

Abstract
The present study provides spectroscopic and experimental evidence demonstrating that degenerate metathesis is critical to the effectiveness of this emerging class of chiral catalysts. Isolation and characterization (X-ray) of both diastereomeric complexes, as well as examination of the reactivity and enantioselectivity patterns exhibited by such initiating neophylidenes in promoting RCM processes, are disclosed. Only when sufficient amounts of ethylene are generated and inversion at Mo through degenerate processes occurs at a sufficiently rapid rate, is high enantioselectivity achieved, irrespective of the stereochemical identity of the initiating alkylidene (Curtin-Hammett kinetics). With diastereomeric metal complexes that undergo rapid interconversion, stereomutation at the metal center becomes inconsequential and stereoselective synthesis of a chiral catalyst is not required. 
We recently disclosed a class of chiral Mo-based complexes (e.g., S-1 and S-2, Figure 1) that are prepared diastereoselectively (7.0:1 and 2.2:1, respectively) and used in situ to promote olefin metathesis1,2 with high reactivity and enantioselectivity.3 A notable feature of the new catalysts is the presence of a donor (pyrrolide) and an acceptor (aryloxide or alkoxide) ligand (vs two acceptor ligands utilized previously)2a which, based on initial theoretical studies,4 is likely critical to achieving high efficiency. The fluxional pyrrolide-aryloxides bear only monodentate ligands and a stereogenic metal center, which undergoes inversion with every sequence that involves formation of a metallacyclobutane (MoR=C + substrate olefin →metallacyclobutane→C=MoS + product olefin).5 In the simplest analysis, each olefin metathesis catalytic cycle includes two inversion processes: cross-metathesis leading to substrate-derived alkylidene and a subsequent ring-closing, ring-opening, or cross-metathesis. Under such a regime, a stereogenic-at-metal complex emerges from a reaction as the same stereoisomer that begins the process (net retention). Inversion at the metal might, however, occur beyond the boundaries of a productive catalytic cycle due to degenerate metathesis; such isomerizations can have significant consequences on reaction efficiency as well as enantioselectivity. Herein, we provide evidence demonstrating that degenerate metathesis, prevalent in transformations catalyzed by stereogenic-at-metal complexes, is critical to the effectiveness of the enantioselective ring-closing metathesis (RCM) processes.
Figure 1.

Stereogenic-at-Mo Complexes (Major Diastereomers)
We began with determination of the structure of R-1, generated as the minor diastereomer (7:1 S:R). In spite of being the minor component, R-1 was isolated in sufficient quantities to allow us to secure its X-ray structure (Scheme 1; X-ray of S-13a also illustrated).6 Earlier studies had indicated that ≥95% of R-1 remains intact in an RCM reaction (Table 1) that proceeds to completion in 30 min with a 7:1 mixture of S-1:R-1.3a A comparison of the structures of S-1 and R-1 points to a rationale for such reactivity differences. Approach of an alkene to S-1 trans to the pyrrolide7 is hindered by a Br, moved out of the way by a slight rotation about the Mo–O bond. In R-1, however, olefin coordination is blocked by the larger (vs Br) tetrahydronaphthyl ring of the aryloxide ligand. Furthermore, a bromide substituent of the aryloxide ligand resides within 3.04 Å of the Mo in R-1, suggesting a Mo–Br interaction.8 Such association discourages rotation around the Mo–O, which is required if the metal center is to be accessible to a substrate molecule.
Scheme 1.

Isolation and X-ray Crystal Structure of R-1
Table 1.
Time Dependence of Conversion and Selectivity in Catalytic RCM with Pure R-1 (Minor Diastereomer)a
See the Supporting Information (SI) for all experimental details, including spectroscopic data on R-1.
Based on 400 MHz 1H NMR analysis of unpurified mixtures.
Based on HPLC analysis (see the SI for details).
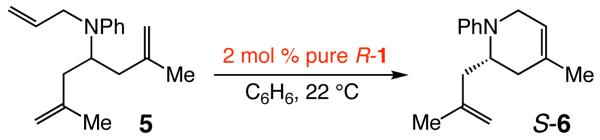
With a pure sample of R-1 available, we examined the ability of this diastereomer to promote enantioselective RCM reactions. As the data summarized in Table 1 indicate, R-1 does promote RCM of triene 5, but at a slower rate than when S-1 is used (180 vs 20 min for >98% conv; Tables 1-2). Surprisingly, however, the RCM with R-1 delivers the same major product enantiomer (S-6) as obtained when S-1 is employed and with nearly the same enantioselectivity (96:4 vs 96.5:3.5 er; see Tables 1-2).
Table 2.
Time Dependence of Conversion and Selectivity in Catalytic RCM with Pure S-1 (Major Diastereomer)a

