstrate that damage to the VH during a critical period of early development interacts with maturational variables at or around the time of puberty to produce a permanent dysregulation in a circuit involving the prefrontal cortex, the nucleus accumbens and basal forebrain cholinergic projections to the PFC. This abnormal limbic-cortical interaction is likely to contribute significantly to the attentional deficits evident in schizophrenia. These results, including the ability of low-dose clozapine to restore aspects of the functionality of this circuit, suggests that this animal model may be useful for studying the neuronal mechanisms mediating the cognitive impairments of schizophrenia and the efficacy of putative treatments for these impairments.
Acknowledgments
The authors’ research was supported by research grants from the National Institute of Health (MH057436 to J.P.B. & M.S.; MH080426 and MH 080332 to M.S.).
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors have nothing to disclose nor are there conflicts of interest in matters associated with the experiments presented in this manuscript.
References
- Arnold HM, Nelson CL, Neigh GH, Sarter M, Bruno JP. Systemic and intra-accumbens administration of amphetamine differentially affects cortical acetylcholine release. Neuroscience. 2000;96:675–85. doi: 10.1016/s0306-4522(99)00590-4. [DOI] [PubMed] [Google Scholar]
- Barch DM, Carter CS, Arnsten A, Buchanan RW, Cohen JD, Geyer M, et al. Selecting paradigms from cognitive neuroscience for translation into use in clinical trials: proceedings of the third CNTRICS meeting. Schizophr Bull. 2009;35:109–114. doi: 10.1093/schbul/sbn163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj SK, Beaudry G, Quirion R, Levesque D, Srivastava LK. Neonatal ventral hippocampus lesion leads to reductions in nerve growth factor inducible-B mRNA in the prefrontal cortex and increased amphetamine response in the nucleus accumbens and dorsal striatum. Neuroscience. 2003;122:669–676. doi: 10.1016/j.neuroscience.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Brady AM, O’Donnell P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. Journal of Neuroscience. 2004;24:1040–49. doi: 10.1523/JNEUROSCI.4178-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, Light GA. Preattentional and attentional cognitive deficits as targets for treating schizophrenia. Psychopharmacology (Berl) 2004;174:75–85. doi: 10.1007/s00213-004-1848-0. [DOI] [PubMed] [Google Scholar]
- Brooks JM, Sarter M, Bruno JP. D2-like receptors in nucleus accumbens negatively modulate acetylcholine release in prefrontal cortex. Neuropharmacology. 2007;53:455–463. doi: 10.1016/j.neuropharm.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JM, Sarter M, Bruno JP. Neuroscience Meeting Planner. Washington, DC: Society for Neuroscience; 2008. Semi-chronic clozapine treatment normalizes top-down regulation of cortical acetylcholine release in an animal model of schizophrenia. Program No. 657.8. Online. [Google Scholar]
- Chambers RA, Moore J, McEvoy JP, Levin ED. Cognitive effects of neonatal hippocampal lesions in a rat model of schizophrenia. Neuropsychopharmacology. 1996;15:587–94. doi: 10.1016/S0893-133X(96)00132-7. [DOI] [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW. Psychopharmacological approaches to modulating attention in the five-choice serial reaction time task: implications for schizophrenia. Psychopharmacology (Berl) 2004;174:86–98. doi: 10.1007/s00213-004-1805-y. [DOI] [PubMed] [Google Scholar]
- El-Rawas R, Saade NE, Thiriet N, Atweh S, Jaber M, Al-Amin HA. Developmental changes in the mRNA expression of neuropeptides and dopamine and glutamate receptors in neonates and adult rats after hippocampal lesion. Schizophrenia Research. 2009 doi: 10.1016/j.schres.2009.05.009. Epub. [DOI] [PubMed] [Google Scholar]
