

SUMMARY
Severe congenital neutropenia (SCN) is a genetically heterogeneous syndrome associated with mutations of ELANE (ELA2), HAX1, GFI1, WAS, CSF3R or G6PC3. We investigated the prevalence of mutations of ELANE in a cohort of 162 SCN patients for whom blood or bone marrow samples were submitted to the North American Severe Chronic Neutropenia Tissue Repository. Mutations of ELANE were found in 90 of 162 patients (55.6%). Subsequently, we conducted an analysis of a subset of 73 of these cases utilizing a high throughput sequencing approach to determine the prevalence of other mutations associated with SCN. Among the 73 patients, mutations of ELANE were detected in 28. In the remaining 45 patients with wild type ELANE alleles, 5 patients had mutations: GFI1 (1), SBDS (1), WAS (1) and G6PC3 (2); no mutations of HAX1 were detected. In approximately 40% of our cases, the genetic basis of SCN remains unknown. These data suggest that for genetic diagnosis of SCN, ELANE genotyping should first be performed. In patients without ELANE mutations, other known SCN-associated gene mutations will be found rarely and genotyping can be guided by the clinical features of each patient.
Keywords: bone marrow failure, chronic neutropenia, DNA mutation
INTRODUCTION
Severe congenital neutropenia (SCN) is a bone marrow failure syndrome characterized by severe neutropenia present from birth, an arrest of neutrophil differentiation at the promyelocyte/myelocyte stage, and a marked propensity to develop myelodysplasia and acute myeloid leukemia. SCN demonstrates multiple modes of inheritance including autosomal recessive, autosomal dominant, X-linked, and sporadic patterns. Accordingly, recent genetic studies have identified multiple gene mutations in SCN. By far the most common mutations affect the ELANE (ELA2) gene encoding neutrophil elastase (all in autosomal dominant or sporadic SCN) (Ancliff, et al 2001, Ancliff, et al 2002, Bellanne-Chantelot, et al 2004, Dale, et al 2000, Germeshausen, et al 2001, Horwitz, et al 1999). Recent studies suggest that mutations of HAX1 and G6PC3 are responsible for cases of autosomal recessive SCN (Carlsson, et al 2008, Germeshausen, et al 2008, Ishikawa, et al 2008, Klein, et al 2007). Mutations of WAS, encoding the Wiskott-Aldrich syndrome protein, are associated with X-linked inherited SCN (Ancliff, et al 2006, Devriendt, et al 2001). There are also case reports of mutations of GFI1 (Person, et al 2003), and CSF3R (Dror, et al 2000, Druhan, et al 2005, Sinha, et al 2003) (encoding the granulocyte colony-stimulating factor (G-SCF) receptor) in SCN.
In this study, we first investigated the frequency of ELANE mutations in a cohort of 162 SCN patients for whom bone marrow samples were available in the North American Severe Chronic Neutropenia Tissue Repository. In a subset of 73 cases, we subsequently screened for mutations in ELANE, HAX1, WAS, SBDS, GFI1, and G6PC3 (Figure 1). SBDS was included, because mutations of this gene have been associated with severe neutropenia (Boocock, et al 2003, Calado, et al 2007). A high-throughput sequencing pipeline using exon-based re-sequencing of whole-genome-amplified genomic DNA and a semi-automated method was used to detect mutations. Our goal was to define the incidence of mutations in these genes in a North American population of patients with clinically defined SCN.
Figure 1. Study design.

A total of 162 individuals with SCN and DNA samples in the Severe Chronic Neutropenia International Registry (SCNIR) tissue repository underwent ELANE genotyping. ELANE sequence variants were detected in 90 (55.6%). Of the 72 SCN samples with normal ELANE, 45 had sufficient DNA and appropriate informed consent to enable additional genotyping at Washington University. These 45 samples, along with 28 of the ELANE variant samples, underwent sequencing of their ELANE, HAX1, WAS, SBDS, GFI1, and G6PC3 genes.
MATERIALS AND METHODS
Human subjects
A total of 162 patients diagnosed with SCN were included in this study. All of these patients had a bone marrow sample suitable for genotyping available in the North American Severe Chronic Neutropenia Tissue Repository. This study was conducted with approval of the Human Subjects Committee, University of Washington, and the institutional review boards of other participating institutions for the study. Informed consent was obtained in accordance with the Declaration of Helsinki. All patients met clinical criteria for SCN, including a minimum of 3 documented absolute neutrophil counts (ANCs) < 0.5 ×109/l over a 3-month period and onset of neutropenia within the first few months of life. Patients with cyclic neutropenia and syndromic neutropenia (e.g., Shwachman-Diamond syndrome and Barth syndrome) were specifically excluded from this study.
