

INTRODUCTION
Although allogeneic hematopoietic progenitor cell transplant (HPCT) is curative therapy for many disorders of lymphohematopoiesis, it is associated with significant morbidity and mortality.1–5 Much of this risk can be traced to graft-versus-host disease (GVHD) and the immunosuppressive measures required for its prevention and/or treatment.6,7 T cell depletion is an effective means of reducing or eliminating GVHD, but if too rigorous, can also result in delayed immune reconstitution, Whether the immune suppression is pharmacologic or secondary to graft manipulation, the graft recipient is left quantitatively or functionally lymphopenic and at increased risk of life threatening/fatal opportunistic infection. T lymphocytes and in particular CD4+ T lymphocytes, are the most affected and their absence leaves the patient at particular risk for viral infection.
Refractory viral diseases in the immunocompromised host have been treated by infusion of virus specific lymphocytes. Infusion of ex-vivo activated and/or expanded anti-virus T cells has been used for the treatment of CMV and EBV. 8, 9 Donor lymphocyte infusion (DLI) therapy has been used after transplant for a variety of infections including persistent adenovirus,10 CMV,11 EBV-LPD,12, 13 and hepatitis.14, 15 Both virus specific and unmanipulated donor lymphocytes have been documented to help restore T cell numbers16, 17, and to successfully treat infection or relapse17–19. It has been shown that recipients of allogeneic HPCT who are deficient in CD8+ cytotoxic T lymphocytes specific for CMV are specifically at risk for active infection with CMV. In one study, patients who had received CMV-specific clones of CD8+ T cells derived from donor bone marrow had improved reconstitution of their cellular immunity against CMV20. Although the infusions of the donor-derived CMV-specific clones were well tolerated and all patients had reconstituted CMV-specific cytotoxic T lymphocytes by day 49 following HPCT, the virus specific CD8+ CTL response persisted only in those patients in whom CD4+ helper responses recovered. Virus-specific CTLs have been found to be effective therapy for prevention of other post transplant complications including EBV lymphoproliferative disease.21,22 Unfortunately, the infrastructure and resources required to make this type of approach broadly applicable in the clinical setting are not widely available and the use of unmanipulated DLI therapy is often complicated by the development of GVHD. Alternative approaches are needed.
L-leucyl-L-leucine methyl ester (LLME) is a compound taken up by cells through saturable facilitated transport23,24 and once intracellular, accumulates in lysosomes and endosomes where dipeptidyl peptidase I (DPPI) converts LLME to pro-apoptotic metabolites. Since DPPI is expressed primarily by cytotoxic granule-containing leukocytes, LLME induces programmed cell death of NK cells, monocytes, granulocytes, the majority of CD8+ T cells, and a small fraction of CD4+ T cells.24 LLME has been effective in preventing GVHD in mouse and man, but its human application had been limited by toxicity to hematopoietic stem cells when unseparated marrow was treated prior to infusion.25–27 In a phase I trial, HLA-matched or mismatched marrow grafts were incubated ex vivo with LLME prior to infusion.26 Although LLME was effective in preventing GVHD, toxicity to marrow CFUs was noted, and one patient died from secondary graft failure. The study was therefore terminated. Aside from the adverse effects of LLME on hematopoietic stem cells directly exposed to the agent, other infusional or delayed toxicities were not observed. Use of donor lymphocyte infusions (DLI) following T-cell depleted transplantation circumvents the problem of hematopoietic stem cell exposure to LLME. Additionally, this facilitates administration of precisely defined doses of LLME-treated T cells, which may allow for further reduction in GVHD risk, as well as accelerated CD4+ T cell reconstitution.
We have undertaken a study of the use of LLME treated DLI following T-cell depleted allogeneic HPCT specifically to aid with immune reconstitution. In this clinical trial, we have demonstrated the rapid emergence of virus-specific responses following LLME DLI with minimal associated GVHD. This paper presents data detailing the pace of immune recovery and the rapid development of anti-viral responses in the six patients from the larger cohort who developed viral infections during the time period immediately preceding or coincident with the administration of the LLME DLI.
MATERIALS AND METHODS
Eligibility
From February 2002 through July 2007, all patients at Thomas Jefferson University Hospital (TJUH) who had received a T cell depleted (CD34 enriched) HPCT were offered participation in a phase I study of LLME treated DLI to improve immune reconstitution. The study was subsequently opened to patients at the Medical College of Wisconsin. Patients were eligible if they had an HLA matched sibling donor, HLA matched unrelated donor or an HLA partially matched (single antigen mismatch, two antigen mismatch or haplodisparate) related donor. Patients were required to have undergone HPCT at least 28 days earlier and have a non-detectable ATG level (less than 2 mcg/ml of serum for rabbit ATG) within the prior 21 days. To be enrolled, patients must have demonstrated neutrophil engraftment as defined by an absolute neutrophil count greater than 500 cells/uL for three consecutive days, but have persistent CD4 lymphopenia (absolute CD4+ T cell count < 100 cells/μL). If a CD4+ T cell count was not obtainable, an absolute lymphocyte count <100 cells/ul was considered prime facie evidence of CD4+ T cells <100 cells/ul. Patients were excluded if there was evidence of overt relapse of the underlying malignancy or persistent disease or active GVHD. The presence of opportunistic infection was not a contraindication to enrollment on this study. The study was approved by the Institutional Review Boards at Thomas Jefferson University and the Medical College of Wisconsin and informed consent was obtained for all participating patients.