- Flores G, Alquicer G, Silva-Gomez AB, Zaldivar G, Stewart J, Quirion R, Srivastava LK. Alterations in dendritic morphology of prefrontal cortical and nucleus accumbens neurons in post-pubertal rats after neonatal excitotoxic lesions of the ventral hippocampus. Neuroscience. 2005;133:463–70. doi: 10.1016/j.neuroscience.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Gaykema RP, Zaborszky L. Direct catecholaminergic-cholinergic interactions in the basal forebrain. II. Substantia nigra-ventral tegmental area projections to cholinergic neurons. Journal of Comparative Neurology. 1996;374:555–77. doi: 10.1002/(SICI)1096-9861(19961028)374:4<555::AID-CNE6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Goto Y, O’Donnell P. Delayed mesolimbic system alteration in a developmental animal model of schizophrenia. Journal of Neuroscience. 2002;22:9070–77. doi: 10.1523/JNEUROSCI.22-20-09070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, O’Donnell P. Prefrontal lesion reverses abnormal mesoaccumbens response in an animal model of schizophrenia. Biological Psychiatry. 2004;55:172–76. doi: 10.1016/s0006-3223(03)00783-2. [DOI] [PubMed] [Google Scholar]
- Grace AA. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Research Reviews. 2000;31:330–41. doi: 10.1016/s0165-0173(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ, Vermeulenvanderzee E, Kortschot AT, Witter MP. Organization of the projections from the subiculum to the ventral striatum in the rat- a study using anterograde transport of phaseolus-vulgaris leukoagglutinin. Neuroscience. 1987;23:103–20. doi: 10.1016/0306-4522(87)90275-2. [DOI] [PubMed] [Google Scholar]
- Gruber AJ, O’Donnell P. Bursting activation of prefrontal cortex drives sustained up states in nucleus accumbens spiny neurons in vivo. Synapse. 2009;63:173–80. doi: 10.1002/syn.20593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan JJ, Jones DN. Predicting drug efficacy for cognitive deficits in schizophrenia. Schizophr Bull. 2005;31:830–853. doi: 10.1093/schbul/sbi058. [DOI] [PubMed] [Google Scholar]
- Henny P, Jones BE. Projections from basal forebrain to prefrontal cortex comprise cholinergic, GABAergic and glutamatergic inputs to pyramidal cells or interneurons. European Journal of Neuroscience. 2008;27:654–70. doi: 10.1111/j.1460-9568.2008.06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelheber AM, Sarter M, Bruno JP. Increases in cortical acetylcholine release during sustained attention performance in rats. Cognitive Brain Research. 2000;9:313–325. doi: 10.1016/s0926-6410(00)00012-4. [DOI] [PubMed] [Google Scholar]
- Huang T, Yang L, Gitzen J, Kissinger PT, Vreeke M, Heller A. Detection of basal acetylcholine in rat brain microdialysates. Journal of Chromatography. 1995;670:323–27. doi: 10.1016/0378-4347(95)00181-6. [DOI] [PubMed] [Google Scholar]
- Hyde TM, Crook JM. Cholinergic systems and schizophrenia: primary pathology or epiphenomena? Journal of Chemical Neuroanatomy. 2001;22:53–63. doi: 10.1016/s0891-0618(01)00101-6. [DOI] [PubMed] [Google Scholar]
- Ichikawa J, Dai J, O’Laughlin IA, Fowler WL, Meltzer HY. Atypical, but not typical, antipsychotic drugs increase cortical acetylcholine release without an effect in the nucleus accumbens or striatum. Neuropsychopharmacology. 2002;26:325–339. doi: 10.1016/S0893-133X(01)00312-8. [DOI] [PubMed] [Google Scholar]
- Ingham CA, Bolam JP, Smith AD. GABA-immunoreactive synaptic boutons in the rat basal forebrain: comparison of neurons that project to the neocortex with pallidosubthalamic neurons. Journal of Comparative Neurology. 1988;273:263–82. doi: 10.1002/cne.902730210. [DOI] [PubMed] [Google Scholar]
- Kapur S, VanderSpek SC, Brownlee BA, Nobrega JN. Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J Pharmacol Exp Ther. 2003;305:625–631. doi: 10.1124/jpet.102.046987. [DOI] [PubMed] [Google Scholar]
