

Abstract
Spinal cord microglial toll-like receptor-4 (TLR4) has been implicated in enhancing neuropathic pain and opposing morphine analgesia. The present study was initiated to explore TLR4-mediated pain modulation by intrathecal lipopolysaccharide, a classic TLR4 agonist. However, our initial study revealed that intrathecal lipopolysaccharide failed to induce low-threshold mechanical allodynia in naive rats, suggestive that TLR4 agonism may be insufficient to enhance pain. These studies explore the possibility that a second signal is required; namely, heat shock protein-90 (HSP90). This candidate was chosen for study given its known importance as a regulator of TLR4 signaling. A combination of in vitro TLR4 cell signaling and in vivo behavioral studies of pain modulation suggest that TLR4-enhancement of neuropathic pain and TLR4-suppression of morphine analgesia each likely require HSP90 as a cofactor for the effects observed. In vitro studies revealed that DMSO enhances HSP90 release, suggestive that this may be a means by which DMSO enhances TLR4 signaling. While 2 µg and 100 µg lipopolysaccharide intrathecally did not induce mechanical allodynia across the time course tested, co-administration of 1 µg lipopolysaccharide with a drug that enhances HSP90-mediated TLR4 signaling now induced robust allodynia. In support of this allodynia being mediated via a TLR4/HSP90 pathway, it was prevented or reversed by intrathecal co-administration of a HSP90 inhibitor, a TLR4 inhibitor, a microglia/monocyte activation inhibitor (as monocytes-derived cells are the predominant cell type expressing TLR4), and interleukin-1 receptor antagonist (as this proinflammatory cytokine is a downstream consequence of TLR4 activation). Together, these results suggest for the first time that TLR4 activation is necessary but not sufficient to induce spinally mediated pain enhancement. Rather, the data suggest that TLR4-dependent pain phenomena may require contributions by multiple components of the TLR4 receptor complex.
Keywords: HEK293-TLR4, lipopolysaccharide, chronic constriction injury, morphine, mechanical allodynia, analgesia
Peripheral nerve injury activates spinal microglia and astrocytes, as reflected by activation marker upregulation (Watkins et al., 2005). Microglial activation is generally thought to occur prior to astrocyte activation (Raghavendra et al., 2003a). Such activation causes the release of proinflammatory products including interleukin-1, which enhances pain (Kwon et al., 2005). Indeed, blockade of microglial activation with minocycline prevents neuropathic pain development (Mika, 2008) and blockade of proinflammatory mediators such as interleukin-1 which reverses neuropathic pain (Watkins et al., 2005).
While glia are important for the development and maintenance of neuropathic pain, how peripheral nerve injury triggers spinal glial activation is not clear. Numerous candidate neuron-to-glia signals have been proposed for initiating glial activation, including neurotransmitters, neuromodulators, and neuronally derived chemokines such as fractalkine and monocyte chemotactic protein-1 (Watkins et al., 2007, White et al., 2007).
Recently, a very intriguing mechanism has been proposed for spinal microglial activation in response to peripheral nerve injury; that is, activation of toll-like receptor 4 (TLR4) (Tanga et al., 2005, Hutchinson et al., 2008b). Within the central nervous system, TLR4 is expressed pre-dominantly by microglia and by resident and recruited macrophages (Olson and Miller, 2004). To our knowledge, there are no known reports of TLR4 expression by neurons within the spinal cord, the anatomical region of focus here. TLR4 activation leads to the production of proinflammatory mediators implicated in neuropathic pain, including interleukin-1 (Olson and Miller, 2004). While TLR4 is classically thought of as the receptor activated by endotoxin (lipopolysaccharide [LPS] from gram negative bacteria), TLR4 has recently been recognized as also becoming activated in response to “endogenous danger signals” (Osterloh and Breloer, 2008). These are substances released by host cells by cellular stress or damage. In neuropathic pain, these could arise, for example, from neuronal components such as heat shock proteins (Costigan et al., 1998) or degraded cell membrane components (Osterloh and Breloer, 2008) released by stressed or dying sensory afferents, by alterations in the blood-brain barrier (Gordh et al., 2006) allowing entry into the neuropil of blood components normally prevented from doing so (Ward et al., 2006), or other such sources. While the identity of the endogenous danger signal(s) activating TLR4 in response to peripheral nerve damage remains unknown, what is clear is that: (a) peripheral nerve injury upregulates spinal TLR4 mRNA (Tanga et al., 2005, Hutchinson et al., 2008b), (b) development of neuropathic pain (mechanical allodynia) is suppressed in TLR4 knockout/knockdown mice (Tanga et al., 2005), and (c) established neuropathic pain (mechanical allodynia) is reversed by intrathecal classical LPS-derived TLR4 antagonists or the recently discovered novel TLR4 antagonists (+)- or (−)-naloxone (Hutchinson et al., 2008b).
The present study was originally initiated to explore TLR4-mediated pain enhancement by intrathecal LPS, a classic TLR4 agonist. However, our initial pilot studies and the data included in Experiment 1 both revealed that a wide range of intrathecal LPS doses failed to induce mechanical allodynia in naive (non-neuropathic) rats. The clear involvement of TLR4 under neuropathic pain conditions, yet failure of TLR4 activation to enhance pain in normal rats, suggests that a pure TLR4 signal may not be sufficient to enhance pain. That is, it suggests that a second signal is required. Hence the present studies explore this issue for TLR4 receptor activation in vitro and for enhancing pain in vivo.
Methods and Materials
Subjects
Pathogen-free adult male Sprague–Dawley rats (n=6 rats/group for each experiment; 300–375 gm; Harlan Labs, Madison, WI, USA) were used in all experiments. Rats were housed in temperature (23±3°C) and light (12 hr:12 hr light:dark cycle; lights on at 0700) controlled rooms with standard rodent chow and water available ad libitum. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado at Boulder.
Drugs
(+)-Naloxone was obtained from the National Institute on Drug Abuse (Research Triangle Park, NC and Bethesda, MD, USA). Sterile endotoxin-free isotonic saline (Abbott Laboratories, North Chicago, IL, USA) was its vehicle. Lipopolysaccharide (LPS; Escherichia Coli; Serotype: 0111:B4, Sigma, St. Louis, MO, USA), 17-dimethylaminoethylamino-17-desmethoxygeldanamycin (17-DMAG; Calbiochem, San Diego, CA, USA), geldanamycin (Sigma), dimethyl sulfoxide (DMSO; Sigma), and minocycline (Sigma) were obtained commercially. Morphine was gifted from Mallinkrodt (St. Louis, MO, USA). Interleukin-1 receptor antagonist (IL-1ra) and its vehicle were gifted from Amgen. Where applicable, drugs were prepared and are reported as free base concentrations. Vehicles were administered equivolume to the drugs under test. Stock solutions of each compound (except LPS) were negative for endotoxin contamination using a non-TLR4 dependent endotoxin test (Limulus amebocyte lysate assay).
Behavioral assessment of responsivity to mechanical stimuli and radiant heat
All testing was conducted blind with respect to group assignment according to our previously published procedures.
Von Frey Test
The von Frey test (Chaplan et al., 1994) was performed (Experiments 1, 2, 3, 7 and 9) within the sciatic innervation region of the hindpaws as previously described in detail (Chacur et al., 2001, Milligan et al., 2001). Von Frey assessments were made prior to (baseline) and at specific times after experimental manipulations, as detailed in each experiment. The behavioral responses were used to calculate absolute threshold (Harvey, 1986, Treutwein and Strasburger, 1999), as described in detail previously (Milligan et al., 2000, Milligan et al., 2001).
Hargreaves Test
Thresholds for behavioral response to heat stimuli applied to the tail were assessed in Experiment 4 using a modified Hargreaves test (Hargreaves et al., 1988) as described previously (Hutchinson et al 2008a).
Chronic constriction injury (CCI)
Neuropathic pain was induced using the CCI model of partial sciatic nerve injury (Bennett and Xie, 1988). CCI was performed at mid-thigh level of the left hindleg as previously described (Hutchinson et al 2008a). Drug testing was delayed until 10–14 days after surgery to ensure establishment of neuropathic pain prior to the initiation of drug delivery.