ELANE genotyping was initially performed in all patients as previously described and reported for some of these patients (Dale, et al 2000, Rosenberg, et al 2008). To investigate the frequency of mutations in genes other than ELANE, a subset of 73 patients with SCN were chosen for further sequencing based on the following criteria: 1) Informed consent allowing for resequencing at Washington University was obtained; 2) Adequate genomic DNA for resequencing was available. Priority was given to those cases with wild type ELANE alleles; of the 73 cases, no ELANE variants were detected in 45. Genomic DNA was prepared from patients’ blood or bone marrow using standard protocols.
Sequencing
Initially, Phi 29-based whole-genome amplification was performed on genomic DNA samples using the Qiagen Repli-G service (Qiagen, Valencia, CA). Coding exons, including the exon/intron boundaries, were amplified by polymerase chain reaction (PCR) to generate amplicons for resequencing, because of the limited starting material available for many of the samples. Universal tails were added to the 5′ ends of amplification primers to serve as the sequencing primer sites. Amplicons were purified by Exonuclease/SAP treatment and then directly sequenced using Big Dye Terminator chemistry on an ABI3730 automated sequencer (Applied Biosystems, Foster City, CA). All coding exons of ELANE, HAX1, SBDS, GFI1, and G6PC3 were sequenced. Due to its large size, only those exons previously shown to harbour mutations were sequenced for WAS (exons 7–12). All primer sequences are provided in Table S1 and the nucleotide sequence of all novel genetic variants is provided in Table S2. The semiautomated sequence analysis was performed as previously described (Link, et al 2007).
RESULTS
ELANE genotyping
ELANE genotyping data was obtained on a total of 162 patients with SCN; results for some of these patients have been previously reported (Dale, et al 2000, Rosenberg et al 2008). ELANE sequence variants were observed in 90 (55.6%) of cases of SCN. A total of 43 distinct ELANE variants were identified (Figure 2). Consistent with previous reports, these putative pathogenic mutations were distributed throughout the entire gene, most of which were missense, with a few nonsense, indel, and splice variants (Ancliff, et al 2001, Ancliff, et al 2002, Bellanne-Chantelot, et al 2004, Dale, et al 2000, Germeshausen, et al 2001, Horwitz, et al 1999). Seventeen of the missense and nonsense variants have not been reported previously, including G27R, I31M, A32G, M37R, V54D, G56R, R74P, I91N, L92F, I100del, L143P, V157I, R162S, R164Q, D172TfsX10, C179X, G181W. Together with previous studies, this brings the total number of reported ELANE putative mutations in SCN to 73 (Ancliff, et al 2001, Bellanne-Chantelot, et al 2004, Carlsson, et al 2006, Dale, et al 2000, Germeshausen, et al 2001, Horwitz, et al 1999, Ishikawa, et al 2008, Kawaguchi, et al 2003, Salipante, et al 2007, Sera, et al 2005).
Figure 2. Summary of ELANE sequence variants.

Sequence variants detected by genotyping 162 patients with SCN in the North American SCNIR are summarized. Three cases with putative splice mutants [IVS4+5 G>A (2) and IVS3-8 (Exon4) C>A] are not included in this figure, because their effect on neutrophil elastase (NE) protein has not been experimentally validated. Multiple patients with the same mutation are indicated in parentheses. Novel variants are bolded.
Selection of samples for high-throughput sequencing
To investigate the relative frequency of other SCN-associated gene mutations, a high-throughput sequencing strategy was employed to screen for mutations in HAX1, WAS, SBDS, GFI1, G6PC3 and ELANE (Figure 1). In the cohort of 162 patients with SCN, no sequence variants of ELANE were detected in 72. Of these, adequate genomic DNA enabling sequencing at Washington University was available for 45. We also sequenced 28 cases with known ELANE variants to assess the fidelity of our sequencing pipeline and to determine whether ELANE variants co-existed with other SCN-associated gene mutations. The clinical characteristics of the 28 patients with ELANE variants and 45 with normal ELANE alleles are summarized in Tables 1 and 2, respectively.
Table 1.