Conditioning Regimen and Supportive Care
All six patients reviewed in this paper received a CD34 enriched HPCT following a preparative regimen of fludarabine (30 mg/m2, days -6 through -2), cytarabine (2 grams/m2, days -6 though -2), and melphalan (200 mg/m2, day -1). All patients received rabbit ATG (Thymoglobulin ®, SangStat) during the peritransplant period. Prophylactic antimicrobials consisted of an amphotericin product, valacyclovir and trimethoprim-sulfamethoxazole. All patients received transfusion support with leukodepleted and irradiated cellular blood products. Patients who were CMV-seronegative received CMV-seronegative blood products.
ATG Levels
Patients had blood drawn for ATG levels prior to ATG administration, on the day of transplant, on day 7 following HPCT and generally weekly thereafter as described previously.28 At the beginning off this trial, patients became eligible to receive LLME DLI when the ATG level was <1.0 mcg/ml. Subsequently, based on further information about the pharmacokinetics and mechanisms of action of ATG, the protocol was revised to allow for inclusion when the ATG level became < 2.0 mcg/ml.
LLME Treatment
LLME was synthesized under GMP conditions by Bachem (Switzerland). Immediately prior to use the dry LLME powder was prepared in Normosol R at a 500 uM concentration. Washed cells (blood or apheresis product) were incubated with LLME at a concentration of 10 × 106 cells/ml for sixty minutes. Treated cells were washed once at 4°C in Normosol, and then twice at room temperature. Only products with ≥80% depletion of NK cells (CD 16/56+, CD3−) determined by flow cytometry were released for infusion. The treatment dose was based on the number of viable CD3+ cells.
Immunophenotyping
Intracellular staining for perforin and surface staining for lymphocyte subsets were performed, using standard staining techniques as previously described.29,30 Flow cytometric analysis was performed using the FACScan (Becton Dickinson).
Anti-Viral Testing
Ex vivo ELISPOT assay
PBMC were separated from heparinized whole blood or buffy coats using Ficoll-Hypaque density gradient centrifugation, and cells were used either fresh or thawed from cryopreserved aliquots. The ex vivo ELISPOT assay for detection of IFN-γ was based on the protocol previously described.31 96-well polyvinylidene difluoride backed plates (Millipore, Bedford, MA) were coated overnight at 4°C with 15 μg/ml of anti-IFN-γ monoclonal antibody (MAb) 1-D1K (Mabtech, Stockholm, Sweden). Wells were washed and blocked with RPMI supplemented with 10% FCS, penicillin, streptomycin, glutamine, and 10mM HEPES (R10) for 1 hr at 37°C. Next, each antigen was incubated with 250,000 PBMCs in 100 μl R10 in triplicate wells for 6 to 48 hrs at 37°C in 5% CO2. Wells were washed extensively with PBS, 0.05% Tween 20, 1 μg/ml of biotinylated anti-IFN-γ MAb 7-B6-1 (Mabtech) was added, and plates incubated at RT for 3 hrs. Next, wells were washed and incubated with a 1:1000 dilution of alkaline phosphatase conjugate (Mabtech) for 2hrs. Wells were washed again and incubated with alkaline phosphatase substrate (Vector Labs, Burlingame, CA) for 30 minutes; the reaction was stopped with tap water. After wells were air-dried, large spots with fuzzy borders were counted under a dissecting microscope. The number of spot-forming cells (SFCs) per 106 PBMC was calculated as the mean number of SFCs in triplicate microwells × 4. Antigen-specific precursor frequencies were calculated as the mean number of SFCs in the antigen-stimulated microwells minus the mean number of SFCs in the control microwells.
Viral Titers
Whole blood samples were sent on all patients weekly to screen for the reemergence of CMV using quantitative real time DNA PCR as previously described.32–35 Assays were performed by the clinical laboratory at TJUH or at Viracor (Lee’s Summit, MO). When clinically indicated, whole blood samples were also analyzed for adenovirus, EBV and BK virus also using quantitative real time DNA PCR.36–39 In patients who developed reactivation of any virus as demonstrated by a rising PCR titer, samples were followed weekly until resolution, as evidenced by at least two consecutive undetectable titers.
CDR3-Size Spectratype Analysis/Spectratype Complexity Index
As previously described,40 peripheral blood lymphocytes (PBL) were enriched from donor and patient peripheral blood samples by centrifugation over Ficoll-Histopaque. CD4+ and CD8+ subsets were separated by standard antibody-panning techniques and the enriched subset populations were solubilized in Ultraspec (Biotex Laboratories, Houston, TX). Vβ spectratype analysis of separated PBL from untreated donor samples served as the reference point for full repertoire complexity, and was used as the standard of comparison for the LLME-treated, as well as post-transplant samples. At the time of the post-transplant analysis, all patients exhibited >90% donor chimerism, as determined by molecular analysis of short tandem repeats.41
Total RNA was isolated from Ultraspec samples and cDNA was prepared, as previously described.40 Semi-nested PCR was performed using a panel of human Vβ sense oligoprimers and 2 Cβ antisense oligoprimers; the second Cβ being fluorescently labeled. The PCR products were run on a sequencing gel and analyzed by the Genotyper Genescan software program (Applied Biosystems, Foster City, CA). The complexity index within a Vβ family was determined as a percentage of the number of peaks found in its spectratype histogram in relation to the number of peaks in the corresponding donor Vβ family histogram. Any Vβ family with a complexity index of ≥85% was considered to be fully complex. Histogram peaks were identified by the Genotyper Genescan program. Any donor or patient sample exhibiting fewer than 12 evaluable Vβ family spectratypes was excluded.