Catheter implantation and intrathecal drug administration
Acute intrathecal drug administration was based on that described previously. Catheters were preloaded with drugs at the distal end in a total volume of no greater than 25 µl and the drugs were administered over 20–30 sec. In studies where drug injection was delayed until after recovery from anesthesia, the guide needle was removed after catheter placement, the catheter was sutured to the superficial musculature of the lower back, and the exterior end led subcutaneously to exit through a small incision at the nape of the neck. In this case, the catheters were 90 cm in length, allowing remote drug delivery 2 hr after catheter placement, without touching or otherwise disturbing the rats during the testing.
TLR4 cell line culture and reporter protein assay
A human embryonic kidney-293 (HEK293) cell line stably transfected to express human TLR4 at high levels was purchased from Invivogen (293-htlr4a-md2cd14; here referred to as HEK-TLR4) and cultured and tested as previously described in detail (Hutchinson et al., 2008b; Hutchinson et al., 2009)
Cell fractionation and HSP90 expression quantification
Cultures of HEK-TLR4 cells were processed using a compartmental protein extraction kit (BioChain Institute, Hayward, CA, USA), to separate cytoplasmic proteins from membrane proteins. Additionally, the supernatant from each culture was lyophilized and re-suspended in a small volume of water in order to shift supernatant protein concentrations within a detectable range (at least 2 mg/ml). For each culture, the protein concentration of the cytoplasmic, membrane, and supernatant fraction was measured using the bicinchoninic acid method (Smith et al., 1985). Equivalent amounts of protein were pipetted blind as to group assignment into NuPAGE Bis-Tris (4–12%) gels (Invitrogen, Carlsbad, CA, USA ), which were then run at 160 V for 1 hr. Following electrophoresis, proteins were transferred to a nitrocellulose membrane electrophoretically at 40 V for 1 hr. Non-specific binding sites on the membrane were blocked with 5% non-fat milk in TBS containing 0.5% tween-20. Membranes were incubated overnight at 4°C with polyclonal primary antibodies against HSP90 (Cell Signaling, Danvers, MA, USA). After washing, the antibody-protein complexes were probed with appropriate secondary antibodies tagged with horseradish peroxidase for 1 hr at room temperature and detected with chemiluminescent reagents. Protein bands were quantified using the software Quantity One (Bio-Rad, Hercules, CA, USA). In order to re-probe membranes for loading controls, membranes were stripped with the Re-Blot Western Blot Recycling Kit (Millipore, Billerica, MA, USA).
GAPDH was used as a loading control for cytoplasmic HSP90 expression. We sought to measure Na+/K+-ATPase as a loading control for membrane HSP90 expression but found that Na+/K+-ATPase expression itself was changed by treatment with LPS or DMSO. From the literature, other possible loading controls specifically expressed in the membrane (e.g., cadherin) also are affected by LPS (Simiantonaki et al., 2007; Veszelka et al., 2006). Membrane HSP90 expression was therefore quantified without a loading control.
In Western blots of cytoplasmic and membrane protein fractions probed for HSP90, a single band at approximately 90 kDa was observed, corresponding to the known molecular weight of HSP90. When analyzing HSP90 expression in supernatants, however, we observed 2 bands per sample (at approximately 55 and 65 kDa), suggesting that HSP90 was degraded during the lyophilization process. For each supernatant sample, both of these bands were used for quantification.
Experiment 1. Effect of intrathecal lipopolysaccharide (LPS) on pain threshold
After recording of baseline (BL) withdrawal thresholds (von Frey test), rats were injected intrathecally over lumbosacral spinal cord with either 0 (saline vehicle), 2 or 100 µg LPS (n=6/group). Withdrawal thresholds of the vehicle and 2 µg LPS groups were then retested 3 and 24 hr later. As the 100 µg LPS data were collected for comparison with the data in Experiments 7 and 8, below, von Frey testing was performed at BL and 24 hr.
Experiment 2. Effect of a systemically delivered HSP90 inhibitor (geldanamycin) on CCI-induced allodynia
The HSP90 inhibitor geldanamycin (Taldone et al., 2008) was examined for its effect on CCI-induced mechanical allodynia, so to explore whether a HSP cofactor may be required for TLR4-mediated neuropathic pain. After recording pre-surgical (pre-CCI) withdrawal thresholds (von Frey test), sciatic nerve injury (CCI) was performed. Ten days later, post-CCI pre-drug BL withdrawal thresholds were recorded. Rats were then injected subcutaneously at the nape of the neck with either 0 (vehicle), 20 or 50 µg/kg geldanamycin (n= 6/group). Withdrawal thresholds were then retested 3 hr later. Geldanamycin was used here, rather than 17-DMAG as in subsequent studies, as geldanamycin (but not 17-DMAG) is blood brain barrier permeable.
Experiment 3. Effect of an intrathecally delivered HSP90 inhibitor (17-DMAG) on CCI-induced allodynia
To define if HSP90 involvement in CCI-induced allodynia occurs within the spinal cord, pre-surgical (pre-CCI) withdrawal thresholds (von Frey test) were first recorded, followed by sciatic damage (CCI). Ten days later, post-CCI pre-drug BL withdrawal thresholds were again recorded. Rats were then injected intrathecally over lumbosacral spinal cord with either 0 (vehicle) or 10 µg 17-DMAG (n= 6/group), a second generation geldanamycin derivative (Taldone et al., 2008) used here to confirm that similar results could be obtained with a second, structurally distinct, HSP90 inhibitor. Withdrawal thresholds were then retested 1 and 3 hr later, with the 1 hr timepoint added to the time course followed in the prior study, given the anticipated faster onset of effect with intrathecal drug delivery.
Experiment 4. Effect of intrathecal 17-DMAG on intrathecal morphine analgesia
To define whether a HSP90 inhibitor could potentiate morphine-induced analgesia (a phenomenon previously reported to be enhanced by TLR4 inhibitors (Hutchinson et al., 2007; Watkins et al., 2009), baseline Hargreaves response latencies were recorded followed by intrathecal delivery of 15 µg morphine plus either 17-DMAG (4 µg) or vehicle (n= 6/group). Withdrawal thresholds were then retested across a time course through 175 min post-drug.
Experiment 5. Effect of dimethyl sulfoxide (DMSO) on HSP90 expression and, on LPS-induced TLR4 signaling in vitro
Prior to the undertaking of Experiment 6, it was first necessary to define whether DMSO could increase HSP90 expression and, in turn, TLR4 signaling in response to LPS. DMSO, rather than HSP90 itself was used here and in studies below based on the problematic history HSPs have had with LPS contamination that would confound interpretation of the results (Marincek et al., 2008). Thus, the induction of HSP90 expression by DMSO was tested using HEK-TLR4 cells incubated with either aCSF alone, 2% DMSO in aCSF, LPS (100 ng/ml) in aCSF or 2% DMSO + LPS (100 ng/ml) for 24 hr, then supernatant collected and cells fractionated followed by quantification of HSP90 protein (n=8–9 per condition). For DMSO induced TLR4 signaling, HEK-TLR4 cells were incubated (each condition in triplicate) with either 0 (media), 0.00000001, 0.0000001, 0.000001, 0.00001, 0.0001, 0.001, 0.01, 0.1, 1, 10 or 100 ng/ml LPS combined with media containing either 0, 0.1%, 1% or 2% DMSO. Supernatants were collected and assayed for secreted alkaline phosphatase (SEAP) activity 24 hr later.
Experiment 6. Effect of 17-DMAG on in vitro potentiation of LPS-induced TLR4 signaling by DMSO
To define whether a HSP90 inhibitor would block the enhancement of LPS-induced signaling by DMSO, HEK-TLR4 cells were incubated in media with LPS (0, 1, 10 or 100 ng/ml), DMSO (0 or 2%), and 17-DMAG (0, 0.01, 0.1, or 1 µg) (all conditions in triplicate). Supernatants were collected and assayed for SEAP activity 24 hr later.
Experiment 7. Effect of intrathecal LPS combined with intrathecal DMSO on pain threshold
To test whether co-administration of DMSO with LPS could induce mechanical allodynia, rats were first assessed for BL withdrawal thresholds (von Frey test) and then injected intrathecally with either 1 µg LPS, 4 µl DMSO, or the combination of 1 µg LPS plus 4 µl DMSO (n= 6/group). Withdrawal thresholds were then retested 3 and 24 hr later.