Clinical characteristics for 28 SCN patients with mutant ELANE allele
| Pre-G-CSF Treatment | Post- G-CSF ANC | Post- G-CSF AMC | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Gender | AML/MDS | WBC ×109/l | RBC ×1012/l | MCV fl | Platelets ×109/l | ANC ×109/l | AMC ×109/l | ALC ×109/l | ||
| 3 | F | 10.1 | 4.3 | 79 | 464 | 0.0 | 2.42 | 6.26 | 0.78 | 0.7 | |
| 4 | M | 8.5 | 5.0 | 78 | 275 | 0.09 | 3.42 | 4.60 | 2.24 | 1.4 | |
| 12 | F | 12.5 | 4.5 | 71 | 798 | 0.00 | 1.41 | 8.52 | 1.44 | 4.65 | |
| 14 | M | NA | NA | NA | NA | NA | NA | NA | 1.17 | 3.70 | |
| 20 | M | 15.5 | 3.4 | 92 | 658 | 0.08 | 5.84 | 8.51 | 1.50 | 1.55 | |
| 31 | M | AML | 5.5 | 4.5 | 80 | 425 | 0.10 | 1.90 | 3.96 | 2.26 | 1.53 |
| 35 | F | AML | 7.8 | 4.1 | 79 | 411 | 0.05 | 0.15 | 7.04 | 1.61 | 1.31 |
| 36 | M | MDS | 5.4 | 4.0 | 94 | 375 | 0.27 | 1.12 | 2.67 | 0.00 | 1.76 |
| 38 | M | MDS | 2.7 | 4.5 | NA | 589 | 0.00 | 0.54 | 1.65 | 3.40 | 0.81 |
| 40 | M | AML | 13.6 | 4.0 | 64 | 722 | 0.08 | 3.51 | 8.63 | 0.28 | 0.68 |
| 42 | M | 2.7 | 4.9 | 75 | 331 | 0.09 | 0.67 | 1.11 | 1.51 | 1.19 | |
| 47 | F | AML | 11.3 | 4.0 | 72 | 328 | 0.05 | 3.97 | 5.89 | 0.88 | 2.21 |
| 48 | F | AML | 7.2 | 3.9 | 73 | 580 | 0.00 | 1.59 | 5.20 | 0.57 | 1.04 |
| 49 | M | AML | 9.1 | 4.1 | 79 | 423 | 0.26 | 3.37 | 4.60 | 1.13 | 4.53 |
| 50 | M | 5.4 | 4.7 | NA | 304 | 0.00 | 1.40 | 3.90 | 0.78 | 1.47 | |
| 51 | F | 14.9 | 3.0 | 91 | 369 | 0.00 | 4.47 | 7.15 | 2.25 | 2.97 | |
| 52 | F | 6.5 | 3.2 | NA | 235 | 0.00 | 2.02 | 4.49 | 0.80 | 3.97 | |
| 53 | M | 8.0 | 4.7 | 78 | 444 | 0.19 | 1.33 | 6.14 | 1.62 | 1.20 | |
| 54 | M | NA | NA | NA | NA | NA | NA | NA | 0.10 | 1.90 | |
| 55 | F | 9.7 | 3.7 | 81 | 547 | 0.09 | 1.38 | 8.15 | 0.81 | 1.78 | |
| 56 | F | 10.2 | 5.2 | 96 | 537 | 0.00 | 2.02 | 6.09 | 1.19 | 2.03 | |
| 57 | F | 5.6 | 4.5 | NA | 434 | 0.06 | 1.12 | 2.03 | 1.59 | 0.90 | |
| 58 | M | 10.0 | 3.2 | NA | 350 | 0.07 | 2.62 | 6.34 | 0.71 | 2.74 | |
| 59 | M | 13.4 | 3.2 | 91 | 656 | 0.07 | 2.33 | 10.43 | 1.67 | 8.70 | |
| 60 | F | 12.0 | 3.1 | 85 | 704 | 0.12 | 1.99 | 9.96 | 1.79 | 0.90 | |
| 61 | F | 15.8 | 3.3 | 92 | 739 | 0.09 | 2.49 | 10.04 | 1.23 | 1.72 | |
| 62 | F | 18.0 | 3.7 | 90 | 277 | 0.18 | 1.62 | 16.11 | 2.05 | 2.17 | |
| 63 | M | 8.2 | 3.7 | 84 | 459 | 0.00 | 2.95 | 5.03 | 2.06 | 1.91 | |
ANC: absolute neutrophil count; AMC: absolute monocyte count; ALC: absolute lymphocyte count
Table 2.
Clinical characteristics for 45 SCN patients with wile type ELANE allele
| Pre-G-CSF Treatment | Post- G-CSF ANC | Post- G-CSF AMC | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Gender | AML/MDS | WBC ×109/l | RBC ×1012/l | MCV fl | Platelets ×109/l | ANC ×109/l | AMC ×109/l | ALC ×109/l | ||
| 1 | M | 5.4 | 5.2 | NA | 389 | 0.36 | 0.77 | 4.33 | NA | NA | |
| 2 | M | 3.3 | 4.6 | NA | 270 | 0.18 | 0.99 | 2.18 | 1.40 | 1.2 | |
| 5 | F | 4.9 | 4.3 | 76 | 404 | 0.28 | 0.65 | 3.50 | 1.22 | 0.50 | |
| 6 | M | 4.2 | 4.3 | 99 | 370 | 0.14 | 0.85 | 2.80 | 0.91 | 0.60 | |
| 7 | F | 3.1 | 3.9 | 82 | 247 | 0.09 | 0.30 | 2.55 | 2.09 | 0.48 | |
| 8 | F | 5.7 | NA | NA | 108 | 0.68 | 0.32 | 4.50 | NA | NA | |
| 9 | F | 4.3 | 4.6 | 100 | 188 | 0.04 | 1.28 | 3.24 | 3.51 | 0.59 | |
| 10 | M | 6.1 | 3.3 | 85 | 319 | 0.25 | 0.59 | 5.13 | NA | NA | |