Statistical methods
Data from the ELISPOT assays were analyzed using a paired t test to compare the mean number of spots in the quadruplicate control and experimental (antigen-stimulated) microwells. The results were compared between responders using the Mann-Whitney U test. Data comparisons for Vβ repertoire analysis were analyzed by the nonparametric Wilcoxon rank-sum analysis. P values of ≤0.05 were considered statistically significant.
RESULTS
Patients
Twenty-three patients with hematologic malignancies were enrolled in a study of LLME treated DLI to accelerate immune reconstitution after receiving CD34 enriched HPCT. Six of these 23 patients represented a unique subset in that they each developed one or more viral infections during the time period immediately before or concurrent with the administration of the LLME DLI. These patients demonstrated a rapid and consistent pattern of response to their viral infections which is the subject of this report.
Five of these six patients received HPCT from HLA identical sibling donors, while one received HPCT from an unrelated donor. One patient (patient #1) was treated with steroids for adrenal insufficiency and idiopathic pneumonia syndrome (IPS) and expired 35 days following LLME DLI (134 days s/p HPCT). A second patient (patient #3) also developed IPS and was treated with steroids. This patient expired 100 days following LLME DLI (148 days s/p HPCT). Although both of these patients have been included in this analysis of anti-viral responses, neither is considered evaluable for immune reconstitution due to the administration of corticosteroids for the treatment of IPS.
LLME Treatment and Lymphocyte Subset Depletion
Following LLME treatment of the DLI product as described above, patients received a targeted number of CD3+ cells/kg (Table 1). Initial cohorts of recipients of unrelated donor grafts received a dose of 105 CD3+ cells/kg, while the first cohort of patients receiving HLA identical sibling stem cell grafts received a dose of 106 CD3+ cells/kg. Dose escalation was allowed for each individual patient if that patient did not develop GVHD and had also not recovered a CD4+ T cell count of > 200/ul. If at least 3 patients in one cohort received LLME treated DLI at a dose without toxicity/GVHD, the next cohort received a dose one log higher. The patients described here received either 106 CD3+ cells/kg (HLA identical unrelated donor – n=1; HLA identical sibling donors, n=2) or 107 CD3+ cells/kg (HLA identical sibling donors, n=3).
Table 1. Patient Characteristics.
Six patients developed viral infections concurrent with or immediately preceding LLME DLI. All had resolution or partial resolution of the viral disease. Four of six developed rapid recovery of CD3+/CD4+ cells. Two patients, who expired at days 35 and 46 s/p LLME DLI of other causes did not.
| Age/Gender | Disease | Graft Type | LLME DLI | Infection | Time to CD4>100 (days) | GVHD | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CD3+ T Cell Dose/Kg | Days s/p HPCT | Virus | Days s/p HPCT | Resolved? | Other Therapy for OI | ||||||
| 1 | 65 M | ALL - 1st CR | Sib | 1 × 106 | 99 | EBV-PTLD | 113 | Partial1 | Rituximab | NA | No |
| 2 | 23 M | CML - 2nd CP | Sib | 1 × 106 | 102 | Adeno, CMV | 91, 105 | Yes | Ganciclovir, Cidofovir, IVIg | 21 | No |
| 3 | 53 M | CML - 2nd CP | Sib | 1 × 107 | 64 | CMV | 28 | Yes | Foscarnet + IVIg | 57 | Grade 32 |
| 4 | 61 M | ALL - Ph+-1st CR | Sib | 1 × 107 | 48 | CMV | 26 | Yes | Ganciclovir/valganciclovir, IVIg | 34 | No |
| 5 | 28 M | AML- 1st relapse | Sib | 1 × 107 | 72 | EBV- PTLD | 75 | Yes | Rituximab | 10 | No |
| 6 | 45 M | AML - 2nd relapse | URD | 1 × 106 | 69 | CMV | 24 | Yes | Valganciclovir | NA | No |
HPCT – Hematopoietic Progenitor Cell Transplant; GVHD – Graft-Versus-Host Disease; ALL – acute lymphoblastic leukemia; CML – chronic myelogenous leukemia; AML – acute myelogenous leukemia; CR – complete remission; CP – chronic phase; Ph+ - Philadelphia Chromosone; Sib – HLA identical sibling donor; IRD – unrelated donor; EBV-PTLD – Epstein Barr Virus related Post-Transplant Lymphoproliferative Disease; Adeno – Adenovirus; CMV – cytomegalovirus; OI – Opportunistic Infection; IVIg – Intravenous Immune Globulin
Patient expired from pulmonary and renal complications prior to complete resolution of EBV-PTLD.