Experiment 8. Characterization of mechanical allodynia induced by intrathecal LPS+DMSO: Effect of treatment with inhibitors of HSP90, TLR, and microglial activation prior to induction of allodynia
To begin to define the mechanisms underlying allodynia induced by co-administration of DMSO and LPS, rats were first assessed for pre-drug BL withdrawal thresholds (von Frey test) and then injected intrathecally over lumbosacral spinal cord with 1 µg LPS plus 4 µl DMSO as in Experiment 7. In addition, at this same time, each rat was co-administered either vehicle, 17-DMAG (10 µg), (+)-naloxone (20 µg), or minocycline (100 µg) (n=6/group). Withdrawal thresholds were then retested 24 hr later (n=6/group).
Experiment 9. Characterization of mechanical allodynia induced by intrathecal LPS+DMSO: Effect of treatment with interleukin-1 (IL-1) inhibitor after induction of allodynia
To test whether IL-1ra would reverse DMSO + LPS induced mechanical allodynia, rats were first assessed for pre-drug BL withdrawal thresholds (von Frey test) and then injected intrathecally over lumbosacral spinal cord with 1 µg LPS plus 4 µl DMSO. After confirming the development of mechanical allodynia 24 hr later, rats were briefly re-anesthetized and injected intrathecally with either vehicle or IL-1ra (100 µg) (n= 6/group). Withdrawal thresholds were then retested 1 hr later.
Statistics
Data from the von Frey test were analyzed as the interpolated 50% thresholds (absolute threshold) in log base 10 of stimulus intensity (monofilament stiffness in milligrams × 10). Data from the Hargreaves test were calculated as the % of maximal possible effect (%MPE) using the following equation: (Carmody, 1995). All analyses and calculations were conducted with Excel 2003 SP2 (Microsoft), R Project version 2.6.1, SPSS 14.0.1 (SPSS) and Prism 5.0 (GraphPad). Significance was set at p<0.05. Pre-drug baseline measures were analyzed by one-way ANOVA. Post-drug time course measures were analyzed by repeated measures two-way ANOVAs followed Bonferroni post-hoc tests, where appropriate. Cell culture data were analyzed by ANOVA. Statistical significance was set at p<0.05.
RESULTS
Experiment 1. Failure of lipopolysaccharide (LPS), a classic TLR4 agonist, to produce mechanical allodynia following intrathecal administration
Given prior studies documenting the importance of spinal cord TLR4 for the initiation and maintenance of neuropathic pain (Tanga et al., 2005, Hutchinson et al., 2008b), we tested whether pain enhancement would also be induced by direct stimulation of spinal TLR4 with LPS, a classic TLR4 agonist. LPS was chosen for use here as the TLR4 agonist, as no study has yet defined which endogenous substance(s), released as a consequence of peripheral nerve injury, are the proximate stimulus for TLR4 activation under conditions of neuropathic pain (Tanga et al., 2005, Hutchinson et al., 2008b). Surprisingly, pilot studies with intrathecal LPS doses ranging from 2 to 100 µg LPS failed to induce mechanical allodynia across time courses up to 24 hr after injection (data not shown). The 2 µg and 100 µg doses were tested with full groups here. Figure 1 illustrates that 2 µg LPS intrathecal dose failed to induce mechanical allodynia across a time course. As the intent of including the 100 µg LPS dose here was to allow comparison of the 100 µg results to data to be presented in Experiments 7 and 8 (where only BL and 24 hr data were assessed), the 100 µg dose was tested only at 24 hr and also failed to affect pain thresholds at this timepoint (Figure 1).
Figure 1. Lack of mechanical allodynia through 24hr after 2 or 100 µg LPS.

Following von Frey testing prior to drug delivery (baseline; BL), rats were intrathecally administered either 0 (saline vehicle), 2 or 100 µg LPS. Von Frey responses were again recorded 3 and/or 24 hr later, with the 24 hr timepoint based on knowledge of the time course of effects known from the data illustrated in Figure 6 and Figure 7. n=6/group
Experiment 2. Chronic Constriction Injury (CCI)-induced allodynia, a TLR4-dependent form of neuropathic pain, is suppressed by a systemically delivered HSP90 inhibitor (geldanamycin)
The failure of intrathecal LPS to enhance pain suggests that TLR4 binding by this classic TLR4 agonist may not be sufficient to produce allodynia following intrathecal administration in naive rats. This raises the question of whether the previously reported TLR4 mediation of neuropathic pain (Tanga et al., 2005, Hutchinson et al., 2008b) implies that such TLR4 signaling is dependent upon a co-factor to enhance TLR4 signaling and, if so, what that co-factor may be. One candidate is HSP90, as it has been documented to be an important co-factor that enhances TLR4 signaling but is not itself a TLR4 receptor agonist (Byrd et al., 1999, Triantafilou et al., 2008). Given that (a) activated glia, as well as tissue stress/trauma, can produce HSPs, including HSP90 (Jeon et al., 2004), and (b) that HSPs are upregulated in, and likely released by, proximal axons of damaged sensory neurons (Costigan et al., 1998, Osterloh and Breloer, 2008), we explored whether HSP90 may be an important co-factor for maintaining a previously documented TLR4-dependent neuropathic pain state; that is, mechanical allodynia induced by CCI.
Prior to CCI, no differences in response thresholds were observed between groups (Figure 2A; Pre-CCI). Ten days after CCI, reliable mechanical allodynia was observed which, again, was comparable between groups (Figure 2A; Baseline). While systemic vehicle failed to affect response thresholds, systemic administration of the blood brain barrier permeable HPS90 inhibitor geldanamycin (Whitesell et al., 1994, Whilesell and Cook, 1996) reliably reduced mechanical allodynia (Figure 2A; 3 hr) (posthoc analyses compared to vehicle: p< 0.01 for 20 µg/kg geldanamycin; p <0.001 for 50 µg/kg geldanamycin).
Figure 2. Reversal of established chronic constriction injury (CCI)-induced mechanical allodynia by 2 inhibitors of HSP90.
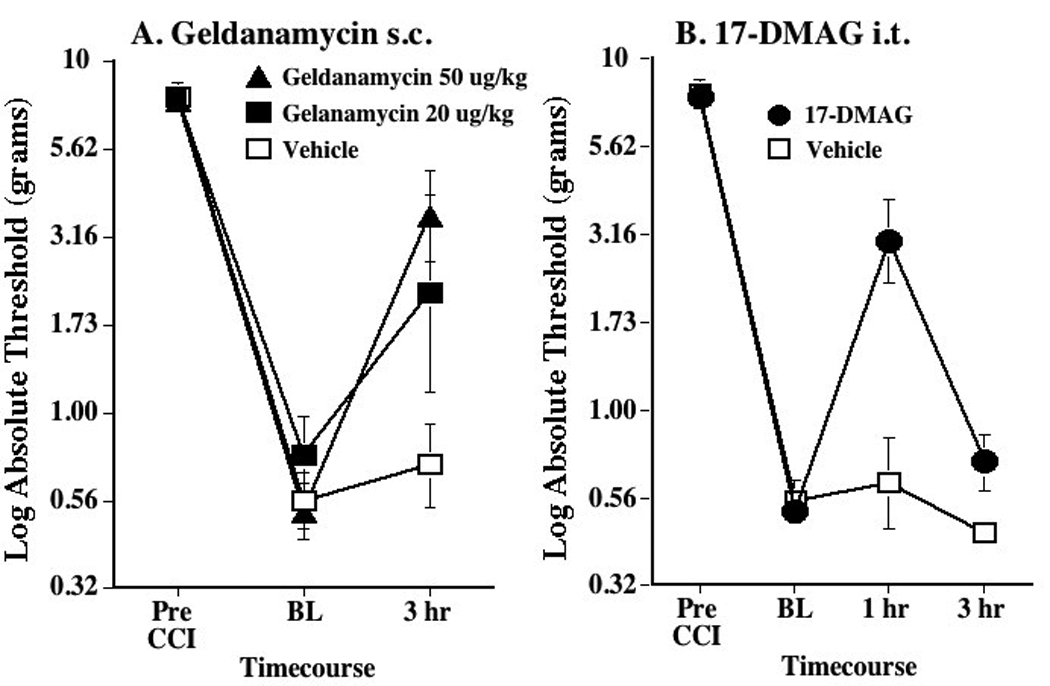
Panel A: Reversal of CCI-induced mechanical allodynia by systemic geldanamycin. Following von Frey testing prior to CCI surgery (Pre-CCI), the sciatic nerve was loosely ligated, causing the fall in pain threshold recorded prior to systemic drug delivery (baseline; BL). Whereas subcutaneous (s.c.) vehicle produced no reliable reversal of allodynia (open squares), both 20 (filled squares) and 50 (filled triangles) µg/kg s.c. geldanamycin reduced allodynia at the 3 hr timepoint. Panel B: Reversal of CCI-induced mechanical allodynia by intrathecal 17-DMAG. Following von Frey testing prior to CCI surgery (Pre-CCI), the sciatic nerve was loosely ligated, causing the fall in pain threshold recorded prior to systemic drug delivery (baseline; BL). Whereas intrathecal (i.t.) vehicle produced no reliable reversal of allodynia (open squares), 10 µg i.t. 17-DMAG (filled circles) reduced allodynia at the 1 hr timepoint. n=6/group
Experiment 3. Chronic Constriction Injury (CCI)-induced allodynia, a TLR4-dependent form of neuropathic pain, is suppressed by an intrathecally delivered HSP90 inhibitor (17-DMAG)
While Experiment 2 suggests an important involvement of HSP90 in TLR4 dependent, CCI-mediated neuropathic pain, the site of HSP90 involvement cannot be defined based on systemic administration, as HSP90 could logically be involved, at minimum, at either the sciatic nerve injury site or spinal cord. Thus, to define whether a spinal site of action of HSP90 is likely, Experiment 2 was repeated using intrathecal delivery of a HSP90 inhibitor. To provide converging lines of evidence for HSP90 involvement, a second HSP90 inhibitor (17-DMAG) (Jez et al., 2003) was employed here.