| 11 | M | 3.3 | NA | NA | 949 | 0.12 | 0.17 | 2.84 | 1.24 | 0.31 | |
| 13 | F | AML | 6.1 | 4.4 | 74 | 201 | NA | NA | NA | NA | NA |
| 15 | M | NA | NA | NA | NA | NA | NA | NA | 0.24 | 1.65 | |
| 16 | M | 3.9 | 3.8 | 79 | 656 | 0.08 | 0.34 | 3.08 | 2.57 | 0.60 | |
| 17 | M | 4.1 | 4.5 | 76 | 376 | 0.12 | 1.86 | 1.79 | 1.12 | 1.74 | |
| 18 | M | 3.6 | 4.0 | NA | 336 | 0.43 | 0.50 | 0.00 | 2.02 | 0.19 | |
| 19 | F | 2.5 | 5.2 | NA | 240 | 0.28 | 0.31 | 1.75 | NA | NA | |
| 21 | F | 6.1 | 3.9 | 79 | 534 | 0.04 | 1.72 | 4.41 | 0.09 | 1.03 | |
| 22 | M | 11.9 | 3.5 | 85 | 532 | 0.57 | 4.69 | 6.19 | 2.42 | 10.71 | |
| 23 | F | 2.3 | 4.4 | 91 | 211 | 0.32 | 0.31 | 0.88 | 1.66 | 0.46 | |
| 24 | M | 6.4 | 3.7 | 86 | NA | 0.00 | 1.85 | 4.74 | 0.51 | 2.84 | |
| 25 | M | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
| 26 | M | 2.8 | 3.7 | 90 | 243 | 0.23 | 0.26 | 2.41 | 0.33 | 0.45 | |
| 27 | F | 3.0 | 3.7 | 84 | 241 | 0.16 | 0.81 | 1.73 | 4.66 | 2.16 | |
| 28 | F | 2.6 | 2.9 | 111 | 263 | 0.41 | 0.31 | 1.69 | 0.06 | 0.45 | |
| 29 | F | 4.0 | 4.3 | 82 | 296 | 0.34 | 0.41 | 2.69 | NA | NA | |
| 30 | F | 4.3 | 4.8 | 81 | 379 | 0.22 | 0.85 | 3.11 | 0.54 | 0.75 | |
| 32 | M | 1.5 | 4.1 | 82 | 368 | 0.14 | 0.15 | 1.05 | 0.14 | 0.06 | |
| 33 | F | AML | 6.6 | 3.9 | 77 | 328 | 0.80 | 0.60 | 4.20 | 6.00 | 1.10 |
| 34 | M | AML | 1.9 | 4.7 | 89 | 148 | 0.22 | 0.04 | 1.46 | 1.98 | 0.08 |
| 37 | F | AML | 3.5 | 4.9 | 77 | 255 | 0.07 | 0.82 | 2.14 | 2.03 | 0.77 |
| 39 | M | AML | 6.4 | 4.8 | NA | 464 | 0.00 | 0.77 | 5.18 | 4.42 | 1.20 |
| 41 | M | MDS | 10.6 | 3.8 | 68 | 564 | 0.18 | 2.65 | 6.89 | 3.44 | 0.68 |
| 43 | M | 11.8 | 4.3 | 80 | 351 | 0.16 | 2.13 | 8.75 | 0.17 | 1.49 | |
| 44 | M | 6.9 | 4.7 | 72 | 390 | 0.15 | 1.40 | 4.46 | 0.07 | 1.15 | |
| 45 | M | 3.2 | 4.4 | 81 | 298 | 0.18 | 0.37 | 2.29 | 1.93 | 0.38 | |
| 46 | F | 4.1 | 5.0 | NA | 376 | 0.04 | 1.39 | 2.01 | 1.48 | 0.99 | |
| 64 | M | 7.9 | 4.3 | 73 | 349 | 0.28 | 1.48 | 5.97 | 4.28 | 1.10 | |
| 65 | M | 6.7 | 3.9 | 73 | 463 | 0.08 | 1.23 | 4.15 | 2.05 | 0.62 | |
| 66 | F | 3.0 | 4.5 | 76 | 238 | 0.59 | 0.47 | 2.15 | 1.05 | 0.38 | |
| 67 | M | 7.4 | 4.2 | 75 | 390 | 0.18 | 1.61 | 5.06 | 3.70 | 2.39 | |
| 68 | M | 4.1 | 4.0 | 76 | 230 | 0.11 | 0.45 | 3.16 | 11.78 | 0.43 | |
| 69 | M | 12.9 | 3.2 | 94 | 524 | 0.00 | 3.15 | 8.39 | 3.67 | 0.23 | |
| 70 | F | 7.2 | 4.2 | 78 | 389 | 0.09 | 1.74 | 5.24 | 0.37 | 0.42 | |
| 71 | M | 5.5 | 4.8 | 107 | 252 | 0.18 | 0.48 | 4.52 | 0.37 | 0.24 | |
| 72 | M | 5.1 | 4.4 | 74 | 417 | 0.09 | 1.08 | 3.38 | 5.77 | 1.73 | |
| 73 | M | 11.0 | 3.98 | 79 | 556 | 0.00 | 3.30 | 7.15 | 1.17 | 0.36 | |
ANC: absolute neutrophil count; AMC: absolute monocyte count; ALC: absolute lymphocyte count
Sequencing strategy
A high-throughput exon-based sequencing strategy utilizing whole-genome amplified genomic DNA isolated from bone marrow or blood leucocytes was used (Link, et al 2007). A semi-automated method to assess sequence quality and coverage was applied to each gene. With the exception of exon 1 of SBDS, sequence quality was very high, with an average of 95 ± 3.0% of samples having adequate coverage (range 81–98%). For a few patient samples, sequence quality/coverage for specific genes was inadequate (white boxes in Figure 3). These samples were eliminated from the final analysis of that gene.