This patient had no GVHD following his first dose of LLME DLI (1 × 10e7/kg) but did not achieve a CD4 count >200/ul. He was therefore offered a second dose of DLI (actual dose 4.1 × 10e7/kg) and received it 43 days after the first dose. He subsequently developed Grade III GVHD within a few days of the second DLI administration. The trial was subsequently changed to allow for further DLI only if the CD4 count was < 100/ul.
Following treatment with LLME, flow cytometric analysis showed as expected, a selective depletion of cells containing perforin. (Figure 1a). The products showed adequate depletions of NK and CD8+ cells with a median 95.2% (range 79.9–100) depletion of NK cells, 76.1% (range 35.1–92.8) depletion of CD3+/CD8+ cells, but only 31.0% (0 – 88.8) depletion of CD3+/CD4+ cells. (Figure 1b)
Figure 1. LLME Depletion.


Flow cytometry of product prior to LLME treatment shows the presence of NK (CD56 or 16 +/CD3−) cells, and CD3+/CD8+ cells, and CD3+/CD4+ cells. However, following LLME treatment, cells which contain perforin, namely the NK cells and a portion of CD8+ cells, are markedly depleted. The CD4 cell population is only minimally depleted in contrast (a). Following treatment with LLME, products showed adequate depletions of NK and CD8+ cells with a median 95.2% (range 79.9–100) depletion of NK cells, 76.1% (range 35.1–92.8) CD3+/CD8+ cells, but only 31.0% (0 – 88.8) CD3+/CD4+ cells (b).
Immune Reconstitution
At the time of the first LLME-treated DLI treatment, all patients demonstrated neutrophil engraftment, but not lymphocyte engraftment. Two patients (#1, #3) were not evaluable based on corticosteroid use as described above. Post treatment flow cytometric analysis showed recovery of NK cells first, as would be expected following T-cell depleted HPCT, including both perforin positive and perforin negative cells, followed by recovery of CD8+ and then CD4+ cells. (Figure 2) Following the first dose of LLME-treated lymphocytes, 4 of 6 patients who were actively fighting viral infections, achieved a CD3+/CD4+ count >100 cells/ul by 27.5 days post infusion (range 10–57 days). (Table 1) This recovery was not only rapid but sustained. Data from a sample patient (#2) who developed adenovirus infection including hepatitis with transaminases 5–10 times the upper limit of normal is shown. (Figure 3a and 3b) Following LLME DLI, reconstitution of lymphocyte subsets was prompt and persistent. By twenty-one days after receiving DLI, he had achieved a CD4 count of 333 cells/ul and has since maintained this count above 100 cells/ul. (Figure 3a) His recovery included reemergence of both memory and naïve T cells (Figure 3b), which have also been sustained. In another patient (#5) who had not achieved 100% donor chimerism at the time he developed EBV-PTLD, the pace of CD4+ T cell recovery was also rapid and sustained, but the CD4 response consisted primarily of memory T cells (CD45 RO+/CD4+). He subsequently converted to 100% donor cells, and maintained a stable number of both memory and naïve CD4+ T cells. (Figure 3c)..
Figure 2. Reconstitution of Perforin Positive Cells.


Following LLME DLI, a product containing perforin negative cells, patients were able to reconstitute both perforin negative and then perforin positive cells. For the sample patient seen here, recovery of CD3+/CD4+ cells was apparent by Day 37 after LLME DLI (Figure 2a). Earlier time points were measured (Figure 2b) showing recovery of the same populations of cells but at lower numbers. The recovery of NK cells heralded (third dot plot & third column) preceded the recovery of CD8 cells. Reconstitution of CD8 cells included both perforin positive and perforin negative cells and preceded the recovery of CD4 positive cells.
Figure 3. Immune Reconstitution Following LLME DLI.



Patients developed rapid and sustained reconstitution of lymphocyte subsets following LLME DLI. As shown in this sample patient, this included CD3+/CD4+, CD3+/CD8+ and CD3−/CD56 or 16+ cells (a) as well as both memory and naïve CD4 cells (b). In one patient who had not converted to 100% donor cells at the time he received LLME DLI, the recovery of memory CD4 cells was more prominent (c)
Anti Viral Responses
We assayed for anti-viral responses in the six patients who developed infections following CD34-enriched HPCT. These included reactivation of Epstein Barr virus (n=2), cytomegalovirus (n=4) and adenovirus (n=1). The infections developed at a median of 51.5 days (range 28–113 days) following the CD34-enriched HPCT. Clinically, five of six patients had a complete response as demonstrated by resolution of the viral pcr titer. One patient (#1) who developed idiopathic pneumonia syndrome and required high dose steroids, had a partial resolution of his EBV titers, but had not completely resolved them prior to his death from multi-system organ failure, 35 days after receiving his first dose of LLME DLI. Sample data are shown for two patients, one (#2) of whom developed adenovirus hepatitis (Figure 4a and 4b) and another (#4) who developed CMV infection (Figure 4c and 4d). In both cases, one can see rapid resolution of the viral pcr titers (Figure 4b and 4d). Similar resolution of viral pcr titers were seen in three other patients (#3, 5, and 6) at a range of 11–32 days (data not shown). The final patient (patient #1) had partial resolution of EBV pcr titer (94% reduction) prior to his death from idiopathic pneumonia syndrome.