Prior to CCI, no differences in response thresholds were observed between groups (Figure 2B; Pre-CCI). Ten days after CCI, reliable mechanical allodynia was observed which, again, was comparable between groups (Figure 2B; Baseline). While intrathecal vehicle failed to affect response thresholds, intrathecal administration of the HPS90 inhibitor 17-DMAG (10 µg) reliably reduced mechanical allodynia (Figure 2C) 1 hr later (posthoc analyses compared to vehicle: p< 0.001 for 17-DMAG at 1 hr), an effect that resolved by 3 hr.
Experiment 4. Intrathecal morphine analgesia, a TLR4-damped form of pain suppression, is enhanced by an intrathecally delivered HSP90 inhibitor (17-DMAG)
As a further test of the hypothesis that spinal cord TLR4-mediated pain enhancement requires a cofactor such as HSP, the intrathecal effect of a HSP90 inhibitor (17-DMAG) was tested for its ability to enhance intrathecal morphine analgesia. This was explored because: (a) morphine is now known to activate TLR4 (Hutchinson et al., 2007; Hutchinson et al., 2009) and release spinal cord IL-1 that opposes, or counter-regulates, morphine analgesia as intrathecal IL-1ra enhances morphine analgesia (Hutchinson et al., 2008a), (b) TLR4 activation opposes morphine’s actions, as inhibiting TLR4 likewise enhances morphine analgesia (Hutchinson et al., 2007; Hutchinson et al., 2008a; Hutchinson et al., 2009). and (c) acute morphine can increase HSPs including HSP90 (Ammon-Treiber et al., 2004, Salas et al., 2007), raising the possibility that TLR4 modulation of morphine’s actions, like neuropathic pain, requires a cofactor such as HSP. If the concept that spinal TLR4 signaling requires a cofactor such as HSP to enhance pain were correct, then it would be predicted that morphine analgesia would be potentiated by blocking HSP90, just as it is by blocking TLR4.
Prior to drugs, all groups exhibited comparable thermal response thresholds (Figure 3). Neither intrathecal vehicle + intrathecal vehicle nor intrathecal 17-DMAG + intrathecal vehicle altered response thresholds through 175 min. While morphine produced analgesia, this effect was reliably potentiated in both magnitude and duration by co-administered 17-DMAG (Figure 3) (posthoc analyses compared to vehicle: p<0.05 85–145 min).
Figure 3. Potentiation of intrathecal (i.t.) morphine by i.t. 17-DMAG, a HSP90 inhibitor.

Following pre-drug baseline (BL) assessment on the Hargreaves test, rats were administered drugs and tested each 10 min for 175 min. While neither i.t. Vehicle + i.t. Vehicle (open squares) nor i.t. 17-DMAG (4 µg) + i.t. vehicle (filled squares) altered pain threshold, analgesia was induced by i.t. vehicle + i.t. morphine (15 µg), with a return to BL levels by 95–115 min. Analgesia produced by this dose of morphine was marked potentiated by coadministration of 4 µg 17-DMAG (filled circles), with a return to BL levels by 155–175 min later. n=6/group
Experiment 5. HSP90 expression and TLR4 signaling in vitro are enhanced by DMSO
The goal of this study was to define whether a drug known to increase HSPs (DMSO) would, as anticipated from the literature (Yufu et al., 1990, Hallare et al., 2004, Bini et al., 2008), enhance TLR4 signaling in vitro (Xing and Remick, 2005, Shuto et al., 2007). If so, and if a known HSP90 antagonist would block this enhanced TLR4 signaling, this would then allow these drugs to be used in subsequent in vivo experiments to test the idea that increasing HSP90 concomitantly with LPS administration would now produced enhanced pain through TLR4 signaling.
As the first step toward defining an appropriate paradigm to approach this issue, a compound previously documented to be capable of inducing HSP (dimethyl sulfoxide; DMSO) (Yufu et al., 1990, Hallare et al., 2004, Bini et al., 2008) was tested in vitro with HEK-TLR4 to examine the changes in expression of HSP90 and the location (supernatant, cytosol or cell membrane) where these changes occurred. There was no significant difference in cytoplasmic HSP90 expression between any of the 4 groups, whereas there was a significant relative decrease in membrane HSP90 in cells treated with DMSO (27 ± 9% decrease; p < 0.05). In contrast, treatment with DMSO (126 ± 42% increase) or LPS + DMSO (133 ± 32% increase) each produced a significant, greater than two-fold relative increase in HSP90 expression in the supernatant, as compared with supernatant from control cultures or cultures treated with LPS alone (p < 0.05), thereby confirming prior evidence of induction of HSP90 expression by DMSO.
Given the DMSO induction of HSP90 expression, HEK-TLR4 cells were then tested to define whether, as predicted, DMSO would enhance TLR4 signaling, as measured by induced SEAP activity. Following 24 hr HEK-TLR4 cell incubation with LPS, with vs. without concomitant exposure to DMSO, analysis of SEAP activity in the supernatants revealed a potentiation of TLR4 activation by DMSO. As seen in Figure 4, the presence of 0.1, 1 or 2% DMSO in the incubation media significantly enhanced SEAP production by the HEK-TLR4 cells in response to LPS. While none of the DMSO doses potentiated SEAP activity in the absence of LPS (see “Media + DMSO” on Figure 4), all DMSO doses elevated SEAP activity both for LPS dose ranges which were apparently ineffective in inducing SEAP in the absence of DMSO (10−8 to 10−3 ng/ml LPS) (p<0.05) as well as for LPS dose ranges where LPS was sufficient to reporter protein induction in the absence of DMSO (10−2 to 103 ng/ml LPS) (p<0.05). This enhancement is also evident in the significant (p<0.0001) increase in the maximal response and significant decrease in the EC50 of LPS with DMSO (0.027 ng/ml LPS + aCSF vs. 0.007 ng/ml LPS + 2% DMSO).
Figure 4. Potentiation of TLR4 signaling in vitro by dimethyl sulfoxide (DMSO).