Figure 3. Summary of genotypes.

Nonsynonymous nucleotide changes and gene deletions or insertions are shown. Known single nucleotide polymorphisms were excluded. White boxes indicate missing sequence data. Sequence variants previously implicated in SCN are highlighted in red. *Homozygous variant.
GFI1
Heterozygous germline mutations of GFI1 (N382S and K403R) each have been reported in a family with SCN (Person, et al 2003). These mutations are thought to act in a dominant-negative fashion to inhibit granulocytic differentiation. A single patient with the GFI1 N382S mutation was detected in our series; no other mutation was detected in this patient (Figure 3). Though the small numbers preclude definitive genotype-phenotype correlations, it is interesting to note that this patient, similar to the previously published case (Person, et al 2003), had a high basal level of circulating monocytes. Moreover, a striking monocytosis was noted after treatment with G-CSF (Table 2). Three other novel sequence variants of GFI1 not in public single nucleotide polymorphism (SNP) databases were detected: R412X, L400F, and P107A. Two of these, L400F and P107A, were seen in patients who also had an ELANE variant.
WAS
Mutations of the WAS gene, encoding the Wiskott-Aldrich syndrome protein (WASP), are associated with X-linked SCN. Three mutations, L270P, I294T, and S270P have been described in patients with SCN (Ancliff, et al 2006, Devriendt, et al 2001). All are thought to disrupt an auto-inhibitory domain in the WASP protein. In the present study, the L270P mutation was identified in a single male patient with SCN (Figure 3). Of note, monocytopenia was noted in our and the previously reported patients with the L270P WAS mutation (Table 2) (Devriendt, et al 2001). A novel WAS sequence variant, P460S, was identified in a female patient with SCN who had no other SCN-associated gene mutation.
SBDS
Compound heterozygous mutations of SBDS (most commonly K62X and 84Cfs3) are present in the majority of cases of SDS (Boocock, et al 2003). These mutations are thought to result in loss-of-function alleles. In the present study, two patients with SCN were found to have heterozygous mutations of SBDS (Figure 3). In addition to the C84YfsX3 mutation, a novel SBDS variant, Q94X, was detected in a single patient with SCN who also carried an ELANE variant.
HAX1 and G6PC3
Recent studies suggest the homozygous or compound heterozygous loss-of-function germline mutations of HAX1 or G6PC3 are responsible for most cases of autosomal recessive inherited SCN (Boztug, et al 2009, Carlsson, et al 2008, Germeshausen, et al 2008, Ishikawa, et al 2008, Klein, et al 2007). Of note, SCN caused by G6PC3 mutations is associated with cardiac and urogenital abnormalities and thrombocytopenia (Boztug, et al 2009). Surprisingly, in our series of patients with SCN, no HAX1 mutations were detected (Figure 3). However, two of our cases were associated with genetic variants of G6PC3. In one patient, the previously described G6PC3 G260R mutation was coupled with the novel genetic variant T118R. This patient has a secundum atrial septal defect, intermittent thrombocytopenia (median 162 × 109/l, range 56–574), but no urogenital abnormality. A second patient carried a homozygous single nucleotide deletion in codon 70 of G6PC3, resulting in a frameshift followed by premature stop codon. This patient also has cardiac abnormalities (atrial septal defect and coronary aneurysm), intermittent thrombocytopenia (median 113 × 109/l, range 42–474), but no urogenital abnormality.