Figure 4. Anti-Viral Responses.




Sample data are shown for two patients. In panels a and b, one sees the data from a patient who developed adenovirus hepatitis. Shortly after receiving the LLME DLI, he began to clear the adenovirus with a 10fold drop in pcr titers by 11 days s/p DLI (113 days s/p HPCT), and complete resolution by 42 days s/p DLI (144 days s/p HPCT). This was associated with an adenovirus specific CD4 IFN-γ response that was evident at 147 and 175 days post DLI. Sample from a second patient, who had developed CMV infection are shown in panels c and d. Once again, the patient had rapid resolution of CMV pcr titers with a significant drop within six days and complete resolution by 20 days s/p DLI (68 days s/p HPCT). He was also found to have developed both a CMV-specific CD8 response (anti-A2 peptide) and CD4 response (CMV ag), but no response to adenovirus.
In addition, ELISPOT data demonstrated the development of viral-specific response following LLME DLI therapy. The patient with adenovirus infection (patient #2) developed an adenovirus-specific IFN-γ response by his CD4+ cells. (Figure 4b) The patient (patient #4) who developed the CMV infection was found to have both a CMV-specific CD8+ T cell response (anti-A2 peptide) and a CD4+ T cell response (CMV ag) (Figure 4d), but no response to adenovirus (not shown). Similarly, ELISPOT data from patient #5 with EBV-PTLD showed an EBV specific response following the LLME DLI (data not shown).
These responses were detected at a median of 11.5 days (range 4–22) following LLME DLI therapy and were associated with regression of the viral infection and resolution of circulating viral DNA by a median of 20 days (range 11–42) in the five patients who did resolve their infections.
LLME DLI was given in conjunction with appropriate pharmacologic therapy in all cases. Patients with EBV-PTLD (n=2) received rituximab, patients with CMV (n=4) or adenovirus (n=1) received foscarnet, ganciclovir or valganciclovir as clinically indicated based on renal insufficiency and count recovery. Three of four patients with CMV also received intravenous immune globulin as adjunctive therapy.
Development of GVHD or Opportunistic Infection
Severe GVHD (Grade III) developed in only one of these six patients (#3), a recipient of an HLA identical sibling graft. This patient had received an initial dose of 107 LLME DLI, but had not reached the target of 200 CD4+ cells/ul, and thus received a second LLME-treated DLI 43 days later. One week following the second dose of LLME DLI, at 4.1 × 107 CD3+ cells/kg (108) he developed GVHD of the skin and gastrointestinal tract.
SPECTRATYPING
CDR3-size spectratype analysis was used to analyze peripheral blood samples to determine if there were any significant differences in the complexity of the reconstituting CD4+ and CD8+ T cell subsets, between donor and recipient (Figure 5). Reconstitution of a complex repertoire was seen in all tested patients following LLME DLI. There were no significant differences in the level of VB family complexity between the donor and “reconstituted” host.
Figure 5. V Beta Spectratyping.
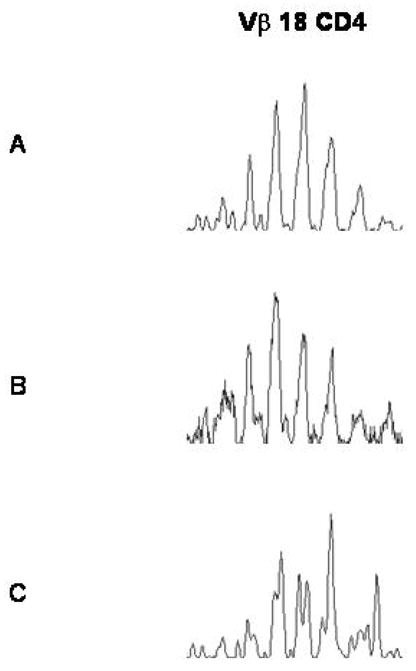
Representative histograms of Vbeta spectratype analysis of the CD4+ T cells enriched from the peripheral blood of one of the patents/pairs. The repertoire complexity of the donor sample before LLME treatment (A), after LLME treatment (B), and reconstituting in the patient post-LLME DLI (C) exhibit similar levels of complexity.
DISCUSSION
Immune recovery following HPCT is slow, particularly following CD34-enriched or T cell depleted HPCT. Prior to immune recovery, patients are susceptible to opportunistic infection including CMV, adenovirus, EBV-PTLD and BK virus. In patients who receive unmanipulated grafts, GVHD pharmacoprophylaxis and treatment may also lead to profound immune suppression and similar types of infections. These infections lead to significant morbidity and mortality following HPCT. In a phase I study using LLME DLI, six patients developed significant viral infections during the immediate pre-DLI time period. Following the LLME DLI, we have seen rapid reconstitution of lymphocyte subsets including CD3+/CD8+, CD3+/CD4+, and both CD45RA+ and CD45RO+. This recovery, unlike that seen with infusion of cultured virus specific CTLs, is sustained. It is associated with the emergence of virus specific clones, but also with the development of a normal repertoire of T cells. Furthermore, it is associated with an acceptable risk of GVHD as compared to unmanipulated DLI.