To test whether DMSO might enhance TLR4 signaling, HEK-TLR4 cells were incubated with 0 (media), 1, 10 or 100 ng/ml LPS, either 0 or 2% DMSO (each condition run in triplicate and either 0 (media), 0.00000001, 0.0000001, 0.000001, 0.00001, 0.0001, 0.001, 0.01, 0.1, 1, 10 or 100 ng/ml LPS combined with either 0, 0.1%, 1% or 2% DMSO in the media. Supernatants were collected and assayed for secreted alkaline phosphatase (SEAP) activity 24 hr later. SEAP activity, across LPS log doses are plotted for media (no DMSO; filled squares), 01% DMSO (open squares), 1% DMSO (open circles), and 2% DMSO (open diamonds). Each DMSO dose enhanced TLR4 signaling, as measured by SEAP activity. n=6/group
Experiment 6. Enhanced TLR4 signaling by DMSO is blocked by the HSP90 inhibitor, 17-DMAG
Given that Experiment 5 supports that DMSO does enhance TLR4 signaling, the next step was to define whether this increased signaling would be blocked by a HSP90 inhibitor (17-DMAG). Following 24 hr HEK-TLR4 cell incubation with LPS, with vs. without concomitant exposure DMSO, and with vs. without concomitant exposure to 17-DMAG, there were clear effects on TLR4-induced SEAP activity. As seen in Figure 5A, only the highest 17-DMAG dose (1 µg) suppressed TLR4 signaling in the absence of DMSO (p<0.01), suggestive of a relatively minor contribution of HSP90 to LPS-induced TLR4 signaling in the absence of DMSO. In contrast, as seen in Figure 5B, all doses of 17-DMAG (0.01, 0.1 and 1 µg) suppressed DMSO enhanced TLR4 signaling in the absence of LPS, returning TLR4 signaling to the levels observed in Figure 5A, in the absence of DMSO. In addition, all doses of 17-DMAG markedly suppressed the DMSO-enhancement of LPS-induced TLR4 signaling across all LPS doses tested (1, 10, and 100 ng/ml) (Figure 5B), suggestive of a robust contribution of HSP90 to the effects observed. These data are also supportive that elevations in HSP70 are unlikely to account for the effects of DMSO observed, despite the fact that DMSO can elevate expression of HSP70 (Hallare et al., 2004) and HSP70 (like HSP90) can facilitate TLR4 signaling (Triantafilou et al., 2001b, Triantafilou and Triantafilou, 2004). This is because HSP90 inhibitors, including 17-DMAG and geldanamycin, elevate HSP70 expression as well (Kwon et al., 2008). Thus, if HSP70 were to importantly contribute to these phenomena, one would have anticipated that 17-DMAG would have created, if anything, a further potentiation of TLR4 signaling instead of the DMSO-selective blockade observed.
Figure 5. Blockade of DMSO-induced potentiation of TLR4 signaling in vitro by the HSP90 inhibitor, 17-DMAG.
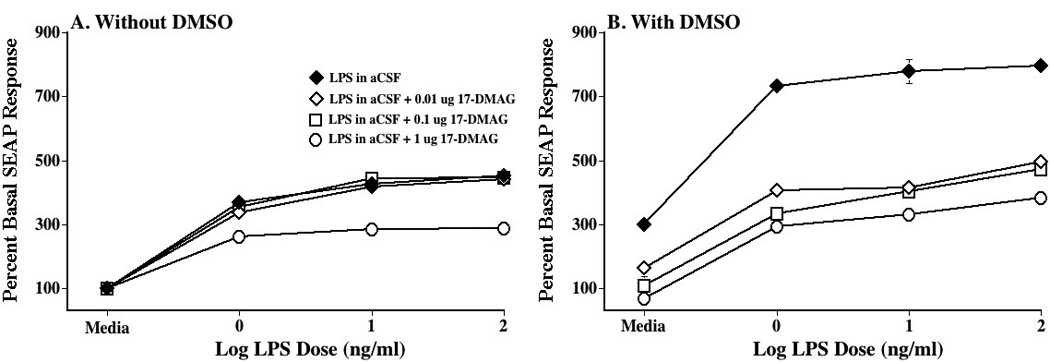
HEK-TLR4 cells were incubated with 0 (media), 1, 10 or 100 ng/ml lipopolysaccharide (LPS), either 0 or 2% DMSO (n= 3 replicates/condition), and either 0 (media; filled diamond), 0.01 (open diamond), 0.1 (open square), or 1 µg (open circle) 17-DMAG. Supernatants were collected and assayed for secreted alkaline phosphatase (SEAP) activity 24 hr later. Panel A: In the absence of DMSO, the HSP90 inhibitor had no effect except at the highest dose, which moderately suppressed TLR4 signaling. Panel B: In the presence of DMSO, TLR4 signaling was robustly enhanced in the absence of the HSP90 inhibitor (filled circle). All 3 17-DMAG doses (0.01 µg: open diamond; 0.1 µg: open square; 1 µg: open circle) markedly suppressed TLR4 signaling, returning SEAP activity to approximately the level obtained in the absence of DMSO (see Panel A). n=triplicate wells per condition
Experiment 7. Intrathecal DMSO co-administered with intrathecal LPS induces mechanical allodynia
As the experiments above indicate that DMSO increases LPS-induced TLR4 signaling in a HSP90-dependent manner, the question arises whether co-administration of DMSO with LPS intrathecally would now result in mechanical allodynia, in contrast to the failure of LPS alone (Experiment 1). This was indeed the result observed. Groups were comparable in mechanical response thresholds prior to intrathecal drugs (Figure 6, BL). Neither LPS alone nor DMSO alone produced more than a transient mild reduction in response thresholds, with little to no effects observed at 24 hr. In contrast, rats coadministered the same doses of LPS and DMSO exhibited reliable, robust allodynia 24 hr later (posthoc analyses compared to vehicle: p<0.05 24 hr post administration).
Figure 6. Induction of mechanical allodynia by intrathecal (i.t.) lipopolysaccharide (LPS) co-administered with i.t. dimethyl sulfoxide (DMSO).

After recording of pre-drug baseline (BL) withdrawal thresholds (von Frey test), rats were injected i.t. over lumbosacral spinal cord with either 1 µg LPS (open diamond), 4 µl DMSO (open square), or the combination of 1 µg LPS plus 4 µl DMSO (filled circle). Withdrawal thresholds were then retested 3 and 24 hr later. No between group differences were observed at BL or 3hr after i.t. injection. At 24 hr, only LPS+DMSO produced mechanical allodynia. n=6/group
Experiment 8. Allodynia induced by intrathecal DMSO co-administered with intrathecal LPS is prevented by blocking HSP90, TLR4 or microglial activation
Experiment 7 documents that intrathecal administration of DMSO converts an ineffective intrathecal dose of LPS into an effective dose for inducing mechanical allodynia. If this effect of DMSO is indeed due to an enhancement of TLR4 signaling, several predictions ensue. Namely, allodynia should be prevented by: (a) a HSP90 inhibitor (17-DMAG), as DMSO is assumed to act via HSP90 induction (Yufu et al., 1990; Hallare et al., 2004; Bini et al., 2008); (b) (+)-naloxone, a recently documented TLR4 antagonist (Hutchinson et al., 2008b); and (c) minocycline, as TLR4 is predominantly expressed by microglial cells and macrophges (Olson and Miller, 2004) and there is no evidence to date that TLR4 is expressed by neurons in spinal cord.
Indeed these were the results obtained. All groups were comparable in their pre-drug baseline withdrawal thresholds (Figure 7A,B,C; BL). In each case, LPS+DMSO+Vehicle induced mechanical allodynia (p<0.05 compared to BL). This allodynia was reliable reduced (p<0.05) by 17-DMAG (Figure 7A), (+)-naloxone (Figure 7B), and minocycline (Figure 7C).
Figure 7. Characterization of mechanical allodynia induced by intrathecal (i.t.) lipopolysaccharide (LPS) co-administered with i.t. dimethyl sulfoxide (DMSO): Effect of treatment with inhibitors of HSP90, TLR, and microglial activation prior to induction of allodynia.

After recording of pre-drug baseline (BL) withdrawal thresholds (von Frey test), rats were injected i.t. over lumbosacral spinal cord with 1 µg LPS plus 4 µl DMSO as in Experiment 7. In addition, each rat was co-administered either vehicle (open square) or 17-DMAG (10 µg; filled square; Panel A), (+)-naloxone (20 µg; filled circle; Panel B), or minocycline (100 µg; filled diamond; Panel C). Withdrawal thresholds were then retested 24 hr later. All 3 test agents reliably reduced mechanical allodynia at 24 hr. n=6/group
Experiment 9. Allodynia induced by intrathecal DMSO co-administered with intrathecal LPS is reversed by blocking IL-1
Based on the results from Experiment 8, a final prediction would be that IL-1 would be a likely candidate for the effects observed. This is based on the production of IL-1 by activated microglia and macrophages (Mika, 2008), its production in response to TLR4 activation (Osterloh and Breloer, 2008), its known ability to enhance pain upon intrathecal administration (Reeve et al., 2000), and its involvement in both TLR4 modulated neuropathic pain (Tanga et al., 2005) and TLR4 modulated attenuation of morphine analgesia (Hutchinson et al., 2007; Hutchinson et al., 2009; Watkins et al., 2009). Hence, the effect of IL-1 receptor antagonist (IL-1ra) was tested for its ability to reverse established LPS+DMSO-induced mechanical allodynia. This would assess whether DMSO+LPS-induced allodynia can be reversed and/or prevented by pharmacological interventions, and explore whether IL-1 mediates the observed allodynia here.