DISSCUSSION
SCN is genetically heterogeneous with multiple genes reported to be associated with this disease, including ELANE, HAX1, WAS, SBDS, GFI1, and G6PC3. There is evidence that the frequency of gene mutations in SCN may depend upon the ethnic composition of the patient population. For example, Rosenberg et al (2008) reported that in 82 North American patients with SCN the frequency of ELANE mutations was 63%. In contrast, a study of 54 patients with SCN from the French Neutropenia Register reported ELANE mutations in only 35% of cases (Bellanne-Chantelot, et al 2004). In the present study, a two-tiered sequencing approach was used to define the mutation frequency of ELANE, HAX1, WAS, SBDS, GFI1, and G6PC3 in a relatively large cohort of patients with SCN. The genotyping data is summarized in Figure 4. Of note, consistent with previous reports (Ancliff, et al 2001, Bellanne-Chantelot, et al 2004, Germeshausen, et al 2001, Rosenberg, et al 2008), we considered all novel ELANE sequence variants as putative pathogenic mutations. It is possible that some of these ELANE variants may represent rare SNPs. Likewise, we considered the biallelic sequence variants of G6PC3 to be likely pathogenic mutations. A more conservative approach was taken for the other SCN-associated genes; for WAS, SBDS, and GFI1 only those variants previously reported in the literature were considered pathogenic mutations. Based on these considerations, and consistent with previous reports (Ancliff, et al 2001, Bellanne-Chantelot, et al 2004, Germeshausen, et al 2001, Rosenberg, et al 2008), ELANE was found to be the most commonly mutated gene in SCN, with a frequency of 55.6% (90 of 162 patients). A total of 5 mutations in the other SCN-associated genes (HAX1, WAS, SBDS, GFI1, and G6PC3) were detected in the 45 evaluable patients with normal ELANE alleles, yielding a combined mutation frequency of 11.1%. However, since only 44.4% of patients with SCN in this study had normal ELA2 alleles, it follows that the combined frequency of HAX1, WAS, SBDS, GFI1, and G6PC3 mutations in our SCN patient population is 4.9% (11.1% × 44.4% = 4.9%). Thus, in nearly 40% of our cases, the genetic basis of SCN remains unknown.
Figure 4. Genotyping summary.
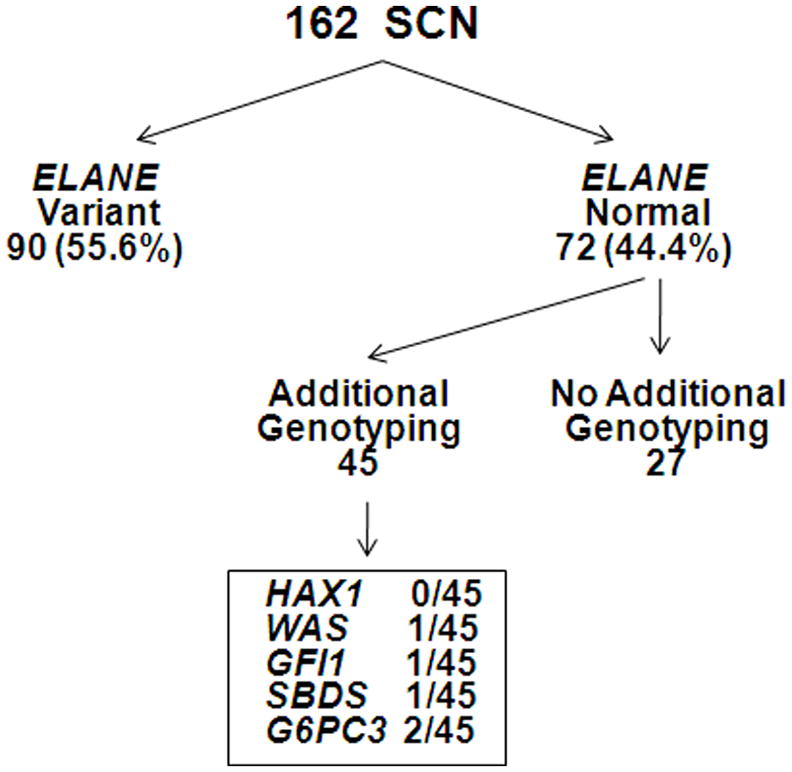
A total of 162 patients with SCN enrolled in the North American SCNIR underwent ELANE genotyping. ELANE sequence variants were detected in 90 (55.6%) of patients. In the subgroup of 45 patients with SCN with normal ELANE that underwent additional genotyping, putative mutations of HAX1, WAS, GFI1, SBDS, and G6PC3 were collectively detected in 5.
A number of novel genetic variants were detected in GFI1, WAS, and SBDS (Figure 3, black type). SIFT (Sorting Intolerant From Tolerant) analysis indicated that the GFI1 R412X, GFI1 L400F, and SBDS Q94X variants were likely to result in functionally deleterious changes (Ng and Henikoff 2003). Interestingly, similar to the GFI N382S mutant, the R412X and L400F variants are predicted to disrupt the final zinc finger of GFI1. Whether these GFI1 variants act as dominant-negative mutants will require further study. Two patients with SCN in this study had heterozygous SBDS mutations. Similar to SBDS C84YfsX3, it is likely that the SBDS Q94X variant results in a loss-of-function allele. Interestingly, a recent report linked heterozygous SBDS C84YfsX3 mutations to aplastic anemia (Calado, et al 2007). On the other hand, neutropenia has not been reported in family members of patients with SDS who are heterozygous for SBDS mutations (Boocock, et al 2003). It is possible that genetic variants of, as yet undefined, genes cooperate with heterozygous SBDS mutations to induce neutropenia. Analysis of a larger series of individuals will be required to define the role of heterozygous SBDS mutations in the pathogenesis of SCN.