LLME-treatment is simple and not labor-intensive. Within a few hours of phlebotomy or leukapheresis, the product is ready for infusion. Thus a patient in need of DLI would have to wait only a few hours longer for this type of product than for an unmanipulated product. This could be a significant advantage over the use of virus-specific CTLs which may take days or weeks to prepare and require facilities not available at many institutions. Infusions of virus specific CTLs can lead to rapid resolution of CMV disease. However, the anti-CMV response is difficult to sustain in patients who are otherwise still immune compromised and lymphopenic. We have shown here that following LLME DLI, the anti-CMV response is rapid and also sustained.
In addition to the inability to maintain the anti-viral response, virus specific CTLs do not provide immune competence against other pathogens. In contrast LLME DLI together with pharmacologic anti-viral therapy (foscarnet, cidofovir, ganciclovir), fosters rapid virus-specific responses that are sustained. In addition, these patients have quantitative evidence of immune recovery across a panel of lymphocyte subsets including cytotoxic cells, memory and naïve cells.
The LLME specifically depletes the stem cell graft of perforin containing cells capable of mediating cytotoxocity, providing a product that is enriched for CD4+ cells, but depleted of NK cells, and perforin containing CD8+ cells. Thus, the infused product is relatively depleted of cytotoxic cells that might cause GVHD or be targeted against specific pathogens while it maintains T cells with helper activity. Although CD8+ T cells mediate direct cytotoxicity against viral infected targets, their generation in vivo is dependent upon viral specific CD4+ helper T cells. Thus, in the presence of replicating viral pathogens, our CD4+ T cell deficient patients receiving LLME DLI were able to mount an appropriate immune response within several days after the infusion.
In the setting of active viral infection, immune recovery following LLME DLI is relatively rapid. The median time to recovery of a CD4 count > 100/ul was 27.5 days in these patients. This contrasts with the slower recovery of patients treated with LLME DLI who were not infected with viruses (data not shown). The more rapid recovery kinetics of virally infected patients likely reflects an initial reactive lymphocytosis directed toward the specific viral pathogen while these patients as well as the uninfected patients subsequently undergo a broader, infection independent expansion of the infused T lymphocytes. In addition, the response was durable. Patients did not relapse in terms dropping lymphocyte counts. More importantly, no patient had relapse of viral disease at any time following LLME DLI. This contrasts with patients who have received only pharmacologic therapy or virus-specific CTLs who unfortunately often suffer relapse of the viral disease9.
While GVHD has long been associated with DLI, in this small group of patients this has not been an issue, except for one patient who did receive a higher dose of cells and likely was developing GVHD from the initial DLI just as a second larger DLI was administered. These patients were treated as part of a study which had as its primary endpoint the development of a CD4 count >100 and as a secondary endpoint the development of GVHD. It would be premature to comment on the risk of GVHD with LLME DLI until a larger cohort of patients have received this therapy. However, in the small number of patients described here, we have seen encouraging results with very little GVHD.
In the absence of severe GVHD, LLME treated DLI appears to be a viable treatment option which has simplicity along with a rapid and durable immune recovery as major advantages. In addition, the rapid reconstitution of more broad based T cell immunity including a complex T cell repertoire is also an advantage. Further trials are required to assess the utility of this therapy in the treatment of established viral infections.
Acknowledgments
The authors gratefully acknowledge the support of the nurses, nurse practitioners, and physician assistants of the Thomas Jefferson Blood & Marrow Transplant Program, the staff of the Blood & Marrow Transplant Clinical Laboratory and the assistance of Victoria Ward in preparing figures for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Champlin R, Khouri I, Giralt S. Use of nonmyeloablative preparative regimens for allogeneic blood stem cell transplantation: induction of graft-vs.-malignancy as treatment for malignant diseases. J Clin Apheresis. 1999;14:45–49. doi: 10.1002/(sici)1098-1101(1999)14:1<45::aid-jca9>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 2.Copelan EA. Hematopoietic stem-cell transplantation. [Review] [99 refs] N Engl J Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 3.Forman SJ. Innovations in autologous transplantation for hematologic malignancy. [Review] [56 refs] Biology of Blood & Marrow Transplantation. 2005;11:28–33. doi: 10.1016/j.bbmt.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 4.Thomas ED. Bone marrow transplantation from bench to bedside. [Review] [11 refs] Ann N Y Acad Sci. 1995;770:34–41. doi: 10.1111/j.1749-6632.1995.tb31041.x. [DOI] [PubMed] [Google Scholar]