Indeed these were the results obtained. Groups were comparable in their pre-drug baseline withdrawal thresholds (Figure 8; BL). Again, LPS+DMSO induced comparable mechanical allodynia in both groups prior to administration of IL-1ra or vehicle (for each, p<0.05 compared to BL). While vehicle failed to affect this allodynia, allodynia was reliably reversed by IL-1ra (Figure 8; p<0.05).
Figure 8. Characterization of mechanical allodynia induced by intrathecal LPS+DMSO: Effect of treatment with IL-1 inhibitor after induction of allodynia.

After recording of pre-drug baseline (BL) withdrawal thresholds (von Frey test), rats were injected intrathecally over lumbosacral spinal cord with 1 µg LPS plus 4 µl DMSO. After confirming the development of mechanical allodynia 24hr later, rats were injected intrathecally with either vehicle (open square) or IL-1ra (100 µg; filled square). Withdrawal thresholds were then retested 1hr later. IL-1ra reversed the LPS+DMSO induced mechanical allodynia. n=6/group
Discussion
While previous reports demonstrate a role of TLR4 in neuropathic pain (Tanga et al., 2005, Hutchinson et al., 2008b) and in opposing morphine analgesia (Hutchinson et al., 2007, Hutchinson et al., 2008a, Watkins et al., 2009), both pilot studies of intrathecal LPS (2–100 µg) and Experiment 1 (2 and 100 µg intrathecal LPS) document that this classic TLR4 agonist failed to produce allodynia through 24 hr. These results complement, and are supported by, prior publications that document a failure to observe pain enhancement following intrathecal LPS (Meller et al., 1994). This result suggests that, under conditions of neuropathic pain and opioid exposure, a cofactor may facilitate TLR4 signaling (Hutchinson et al., 2009). As HSP90 has been previously implicated in such a role despite not being a TLR4 receptor agonist on its own (Byrd et al., 1999, Triantafilou et al., 2001a, Triantafilou and Triantafilou, 2004, Triantafilou et al., 2008), its potential involvement in TLR4-mediated neuropathic pain and morphine analgesia was explored. Indeed, systemic and intrathecal HSP90 inhibitors suppressed neuropathic pain induced by chronic constriction nerve injury (CCI), a model dependent on spinal TLR4 for expression of allodynia (Hutchinson et al., 2008b). A second TLR4-dependent phenomenon was then examined to define whether HSP90 involvement may be generalizable across models. The effect of an intrathecal HSP90 inhibitor was tested on intrathecal morphine analgesia, as morphine analgesia is potentiated by TLR4 antagonists (Hutchinson et al., 2008b) and morphine can elevate HSP90 (Salas et al., 2007). Intrathecal HSP90 inhibition produced a marked potentiation of analgesia, as do TLR4 antagonists (Hutchinson et al., 2007, Hutchinson et al., 2008a, Hutchinson et al., 2008b). This predicted that LPS should produce allodynia, were one to be able to enhance HSP90. This was the effect observed. After defining in vitro that DMSO increases HSP90 expression within the supernatant and that TLR4 signaling is blocked by HSP90 inhibition, this same paradigm was tested intrathecally. While LPS (2 µg to 100 µg) fails to produce mechanical allodynia, 1 µg LPS co-administered intrathecally with 4 µl DMSO produced robust allodynia. This occurred via a HSP90-TLR4 mechanism, as it was blocked or reversed by intrathecal co-administration of a HSP90 inhibitor, a TLR4 inhibitor, a microglia/macrophage activation inhibitor (as microglia/macrophages are the predominant cell types expressing TLR4) (Olson and Miller, 2004), and interleukin-1 receptor antagonist (as this proinflammatory cytokine is a downstream consequence of TLR4 activation) (Osterloh and Breloer, 2008). Together, these results suggest for the first time that TLR4 activation is necessary but not sufficient to induce spinally mediated pain enhancement. Rather, the data suggest that TLR4-dependent pain phenomena require contributions by multiple components of the TLR4 receptor complex.
It is unlikely that the sensitization of LPS-induced mechanical allodynia is due to increased tissue penetrance created by 4 µl DMSO. This is because DMSO is not required for intrathecal efficacy of LPS analogs that are TLR4 antagonists, yet structurally very similar to the TLR4 agonist LPS used here. We have previously studied the effects of intrathecal delivery two such LPS structures, man-made mutant LPS and LPS-RS, a naturally occurring LPS that (like mutant LPS) avidly binds to but does not activate TLR4. Within 1–2 hr of intrathecal delivery in the absence of DMSO, these LPS analogs (20–40 µg) reverse CCI-induced allodynia (Hutchinson et al., 2008b) and potentiate intrathecal morphine analgesia (Hutchinson et al., 2007; Hutchinson et al., 2009; Watkins et al., 2009). Clearly, such chemical structures can penetrate the spinal cord to reach TLR4-modulated pain regulatory sites. The inability of 100 µg of intrathecal LPS to alter pain threshold in the absence of DMSO, versus the potent allodynia induced by 1 µg LPS coad-ministered with 4 µl DMSO suggests that DMSO induces a cofactor that enhances TLR4 signaling, rather than exerting its effects through altered LPS permeability. Based on the data provided here, HSP90 is a high probability cofactor induced by DMSO. Whether other cofactors are also induced is as yet unknown.
The failure of LPS to induce allodynia agrees with Meller et al. (1994), who reported no allodynia with 150 µg intrathecal LPS doses. Interestingly, far greater effects of LPS on pain thresholds have been observed using a “priming” paradigm. Here, a first LPS dose is used to sensitize glial response to a challenge delivered 24 hr later. While 0.2 µg intracerebroventricular (ICV) LPS did not alter pain thresholds, priming with this ICV dose 24 hr prior to testing with a second ICV 0.2 µg LPS dose produced allodynia (Cahill et al., 1998). Immunohistochemistry revealed upregulation of glial activation markers in response to the priming dose 24 hr previously (Cahill et al., 1998). Similarly, 20 µg intrathecal LPS failed to enhance pain yet priming rats intrathecally with 2 µg LPS 24 hr prior to a second 20 µg dose of LPS produced reliable mechanical allodynia (Cahill et al., 2003). Given the results of the present series of studies, it will be intriguing to define whether such “priming” effects may arise, at least in part, via an upregulation of HSP90.
While the mechanisms underlying such priming effects are unknown, it is intriguing that prior LPS upregulates HSP90 (Stanislawska et al., 2004, Narita et al., 2007) and upregulates TLR4 (Stanislawska et al., 2004). Thus one hypothesis would be that the prior LPS challenge upregulates the function of the TLR4 signaling pathway. If true, one would predict that prior LPS challenge that activates spinal cord glia should reduce later morphine analgesia, since TLR4 signaling is activated by opioids and opposes opioid analgesia (Hutchinson et al., 2007, Hutchinson et al., 2008a). And indeed, LPS 24 hr prior to morphine reduces morphine analgesia, an effect blocked by blocking spinal cord glial activation (Johnston and Westbrook, 2005). Similarly, it has been argued that peripheral neuropathy primes glia in the spinal cord to now over-respond to later morphine (Raghavendra et al., 2003b). In agreement with this hypothesis, blocking glial activation returns the analgesic responses of opioid-resistant neuropathic animals to normal ranges (Raghavendra et al., 2003b). Neuropathy (Tanga et al., 2005, Hutchinson et al., 2008b), like LPS cited above, upregulates TLR4. Whether HSPs are similarly upregulated is unknown but predicted from such results.