Based on this data, we recommend that ELANE genotyping be performed in all patients with suspected SCN. For those patients with ELANE mutations, no further genotyping studies are needed. For those without ELANE mutations, a careful search for associated clinical features and review of the family history to ascertain the pattern of inheritance should be performed. Genotyping of G6PC3 and HAX1 should be performed in all patients with autosomal recessive inherited SCN, especially if associated with cardiac (G6PC3) or neuropsychological abnormalities (HAX1). WAS genotyping should be performed in patients with an X-linked pattern of inheritance, especially if associated with monocytopenia. Finally, GFI1 genotyping should be considered in patients with SCN who exhibit extreme monocytosis.
Acknowledgments
The authors gratefully acknowledge the provision of clinical data and biological samples by the physicians and patients associated with this study and with the Severe Chronic Neutropenia International Registry. We also thank Ernest Westrup, Jeff Christensen, and Yu Zhao for their expert assistance with the sequence analyses. This work was supported by grants from the National Institutes of Health [R24 A1049393 (DCD) and RO1 HL079562 (DCL] and a gift from the Amgen Foundation. JX, AAB, ER, SS, and AAA designed, performed or analyzed the experiments. DCL and DCD supervised all of the research and edited the manuscript.
References
- Ancliff PJ, Gale RE, Liesner R, Hann IM, Linch DC. Mutations in the ELA2 gene encoding neutrophil elastase are present in most patients with sporadic severe congenital neutropenia but only in some patients with the familial form of the disease. Blood. 2001;98:2645–2650. doi: 10.1182/blood.v98.9.2645. [DOI] [PubMed] [Google Scholar]
- Ancliff PJ, Gale RE, Watts MJ, Liesner R, Hann IM, Strobel S, Linch DC. Paternal mosaicism proves the pathogenic nature of mutations in neutrophil elastase in severe congenital neutropenia. Blood. 2002;100:707–709. doi: 10.1182/blood-2002-01-0060. [DOI] [PubMed] [Google Scholar]
- Bellanne-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, Vaury C, Barkaoui M, Fenneteau O, Maier-Redelsperger M, Chomienne C, Donadieu J. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood. 2004;103:4119–4125. doi: 10.1182/blood-2003-10-3518. [DOI] [PubMed] [Google Scholar]
- Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- Boztug K, Appaswamy G, Ashikov A, Schaffer AA, Salzer U, Diestelhorst J, Germeshausen M, Brandes G, Lee-Gossler J, Noyan F, Gatzke AK, Minkov M, Greil J, Kratz C, Petropoulou T, Pellier I, Bellanne-Chantelot C, Rezaei N, Monkemoller K, Irani-Hakimeh N, Bakker H, Gerardy-Schahn R, Zeidler C, Grimbacher B, Welte K, Klein C. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009;360:32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado RT, Graf SA, Wilkerson KL, Kajigaya S, Ancliff PJ, Dror Y, Chanock SJ, Lansdorp PM, Young NS. Mutations in the SBDS gene in acquired aplastic anemia. Blood. 2007;110:1141. doi: 10.1182/blood-2007-03-080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson G, Aprikyan AA, Ericson KG, Stein S, Makaryan V, Dale DC, Nordenskjold M, Fadeel B, Palmblad J, Hentera JI. Neutrophil elastase and granulocyte colony-stimulating factor receptor mutation analyses and leukemia evolution in severe congenital neutropenia patients belonging to the original Kostmann family in northern Sweden. Haematologica. 2006;91:589–595. [PubMed] [Google Scholar]
- Carlsson G, Van’t Hooft I, Melin M, Entesarian M, Laurencikas E, Nennesmo I, Trebinska A, Grzybowska E, Palmblad J, Dahl N, Nordenskjold M, Fadeel B, Henter JI. Central nervous system involvement in severe congenital neutropenia: neurological and neuropsychological abnormalities associated with specific HAX1 mutations. J Intern Med. 2008;264:388–400. doi: 10.1111/j.1365-2796.2008.01982.x. [DOI] [PubMed] [Google Scholar]
- Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, Boxer LA, Kannourakis G, Zeidler C, Welte K, Benson KF, Horwitz M. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96:2317–2322. [PubMed] [Google Scholar]
- Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, Verhoef GE, Boogaerts MA, Fryns JP, You D, Rosen MK, Vandenberghe P. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27:313–317. doi: 10.1038/85886. [DOI] [PubMed] [Google Scholar]