- 5.Thomas ED. History, current results, and research in marrow transplantation. [Review] [37 refs] Perspectives in Biology & Medicine. 1995;38:230–237. doi: 10.1353/pbm.1995.0001. [DOI] [PubMed] [Google Scholar]
- 6.Couriel DR, Saliba RM, Giralt S, et al. Acute and chronic graft-versus-host disease after ablative and nonmyeloablative conditioning for allogeneic hematopoietic transplantation. Biology of Blood & Marrow Transplantation. 2004;10:178–185. doi: 10.1016/j.bbmt.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Lee SJ. New approaches for preventing and treating chronic graft-versus-host disease. [Review] [103 refs] Blood. 2005;105:4200–4206. doi: 10.1182/blood-2004-10-4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peggs KS, Mackinnon S. Augmentation of virus-specific immunity after hematopoietic stem cell transplantation by adoptive T-cell therapy. Hum Immunol. 2004;65:550–557. doi: 10.1016/j.humimm.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 9.Riddell SR, Greenberg PD. Cellular adoptive immunotherapy after bone marrow transplantation. [Review] [192 refs] Cancer Treatment & Research. 1995;76:337–369. doi: 10.1007/978-1-4615-2013-9_16. [DOI] [PubMed] [Google Scholar]
- 10.Hromas R, Cornetta K, Srour E, Blanke C, Broun ER. Donor leukocyte infusion as therapy of life-threatening adenoviral infections after T-cell-depleted bone marrow transplantation. Blood. 1994;84:1689–1690. [PubMed] [Google Scholar]
- 11.Witt V, Fritsch G, Peters C, Matthes-Martin S, Ladenstein R, Gadner H. Resolution of early cytomegalovirus (CMV) infection after leukocyte transfusion therapy from a CMV seropositive donor. Bone Marrow Transplant. 1998;22:289–292. doi: 10.1038/sj.bmt.1701328. [DOI] [PubMed] [Google Scholar]
- 12.Papadopoulos EB, Ladanyi M, Emanuel D, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation.[see comment] N Engl J Med. 1994;330:1185–1191. doi: 10.1056/NEJM199404283301703. [DOI] [PubMed] [Google Scholar]
- 13.Chakrabarti S, Milligan DW, Pillay D, et al. Reconstitution of the Epstein-Barr virus-specific cytotoxic T-lymphocyte response following T-cell-depleted myeloablative and nonmyeloablative allogeneic stem cell transplantation. Blood. 2003;102:839–842. doi: 10.1182/blood.V102.3.839. [DOI] [PubMed] [Google Scholar]
- 14.Shouval D, Ilan Y. Immunization against hepatitis B through adoptive transfer of immunity. [Review] [37 refs] Intervirology. 1995;38:41–46. doi: 10.1159/000150413. [DOI] [PubMed] [Google Scholar]
- 15.Shouval D, Ilan Y. Transplantation of hepatitis B immune lymphocytes as means for adoptive transfer of immunity to hepatitis B virus. [Review] [18 refs] J Hepatol. 1995;23:98–101. doi: 10.1016/0168-8278(95)80317-3. [DOI] [PubMed] [Google Scholar]
- 16.Lamb LS, Jr, Gee AP, Henslee-Downey PJ, et al. Phenotypic and functional reconstitution of peripheral blood lymphocytes following T cell-depleted bone marrow transplantation from partially mismatched related donors. Bone Marrow Transplant. 1998;21:461–471. doi: 10.1038/sj.bmt.1701110. [DOI] [PubMed] [Google Scholar]
- 17.Dazzi F, Goldman JM. Adoptive immunotherapy following allogeneic bone marrow transplantation. [Review] [71 refs] Annu Rev Med. 1998;49:329–340. doi: 10.1146/annurev.med.49.1.329. [DOI] [PubMed] [Google Scholar]
- 18.Dazzi F, Szydlo RM, Goldman JM. Donor lymphocyte infusions for relapse of chronic myeloid leukemia after allogeneic stem cell transplant: where we now stand. [Review] [90 refs] Exp Hematol. 1999;27:1477–1486. doi: 10.1016/s0301-472x(99)00096-x. [DOI] [PubMed] [Google Scholar]
- 19.Bollard CM, Kuehnle I, Leen A, Rooney CM, Heslop HE. Adoptive immunotherapy for posttransplantation viral infections. [Review] [94 refs] Biology of Blood & Marrow Transplantation. 2004;10:143–155. doi: 10.1016/j.bbmt.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 20.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor.[see comment] N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 21.O’Reilly RJ, Small TN, Papadopoulos E, Lucas K, Lacerda J, Koulova L. Adoptive immunotherapy for Epstein-Barr virus-associated lymphoproliferative disorders complicating marrow allografts. [Review] [228 refs] Springer Semin Immunopathol. 1998;20:455–491. doi: 10.1007/BF00838055. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Savoldo B, Huls H, et al. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the prevention and treatment of EBV-associated post-transplant lymphomas. Recent Results in Cancer Research. 2002;159:123–133. doi: 10.1007/978-3-642-56352-2_15. [DOI] [PubMed] [Google Scholar]
- 23.Thiele DL, Lipsky PE. The action of leucyl-leucine methyl ester on cytotoxic lymphocytes requires uptake by a novel dipeptide-specific facilitated transport system and dipeptidyl peptidase I-mediated conversion to membranolytic products. J Exp Med. 1990;172:183–194. doi: 10.1084/jem.172.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiele DL, Lipsky PE. Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A. 1990;87:83–87. doi: 10.1073/pnas.87.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiele DL, Lipsky PE. The immunosuppressive activity of L-leucyl-L-leucine methyl ester: selective ablation of cytotoxic lymphocytes and monocytes. Journal of Immunology. 1986;136:1038–1048. [PubMed] [Google Scholar]