One intriguing aspect of HSP90 function is as a key component and modulator of the TLR4 receptor complex. While traditionally, HSPs are regarded as intracellular molecules, there has been intense interest in the past few years regarding newly recognized extracellular and membrane-bound functions of these molecules, including HSP90 (Tsan and Gao, 2004, Clayton et al., 2005). Extracellular HSPs, including HSP90, have been shown to induce the production of nitric oxide and proinflammatory cytokines and chemokines by TLR4-responsive cells (Tsan and Gao, 2004). While extracellular receptors other than TLR4, such as CD91, also bind HSP90 (Basu et al., 2001, Beg, 2002), these others are not signaling receptors. Rather, these are receptors that recognize HSPs complexed with foreign proteins requiring internalization and processing (Beg, 2002). Hence they are unlikely to account for the effects observed here. Likewise, a direct agonism of TLR4 by HSP90 does not account for the data as while some HSPs bind to some TLR family members, but there is no report of HSP90 as aTLR4 receptor agonist.
Classically, TLR4 signaling involves LPS binding first to CD14, which facilitates LPS transfer to MD2, an extracellular protein that, once bound to LPS, then itself binds to TLR4 so to induce TLR4 signaling (Akashi-Takamura and Miyake, 2008). In this model, MD2 is the primary LPS recognition molecule, but lacks the ability to signal intracellularly. TLR4 lacks the ability to bind LPS but is triggered to signal in response to LPS as a result of the above cascade (Akashi-Takamura and Miyake, 2008). Recent studies have modified this view to encompass a TLR4 protein complex regulating TLR4 signaling (Triantafilou et al., 2008). In an elegant series of studies, Triantafilou and colleagues have demonstrated that HSP90, as well as HSP70, are constitutively present and functionally integral components of the TLR4 receptor complex, along with TLR4, CD14, and MD2. HSP90, HSP70, and CD14 are present within the lipid raft, whereas TLR4 is recruited to the raft after exposure to LPS so to form a receptor cluster (Triantafilou and Triantafilou, 2004). HSP90 binds LPS and is involved in the TLR4 response, very early in the receptor-mediated cascade and likely at the receptor complex itself (Byrd et al., 1999). HSP90 is proposed, given its binding capacity for LPS, to be a transfer molecule within the lipid raft to facilitate delivery of LPS to the TLR4-MD2 complex (Triantafilou and Triantafilou, 2004). This is in keeping with the consensus that HSP90, as well as HSP70, are potent activators of the innate immune system, in response to LPS (Tsan and Gao, 2004).
A potential alternative explanation for the effects observed has been reported in a single article to date; namely, that geldanamycin can downregulate cell surface expression of CD14 (Vega and De Maio, 2003). It is unknown whether this occurs in response to LPS+DMSO, to morphine, and/or to neuropathic pain. Were this to be true, this would suggest that CD14 must bind to and facilitate transfer of the TLR4 activating molecules associated with, or induced by, LPS+DMSO, morphine, and/or endogenous signals released in the spinal cord in response to peripheral nerve injury. These issues require future study to clarify whether CD14 serves such a role. At present, what is known is that the geldanamycin-induced downregulation of ~50% of cell surface expression of CD14 in vitro requires 2–3 hr (Vega and De Maio, 2003). This suggests that such changes in CD14 expression may be too slow to account for the effects observed within 25 min to 1 hr after morphine or neuropathic pain, respectively, but remains to be explored.
Thus, in summary, the studies presented here suggest that TLR4 in spinal cord is necessary but not sufficient for inducing enhanced pain responses following binding by classic TLR4 receptor agonists such as LPS. Rather, the data are supportive of the conclusion that TLR4-dependent neuropathic pain and opposition to morphine analgesia each require a cofactor to enhance TLR4-dependent signaling. This cofactor is likely to be at minimum HSP90.
Acknowledgments
These studies were supported by an International Association for the Study of Pain International Collaborative grant, American Australian Association Merck Company Foundation Fellowship, National Health and Medical Research Council CJ Martin Fellowship (ID 465423) and NIH Grants DA015642, DA017670, DA024044 and DE017782. A portion of this work was supported by the NIH Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism. We thank Amgen for the gift of IL-1ra and its vehicle and Avigen for the gift of HEK-TLR4 cells.
Abbreviations
- %MPE
percent maximal possible effect
- 17-DMAG
17-dimethylaminoethylamino-17-desmethoxygeldanamycin
- aCSF
artificial cerebrospinal fluid
- ANOVA
analysis of variance
- BL
baseline
- CCI
chronic constriction injury
- CD14
cluster determinant 14
- DMSO
dimethyl sulfoxide
- HEK293
human embryonic kidney-293
- HSP
heat shock protein
- ICV
intracerebroventricularly
- IL-1
interleukin-1
- IL-1ra
interleukin-1 receptor antagonist
- LPS
lipopolysaccharide
- MD2
myeloid differentiation factor 2
- PE
polyethylene
- SEAP
secreted alkaline phosphatase
- TLR
toll like receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Akashi-Takamura S, Miyake K. TLR accessory molecules. Curr Opin Immunol. 2008;20:420–425. doi: 10.1016/j.coi.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Ammon-Treiber S, Grecksch G, Stumm R, Riechert U, Tischmeyer H, Reichenauer A, Hollt V. Rapid, transient, and dose-dependent expression of hsp70 messenger RNA in the rat brain after morphine treatment. Cell Stress Chaperones. 2004;9:182–197. doi: 10.1379/CSC-42.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- Beg AA. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol. 2002;23:509–512. doi: 10.1016/s1471-4906(02)02317-7. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bini R, Olivero G, Rombetta A, Castagna E, Cotogni P. Effects of dimethyl sulfoxide, pyrrolidine dithiocarbamate, and methylprednisolone on nuclear factor-kappaB and heat shock protein 70 in a rat model of hemorrhagic shock. J Trauma. 2008;64:1048–1054. doi: 10.1097/TA.0b013e318059362e. [DOI] [PubMed] [Google Scholar]
- Byrd CA, Bornmann W, Erdjument-Bromage H, Tempst P, Pavletich N, Rosen N, Nathan CF, Ding A. Heat shock protein 90 mediates macrophage activation by Taxol and bacterial lipopolysaccharide. Proc Natl Acad Sci U S A. 1999;96:5645–5650. doi: 10.1073/pnas.96.10.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Dray A, Coderre TJ. Priming enhances endotoxin-induced thermal hyperalgesia and mechanical allodynia in rats. Brain Res. 1998;808:13–22. doi: 10.1016/s0006-8993(98)00786-0. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Dray A, Coderre TJ. Enhanced thermal antinociceptive potency and anti-allodynic effects of morphine following spinal administration of endotoxin. Brain Res. 2003;960:209–218. doi: 10.1016/s0006-8993(02)03885-4. [DOI] [PubMed] [Google Scholar]
- Carmody J. Avoiding fallacies in nociceptive measurements. Pain. 1995;63:136. doi: 10.1016/0304-3959(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, Maier SF, Watkins LR. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain. 2001;94:231–244. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Clayton A, Turkes A, CNavabi H, Mason MD, Tabi Z. Induction of heat shock proteins in B-cell exosomes. J Cell Sci. 2005;118:3631–3638. doi: 10.1242/jcs.02494. [DOI] [PubMed] [Google Scholar]