- Kerns JG, Neuchterlein KH, Braver TS, Barch DM. Executive functioning component mechanisms and schizophrenia. Biological Psychiatry. 2008;64:26–33. doi: 10.1016/j.biopsych.2008.04.027. [DOI] [PubMed] [Google Scholar]
- Kozak R, Martinez V, Young D, Brown H, Bruno JP, Sarter M. Toward a neuro-cognitive animal model of the cognitive symptoms of schizophrenia: disruption of cortical cholinergic neurotransmission following repeated amphetamine exposure in attentional task-performing, but not non-performing, rats. Neuropsychopharmacology. 2007;32:2074–2086. doi: 10.1038/sj.npp.1301352. [DOI] [PubMed] [Google Scholar]
- Kozak R, Bruno JP, Sarter M. Augmented prefrontal acetylcholine release during challeneged attentional performance. Cerebral Cortex. 2006;16:9–17. doi: 10.1093/cercor/bhi079. [DOI] [PubMed] [Google Scholar]
- Kozak R, Martinez V, Young D, Brown H, Bruno JP, Sarter M. Toward a neuro-cognitive animal model of the cognitive symptoms of schizophrenia: disruption of cortical cholinergic neurotransmission following repeated amphetamine exposure in attentional task-performing, but not non-performing, rats. Neuropsychopharmacology. 2007;32:2074–2086. doi: 10.1038/sj.npp.1301352. [DOI] [PubMed] [Google Scholar]
- LaPlante F, Srivastava LK, Quirion R. Alterations in dopaminergic modulation of prefrontal cortical acetylcholine release in post-pubertal rats with neonatal ventral hippocampal lesions. Journal of Neurochemistry. 2004;89:314–23. doi: 10.1111/j.1471-4159.2004.02351.x. [DOI] [PubMed] [Google Scholar]
- Le Pen G, Grottick AJ, Higgins GA, Moreau JL. Phencyclidine exacerbates attentional deficits in a neurodevelopmental rat model of schizophrenia. Neuropsychopharmacology. 2003;28:1799–809. doi: 10.1038/sj.npp.1300208. [DOI] [PubMed] [Google Scholar]
- Le Pen G, Grottick AJ, Higgins GA, Martin JR, Jenck F, Moreau JL. Spatial and associative learning deficits induced by neonatal excitotoxic hippocampal damage in rats: further evaluation of an animal model of schizophrenia. Behavioral Pharmacology. 2000;11:257–268. doi: 10.1097/00008877-200006000-00009. [DOI] [PubMed] [Google Scholar]
- Lewis BL, O’Donnell P. Ventral tegmental area afferents to the prefrontal cortex maintain membrane potential ‘up’ states in pyramidal neurons via D(1) dopamine receptors. Cereb Cortex. 2000;10:1168–75. doi: 10.1093/cercor/10.12.1168. [DOI] [PubMed] [Google Scholar]
- Li Z, Huang M, Ichikawa J, Dai J, Meltzer HY. N-desmethylclozapine, a major metabolite of clozapine, increases cortical acetylcholine and dopamine release in vivo via stimulation of M1 muscarinic receptors. Neuropsychopharmacology. 2005;30:1986–1995. doi: 10.1038/sj.npp.1300768. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Lipsak BK, Swerdlow NR, Geyer MA, Jaskiw GE, Braff DL, Weinberger DR. Neonatal excitotoxic hippocampal damage in rats causes post-pubertal changes in prepulse inhibition of startle and its disruption by apomorphine. Psychopharmacology. 1995;122:35–43. doi: 10.1007/BF02246439. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weickert CS, Weinberger DR. Gene expression in dopamine and GABA systems in an animal model of schizophrenia: effects of antipsychotic drugs. European Journal of Neuroscience. 2003;18:391–402. doi: 10.1046/j.1460-9568.2003.02738.x. [DOI] [PubMed] [Google Scholar]
- Marquis JP, Goulet S, Dore FY. Neonatal ventral hippocampus lesions disrupt extra-dimensional shift and alter dendritic spine density in the medial prefrontal cortex of juvenile rats. Neurobiol Learn Mem. 2008;90:339–346. doi: 10.1016/j.nlm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Martin G, Siggins GR. Electrophysiological evidence for expression of glycine receptors in freshly isolated neurons from nucleus accumbens. Journal of Pharmacology and Experimental Therapeutics. 2002;302:1135–45. doi: 10.1124/jpet.102.033399. [DOI] [PubMed] [Google Scholar]