- 26.Rosenfeld CS, Thiele DL, Shadduck RK, Zeigler ZR, Schindler J. Ex vivo purging of allogeneic marrow with L-Leucyl-L-leucine methyl ester. A phase I study. Transplantation. 1995;60:678–683. doi: 10.1097/00007890-199510150-00011. [DOI] [PubMed] [Google Scholar]
- 27.Pecora AL, Bordignon C, Fumagalli L, et al. Characterization of the in vitro sensitivity of human lymphoid and hematopoietic progenitors to L-leucyl-L-leucine methyl ester. Transplantation. 1991;51:524–531. doi: 10.1097/00007890-199102000-00048. [DOI] [PubMed] [Google Scholar]
- 28.Kakhniashvili I, Filicko J, Kraft WK, Flomenberg N. Heterogeneous clearance of antithymocyte globulin after CD34+-selected allogeneic hematopoietic progenitor cell transplantation. Biology of Blood & Marrow Transplantation. 2005;11:609–618. doi: 10.1016/j.bbmt.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Hameed A, Olsen KJ, Cheng L, Fox WM, 3rd, Hruban RH, Podack ER. Immunohistochemical identification of cytotoxic lymphocytes using human perforin monoclonal antibody. Am J Pathol. 1992;140:1025–1030. [PMC free article] [PubMed] [Google Scholar]
- 30.Hsieh MH, Varadi G, Flomenberg N, Korngold R. Leucyl-leucine methyl ester-treated haploidentical donor lymphocyte infusions can mediate graft-versus-leukemia activity with minimal graft-versus-host disease risk. Biology of Blood & Marrow Transplantation. 2002;8:303–315. [PubMed] [Google Scholar]
- 31.Olive M, Eisenlohr LC, Flomenberg P. Quantitative analysis of adenovirus-specific CD4+ T-cell responses from healthy adults. Viral Immunol. 2001;14:403–413. doi: 10.1089/08828240152716646. [DOI] [PubMed] [Google Scholar]
- 32.Nitsche A, Oswald O, Steuer N, et al. Quantitative real-time PCR compared with pp65 antigen detection for cytomegalovirus (CMV) in 1122 blood specimens from 77 patients after allogeneic stem cell transplantation: which test better predicts CMV disease development? Clin Chem. 2003;49:1683–1685. doi: 10.1373/49.10.1683. [DOI] [PubMed] [Google Scholar]
- 33.Griscelli F, Barrois M, Chauvin S, Lastere S, Bellet D, Bourhis JH. Quantification of human cytomegalovirus DNA in bone marrow transplant recipients by real-time PCR. J Clin Microbiol. 2001;39:4362–4369. doi: 10.1128/JCM.39.12.4362-4369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kearns AM, Guiver M, James V, King J. Development and evaluation of a real-time quantitative PCR for the detection of human cytomegalovirus. J Virol Methods. 2001;95:121–131. doi: 10.1016/s0166-0934(01)00307-x. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida A, Hitomi S, Fukui T, et al. Diagnosis and monitoring of human cytomegalovirus diseases in patients with human immunodeficiency virus infection by use of a real-time PCR assay. Clinical Infectious Diseases. 2001;33:1756–1761. doi: 10.1086/323782. [DOI] [PubMed] [Google Scholar]
- 36.Vats A, Shapiro R, Singh Randhawa P, et al. Quantitative viral load monitoring and cidofovir therapy for the management of BK virus-associated nephropathy in children and adults. Transplantation. 2003;75:105–112. doi: 10.1097/00007890-200301150-00020. [DOI] [PubMed] [Google Scholar]
- 37.Randhawa P, Ho A, Shapiro R, et al. Correlates of quantitative measurement of BK polyomavirus (BKV) DNA with clinical course of BKV infection in renal transplant patients. J Clin Microbiol. 2004;42:1176–1180. doi: 10.1128/JCM.42.3.1176-1180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Esser JW, Niesters HG, Thijsen SF, et al. Molecular quantification of viral load in plasma allows for fast and accurate prediction of response to therapy of Epstein-Barr virus-associated lymphoproliferative disease after allogeneic stem cell transplantation.[see comment] Br J Haematol. 2001;113:814–821. doi: 10.1046/j.1365-2141.2001.02789.x. [DOI] [PubMed] [Google Scholar]
- 39.van Esser JW, van der Holt B, Meijer E, et al. Epstein-Barr virus (EBV) reactivation is a frequent event after allogeneic stem cell transplantation (SCT) and quantitatively predicts EBV-lymphoproliferative disease following T-cell--depleted SCT. Blood. 2001;98:972–978. doi: 10.1182/blood.v98.4.972. [DOI] [PubMed] [Google Scholar]
- 40.Friedman TM, Varadi G, Hopely DD, et al. Nonmyeloablative conditioning allows for more rapid T-cell repertoire reconstitution following allogeneic matched unrelated bone marrow transplantation compared to myeloablative approaches. Biology of Blood & Marrow Transplantation. 2001;7:656–664. doi: 10.1053/bbmt.2001.v7.pm11787528. [DOI] [PubMed] [Google Scholar]
- 41.Friedman TM, Filicko-O’Hara J, Mookerjee B, et al. T cell repertoire complexity is conserved after LLME treatment of donor lymphocyte infusions. Biology of Blood & Marrow Transplantation. 2007;13:1439–1447. doi: 10.1016/j.bbmt.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]