- Dror Y, Ward AC, Touw IP, Freedman MH. Combined corticosteroid/granulocyte colony-stimulating factor (G-CSF) therapy in the treatment of severe congenital neutropenia unresponsive to G-CSF: Activated glucocorticoid receptors synergize with G-CSF signals. Exp Hematol. 2000;28:1381–1389. doi: 10.1016/s0301-472x(00)00544-0. [DOI] [PubMed] [Google Scholar]
- Druhan LJ, Ai J, Massullo P, Kindwall-Keller T, Ranalli MA, Avalos BR. Novel mechanism of G-CSF refractoriness in patients with severe congenital neutropenia. Blood. 2005;105:584–591. doi: 10.1182/blood-2004-07-2613. [DOI] [PubMed] [Google Scholar]
- Germeshausen M, Schulze H, Ballmaier M, Zeidler C, Welte K. Mutations in the gene encoding neutrophil elastase (ELA2) are not sufficient to cause the phenotype of congenital neutropenia. Br J Haematol. 2001;115:222–224. doi: 10.1046/j.1365-2141.2001.03069.x. [DOI] [PubMed] [Google Scholar]
- Germeshausen M, Grudzien M, Zeidler C, Abdollahpour H, Yetgin S, Rezaei N, Ballmaier M, Grimbacher B, Welte K, Klein C. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. 2008;111:4954–4957. doi: 10.1182/blood-2007-11-120667. [DOI] [PubMed] [Google Scholar]
- Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC. Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet. 1999;23:433–436. doi: 10.1038/70544. [DOI] [PubMed] [Google Scholar]
- Ishikawa N, Okada S, Miki M, Shirao K, Kihara H, Tsumura M, Nakamura K, Kawaguchi H, Ohtsubo M, Yasunaga S, Matsubara K, Sako M, Hara J, Shiohara M, Kojima S, Takihara Y, Kobayashi M. Neurodevelopmental abnormalities associated with severe congenital neutropenia due to the R86X mutation in the HAX1 gene. J Med Genet. 2008;45:802–807. doi: 10.1136/jmg.2008.058297. [DOI] [PubMed] [Google Scholar]
- Kawaguchi H, Kobayashi M, Nakamura K, Konishi N, Miyagawa S, Sato T, Toyoda H, Komada Y, Kojima S, Todoroki Y, Ueda K, Katoh O. Dysregulation of transcriptions in primary granule constituents during myeloid proliferation and differentiation in patients with severe congenital neutropenia. J Leukoc Biol. 2003;73:225–234. doi: 10.1189/jlb.0902427. [DOI] [PubMed] [Google Scholar]
- Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, Rathinam C, Boztug K, Schwinzer B, Rezaei N, Bohn G, Melin M, Carlsson G, Fadeel B, Dahl N, Palmblad J, Henter JI, Zeidler C, Grimbacher B, Welte K. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease) Nat Genet. 2007;39:86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- Link DC, Kunter G, Kasai Y, Zhao Y, Miner T, McLellan MD, Ries RE, Kapur D, Nagarajan R, Dale DC, Bolyard AA, Boxer LA, Welte K, Zeidler C, Donadieu J, Bellanne-Chantelot C, Vardiman JW, Caligiuri MA, Bloomfield CD, DiPersio JF, Tomasson MH, Graubert TA, Westervelt P, Watson M, Shannon W, Baty J, Mardis ER, Wilson RK, Ley TJ. Distinct patterns of mutations occurring in de novo AML versus AML arising in the setting of severe congenital neutropenia. Blood. 2007;110:1648–1655. doi: 10.1182/blood-2007-03-081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person RE, Li FQ, Duan Z, Benson KF, Wechsler J, Papadaki HA, Eliopoulos G, Kaufman C, Bertolone SJ, Nakamoto B, Papayannopoulou T, Grimes HL, Horwitz M. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet. 2003;34:308–312. doi: 10.1038/ng1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg PS, Alter BP, Link DC, Stein S, Rodger E, Bolyard AA, Aprikyan AA, Bonilla MA, Dror Y, Kannourakis G, Newburger PE, Boxer LA, Dale DC. Neutrophil elastase mutations and risk of leukaemia in severe congenital neutropenia. Br J Haematol. 2008;140:210–213. doi: 10.1111/j.1365-2141.2007.06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salipante SJ, Benson KF, Luty J, Hadavi V, Kariminejad R, Kariminejad MH, Rezaei N, Horwitz MS. Double de novo mutations of ELA2 in cyclic and severe congenital neutropenia. Hum Mutat. 2007;28:874–881. doi: 10.1002/humu.20529. [DOI] [PubMed] [Google Scholar]
- Sera Y, Kawaguchi H, Nakamura K, Sato T, Habara M, Okada S, Ishikawa N, Kojima S, Katoh O, Kobayashi M. A comparison of the defective granulopoiesis in childhood cyclic neutropenia and in severe congenital neutropenia. Haematologica. 2005;90:1032–1041. [PubMed] [Google Scholar]
- Sinha S, Zhu QS, Romero G, Corey SJ. Deletional mutation of the external domain of the human granulocyte colony-stimulating factor receptor in a patient with severe chronic neutropenia refractory to granulocyte colony-stimulating factor. J Pediatr Hematol Oncol. 2003;25:791–796. doi: 10.1097/00043426-200310000-00010. [DOI] [PubMed] [Google Scholar]