- Martinez V, Sarter M. Detection of the moderately beneficial cognitive effects of low-dose treatment with haloperidol or clozapine in an animal model of the attentional impairments of schizophrenia. Neuropsychopharmacology. 2008;33:2635–2647. doi: 10.1038/sj.npp.1301661. [DOI] [PubMed] [Google Scholar]
- McGaughy J, Dalley JW, Morison CH, Everitt BJ, Robbins TW. Selective behavioral and neurochemical effects of cholinergic lesions produced by intrabasalis infusions of 192 IgG-saporin on attentional performance in a five-choice serial reaction time task. Journal of Neuroscience. 2000;22:1905–1913. doi: 10.1523/JNEUROSCI.22-05-01905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughy J, Kaiser T, Sarter M. Behavioral vigilance following infusions of 192 IgG-saporin into the basal forebrain: selectivity of the behavioral impairment and relation to cortical AChE-positive fiber density. Behavioral Neuroscience. 1996;110:247–265. doi: 10.1037//0735-7044.110.2.247. [DOI] [PubMed] [Google Scholar]
- Mishara AL, Goldberg TE. A meta-analysis and critical review of the effects of conventional neuroleptic treatment on cognition in schizophrenia: opening a closed book. Biol Psychiatry. 2004;55:1013–1022. doi: 10.1016/j.biopsych.2004.01.027. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Barch DM, Gold JM, Goldberg TE, Green MF, Heaton RK. Identification of separable cognitive factors in schizophrenia. Schizophrenia Research. 2004;72:29–39. doi: 10.1016/j.schres.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Dawson ME. Information processing and attentional functioning in the developmental course of schizophrenic disorders. Schizophr Bull. 1984;10:160–203. doi: 10.1093/schbul/10.2.160. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Luck SJ, Lustig C, Sarter M. CNTRICS final task selection: control of attention. Schizophr Bull. 2009;35:182–196. doi: 10.1093/schbul/sbn158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. The Journal of Neuroscience. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, Lewis BL, Weinberger DR, Lipska BK. Neonatal hippocampal damage alters electrophysiological properties of prefrontal cortical neurons in adult rats. Cerebral Cortex. 2002;12:975–982. doi: 10.1093/cercor/12.9.975. [DOI] [PubMed] [Google Scholar]
- Potter PE, Meek JL, Neff NH. Acetylcholine and choline in neuronal tissue measured by HPLC with electrochemical detection. Journal of Neurochemistry. 1983;41:188–194. doi: 10.1111/j.1471-4159.1983.tb13668.x. [DOI] [PubMed] [Google Scholar]
- Raedler TJ, Knable MB, Jones DW, Urbina RA, Gorey JG, Lee KS, et al. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. American Journal of Psychiatry. 2003;160:118–127. doi: 10.1176/appi.ajp.160.1.118. [DOI] [PubMed] [Google Scholar]
- Rueter LE, Ballard ME, Gallagher KB, Basso AM, Curzon P, Kohlhaas KL. Chronic low dose risperidone and clozapine alleviate positive but not negative symptoms in the rat neonatal ventral hippocampal lesion model of schizophrenia. Psychopharmacology (Berl) 2004;176:312–319. doi: 10.1007/s00213-004-1897-4. [DOI] [PubMed] [Google Scholar]
- Sarter M, Gehring WJ, Kozak R. More attention must be paid: the neurobiology of attentional effort. Brain Res Rev. 2006;51:145–160. doi: 10.1016/j.brainresrev.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Sarter M, Hasselmo ME, Bruno JP, Givens B. Unraveling the attentional functions of cortical cholinergic inputs: interactions between signal-driven and cognitive modulation of signal detection. Brain Research Reviews. 2005a;48:98–111. doi: 10.1016/j.brainresrev.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Sarter M, Martinez V, Kozak R. A neurocognitive animal model dissociating between acute illness and remission periods of schizophrenia. Psychopharmacology (Berl) 2009;202:237–258. doi: 10.1007/s00213-008-1216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Nelson CL, Bruno JP. Cortical cholinergic transmission and cortical information processing in schizophrenia. Schizophrenia Bulletin. 2005b;31:117–138. doi: 10.1093/schbul/sbi006. [DOI] [PubMed] [Google Scholar]