- Costigan M, Mannion RJ, Kendall G, Lewis SE, Campagna JA, Coggeshall RE, Meridith-Middleton J, Tate S, Woolf CJ. Heat shock protein 27: developmental regulation and expression after peripheral nerve injury. J Neurosci. 1998;18:5891–5900. doi: 10.1523/JNEUROSCI.18-15-05891.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordh T, Chu H, Sharma HS. Spinal nerve lesion alters blood-spinal cord barrier function and activates astrocytes in the rat. Pain. 2006;124:211–221. doi: 10.1016/j.pain.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Hallare AV, Kohler HR, Triebskorn R. Developmental toxicity and stress protein responses in zebrafish embryos after exposure to diclofenac and its solvent, DMSO. Chemosphere. 2004;2004:659–666. doi: 10.1016/j.chemosphere.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Harvey LOJ. Efficient estimation of sensory thresholds. Behav Res Meth Istrum Comput. 1986;18:623–632. [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence and reward. TheScientificWorldJOURNAL. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Sprunger DB, Baker EM, Somogyi AA, Martin D, Poole S, Judd CM, Maier SF, Watkins LR. Proinflammatory cytokines oppose opioid induced acute and chronic analgesia. Brain, Behavior & Immunity. 2008a doi: 10.1016/j.bbi.2008.05.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown KD, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: Involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008b;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PR, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf HE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Ying H, Rice KC, Watkins LR. Evidence that opioids may have toll like receptor 4 and MD-2 effects. Brain, Behavior & Immunity. 2009 doi: 10.1016/j.bbi.2009.08.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon GS, Park SW, Kim DW, Seo JH, Cho J, Lim SY, Kim SD, Cho SS. Glial expression of the 90-kDa heat shock protein (HSP90) and the 94-kDa glucose-regulated protein (GRP94) following an excitotoxic lesion in the mouse hippocampus. Glia. 2004;48:250–258. doi: 10.1002/glia.20075. [DOI] [PubMed] [Google Scholar]
- Jez JM, Chen JC, Rastelli G, Stroud RM, Santi DV. Crystal structure and molecular modeling of 17-DMAG in complex with human HSP90. Chem Biol. 2003;19:361–368. doi: 10.1016/s1074-5521(03)00075-9. [DOI] [PubMed] [Google Scholar]
- Johnston IN, Westbrook RF. Inhibition of morphine analgesia by LPS: role of opioid and NMDA receptors and spinal glia. Behav Brain Res. 2005;156:75–83. doi: 10.1016/j.bbr.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Kwon HM, Kim Y, Yang SI, Kim YJ, Lee SH, Yoon BW. Geldanamycin protects rat brain through overexpression of HSP70 and reducing brain edema after cerebral focal ischemia. Neurol Res. 2008 doi: 10.1179/174313208X289615. [DOI] [PubMed] [Google Scholar]
- Kwon MS, Shim EJ, Seo YJ, Choi SS, Lee JY, Lee HK, Suh HW. Differential modulatory effects of cholera toxin and pertussis toxin on pain behavior induced by TNF-alpha, interleukin-1beta and interferon-gamma injected intrathecally. Arch Pharm Res. 2005;28:582–586. doi: 10.1007/BF02977762. [DOI] [PubMed] [Google Scholar]
- Marincek BC, Kuhnle MC, Srokowski C, Schild H, Hammerling G, Momburg F. Heat shock protein-antigen fusions lose their enhanced immunostimulatory capacity after endotoxin depletion. Mol Immunol. 2008;46:181–191. doi: 10.1016/j.molimm.2008.07.039. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Mika J. Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacol Rep. 2008;60:297–307. [PubMed] [Google Scholar]
- Milligan ED, Mehmert KK, Hinde JL, Harvey LO, Martin D, Tracey KJ, Maier SF, Watkins LR. Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res. 2000;861:105–116. doi: 10.1016/s0006-8993(00)02050-3. [DOI] [PubMed] [Google Scholar]
- Milligan ED, O'Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema RP, Holguin A, Martin D, Maier SF, Watkins LR. Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. J Neurosci. 2001;21:2808–2819. doi: 10.1523/JNEUROSCI.21-08-02808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O'Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Narita H, Talorete TP, Han J, Funamizu N, Isoda H. Human intestinal cells incubated with activated sludge and lipopolysaccharide express Hsp90b. Environ Sci. 2007;14:35–39. [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Osterloh A, Breloer M. Heat shock proteins: linking danger and pathogen recognition. Med Microbiol Immunol. 2008;197:1–8. doi: 10.1007/s00430-007-0055-0. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003a;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, Rutkowski MD, DeLeo JA. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain. 2003b;104:655–664. doi: 10.1016/S0304-3959(03)00138-6. [DOI] [PubMed] [Google Scholar]
- Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain. 2000;4:247–257. doi: 10.1053/eujp.2000.0177. [DOI] [PubMed] [Google Scholar]
- Salas E, Alonso E, Sevillano J, Herradon G, Bocos C, Morales L, Ramos MP, Alguacil LF. Morphine differentially regulates hsp90beta expression in the nucleus accumbens of Lewis and Fischer 344 rats. Brain Res Bull. 2007;73:325–329. doi: 10.1016/j.brainresbull.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Shuto T, Furuta T, Cheung J, Gruenert DC, Ohira Y, Shimasaki S, Suico MA, Sato K, Kai H. Increased responsiveness to TLR2 and TLR4 ligands during dimethylsulfoxide-induced neutrophil-like differentiation of HL-60 myeloid leukemia cells. Leuk Res. 2007;31:1721–1728. doi: 10.1016/j.leukres.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Simiantonaki N, Kurzik-Dumke U, Karyofylli G, Jayasinghe C, Kirkpatrick CJ. Loss of E-cadherin in the vicinity of necrosis in colorectal carcinomas: association with NFkappaB expression. Int J Oncol. 2007;31:269–275. [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Stanislawska J, Interewicz B, Olszewski WL. Influence of bacterial antigens on activation of human splenic dendritic cells. Ann Transplant. 2004;9:54–57. [PubMed] [Google Scholar]
- Taldone T, Gozman A, Maharaj R, Chiosis G. Targeting Hsp90: small molecule inhibitors and their clinical development. Curr Opin Pharmacol. 2008 doi: 10.1016/j.coph.2008.06.015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutwein B, Strasburger H. Fitting the psychometric function. Percept Psychophys. 1999;61:87–106. doi: 10.3758/bf03211951. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Triantafilou M, Dedrick RL. A CD14-independent LPS receptor cluster. Nat Immunol. 2001a;2:338–345. doi: 10.1038/86342. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Triantafilou M, Ladha S, Mackie A, Dedrick RL, Fernandez N, Cherry R. Fluorescence recovery after photobleaching reveals that LPS rapidly transfers from CD14 to hsp70 and hsp90 on the cell membrane. J Cell Sci. 2001b;114:2535–2545. doi: 10.1242/jcs.114.13.2535. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Sawyer D, Nor A, Vakakis E, Triantafilou K. Cell surface molecular chaperones as endogenous modulators of the innate immune response. Novartis Found Symp. 2008;291:74–79. doi: 10.1002/9780470754030.ch6. discussion 79–85, 137–140. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Triantafilou K. Heat-shock protein 70 and heat-shock protein 90 associate with Toll-like receptor 4 in response to bacterial lipopolysaccharide. Biochem Soc Trans. 2004;32:636–639. doi: 10.1042/BST0320636. [DOI] [PubMed] [Google Scholar]
- Tsan MF, Gao B. Cytokine function of heat shock proteins. Am J Physiol Cell Physiol. 2004;286:C739–C744. doi: 10.1152/ajpcell.00364.2003. [DOI] [PubMed] [Google Scholar]
- Vega VL, De Maio A. Geldanamycin treatment ameliorates the response to LPS in murine macrophages by decreasing CD14 surface expression. Mol Biol Cell. 2003;14:764–773. doi: 10.1091/mbc.E02-08-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veszelka S, Pasztoi M, Farkas AE, Krizbai I, Ngo TK, Niwa M, Abraham CS, Deli MA. Pentosan polysulfate protects brain endothelial cells against bacterial lipopolysaccharide-induced damages. Neurochem Int. 2007;50:219–228. doi: 10.1016/j.neuint.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Ward JR, Dower SK, Whyte MK, Buttle DJ, Sabroe I. Potentiation of TLR4 signalling by plasmin activity. Biochem Biophys Res Commun. 2006;341:299–303. doi: 10.1016/j.bbrc.2005.12.188. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends in Neurosciences. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Milligan ED, Maier SF. "Listening" and "talking" to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev. 2007;56:148–169. doi: 10.1016/j.brainresrev.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. The "toll" of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends in Pharmacological Sciences. 2009 doi: 10.1016/j.tips.2009.08.002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whilesell L, Cook P. Stable and specific binding of heat shock protein 90 by geld-anamycin disrupts glucocorticoid receptor function in intact cells. Mol Endocrinol. 1996;10:705–712. doi: 10.1210/mend.10.6.8776730. [DOI] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, Remick DG. Mechanisms of dimethyl sulfoxide augmentation of IL-1 beta production. J Immunol. 2005;174:6195–6202. doi: 10.4049/jimmunol.174.10.6195. [DOI] [PubMed] [Google Scholar]
- Yufu Y, Nishimura J, Ideguchi H, Nawata H. Enhanced synthesis of heat shock proteins and augmented thermototlerance after induction of differentiation in HL-60 human leukemia cells. FEBS Lett. 1990;268:173–176. doi: 10.1016/0014-5793(90)81001-5. [DOI] [PubMed] [Google Scholar]