- St Peters M, Bruno JP, Sarter M. Accumbens NMDA receptor stimulation enhances attentional performance as a function of demands on top-down control. Society for Neuroscience Annual Meeting; Chicago, IL. 2009. [Google Scholar]
- Tseng KY, Amin F, Lewis BL, O’Donnell P. Altered prefrontal cortical metabolic response to mesocortical activation in adult animals with a neonatal ventral hippocampal lesion. Biological Psychiatry. 2006;60:585–590. doi: 10.1016/j.biopsych.2006.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Lewis BL, Hashimoto T, Sesack SR, Kloc M, Lewis DA, et al. A neonatal ventral hippocampal lesion causes functional deficits in adult prefrontal cortical interneurons. J Neurosci. 2008;28:12691–12699. doi: 10.1523/JNEUROSCI.4166-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Lewis BL, Lipska BK, O’Donnell P. Post-pubertal disruption of medial prefrontal cortical dopamine-glutamate interactions in a developmental animal model of schizophrenia. Biol Psychiatry. 2007;62:730–738. doi: 10.1016/j.biopsych.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuda I, Tanaka K, Chiba T. Efferent projections of the nucleus accumbens in the rat with special reference to subdivision of the nucleus: biotinylated dextran amine study. Brain Research. 1998;797:73–93. doi: 10.1016/s0006-8993(98)00359-x. [DOI] [PubMed] [Google Scholar]
- Vasey MW, Thayer JF. The continuing problem of false positives in repeated measures ANOVA in psychophysiology – a multivariate solution. Psychophysiology. 1987;24:479–489. doi: 10.1111/j.1469-8986.1987.tb00324.x. [DOI] [PubMed] [Google Scholar]
- Wescott S, Gritton H, Parikh V, Bruno JP, Sarter M. Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience; 2009. Nicotine-evoked recruitment of prefrontal, signal detection-mediating mechanisms, are abolished in the neonatal ventral hippocampal lesion model of schizophrenia. Program No. XXX.X. Online. [Google Scholar]
- Yim CY, Mogenson GJ. Response of nucleus accumbens neurons to amygdala stimulation and its modification by dopamine. Brain Research. 1982;239:401–15. doi: 10.1016/0006-8993(82)90518-2. [DOI] [PubMed] [Google Scholar]
- Young JW, Powell SB, Risbrough V, Marston HM, Geyer MA. Using the MATRICS to guide development of a preclinical cognitive test battery for research in schizophrenia. Pharmacol Ther. 2009;122:150–202. doi: 10.1016/j.pharmthera.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Cullinan WE. Projections from the nucleus accumbens to cholinergic neurons of the ventral pallidum: a correlated light and electron microscopic double-immunolabeling study in rat. Brain Research. 1992;570:92–101. doi: 10.1016/0006-8993(92)90568-t. [DOI] [PubMed] [Google Scholar]
- Zaborszky L, Pang K, Somogyi J, Nadasdy Z, Kallo I. The basal forebrain corticopetal system revisited. Annals of the New York Academy of Sciences. 1999;877:339–67. doi: 10.1111/j.1749-6632.1999.tb09276.x. [DOI] [PubMed] [Google Scholar]
- Zmarowski A, Sarter M, Bruno JP. NMDA and dopamine interactions in the nucleus accumbens modulate cortical acetylcholine release. European Journal of Neuroscience. 2005;22:1731–1740. doi: 10.1111/j.1460-9568.2005.04333.x. [DOI] [PubMed] [Google Scholar]
- Zmarowski A, Sarter M, Bruno JP. Glutamate receptors in nucleus accumbens mediate regionally selective increases in cortical acetylcholine release. Synapse. 2007;61:115–123. doi: 10.1002/syn.20354. [DOI] [PubMed] [Google Scholar]