The observations in eq 1, regarding RCM of 7, a key intermediate in the recent enantioselective total synthesis of quebrachamine,3a–b offer an additional example: triene 7 reacts with R-1 significantly more slowly (12 h vs 1 h with S-1) but, as with S-1, R-8 is generated with precisely the same level of enantioselectivity (98:2 er) and sense of absolute stereochemistry (R-8 major).9 The above data imply that the stereochemical identity of the stereogenic-at-metal complex is significant vis-à-vis the rate of initiation of the initial neophylidene, but has no bearing on the eventual stereochemical outcome. Isomerization of the two diastereomers of the chiral complex, therefore, is likely more facile than ring-closure; that is, Curtin-Hammett condition10 applies for a significant duration of the catalytic cycle. Another set of mechanistically critical observations, depicted in Tables 1-2, are that, irrespective of whether pure R-1 or S-1 is used, initial enantioselectivity is substantially lower than that of the final product (see entries 1). A rationale for such findings will be provided below.
 |
(1) |
The observations described above undermine the validity of the simplest form of the catalytic cycle involving two inversions at the metal center. Two other issues detract from the pathway in Scheme 2: (1) The lower activity of alkylidenes bearing an R Mo center should apply to R-9 as well; S-9 is expected to be more reactive. (2) RCM through R-9 would likely afford R-6, the minor enantiomer observed in the reactions shown in Tables 1 or 2.
Scheme 2.

Initial Mechanism Without Mo Center Isomerization Outside-the Main Catalytic Cycle
The results of RCM of tetraene 12 (eq 2) performed with pure S-1, affording S-6 as the predominant enantiomer (90:10 er), further substantiate that additional inversions at the metal center, outside the confines of a catalytic cycle that involves only a double inversion, play a crucial role. The simplest catalytic cycle for conversion of 12 to 6 would include three olefin metathesis reactions (vs two for 5 or 7). In the absence of any additional isomerizations at Mo, the catalyst would have undergone net inversion after formation of every molecule of 6 (from 12), leading to generation of 6 in low enantiomeric purity.
Inversion at the Mo center can also occur through degenerate olefin metathesis. Thus, as illustrated in Scheme 3, reaction of alkylidene R-9 (Scheme 2) with another molecule of 5 might afford the symmetrically substituted metallacyclobutane 13; such a process might furnish S-9, an alkylidene that can deliver the observed major enantiomer S-6 via metallacyclobutane 14. However, an experiment involving reaction of a 1:1 mixture of allylamine 15:d3-15 in the presence of pure S-1 (eq 3), which did not lead to any detectable amounts of deuterium scrambling, serves as evidence against inversion at Mo through a substrate-induced degenerate process.
Scheme 3.

Mo Isomerization by Substrate-Induced Degenerate Metathesis
 |
(2) |
 |
(3) |
Ethylene, although not present at the earliest stages of the RCM process, can readily promote degenerate olefin metathesis as well. As shown in Scheme 2, through the first RCM, S-1 sheds the neophylidene unit of the initial complex, leading to methylidene S-11,11 which may proceed through cross-metathesis with substrate 5 to produce ethylene (and re-generate R-9). Degenerate metathesis and inversion at the Mo center can thus occur by rapid interconversion of S-11 and R-11 via unsubstituted metallacyclobutane 16 (Scheme 4).3e Cross-metathesis of R-11 with 5 would give S-9, which would readily undergo RCM to afford S-6.
Scheme 4.

Mo Isomerization by Ethylene-Induced Degenerate Metathesis
Three additional findings support the proposal that ethylene initiates degenerate metathesis and promotes high enantioselectivity: (1) Treatment of d3-5 with 2 mol % pure S-1 leads to deuterium scrambling within seven minutes (eq 4).
 |
(4) |
(2) As shown in eq 5, when RCM of 5 is performed with 100 mol % diallyl ether (to generate ethylene and promote rapid methylidene generation), high enantioselectivity is observed early in the reaction (95:5 er vs entries 1, Tables 1-2). The more rapid initiation (vs Table 2) points to a more facile formation of methylidenes 11.
(3) RCM of tetraene 12 with 5 mol % pure S-1 delivers 6 in only 60.5:39.5 S:R after ~2% conversion (eq 6; compare to eq 2). Thus, without sufficient ethylene, the catalytic cycle in its simplest form is largely operative (triple inversion at Mo).
 |
(5) |
 |
(6) |
The proposed mechanism, summarized in Scheme 5, offers a rationale for low enantioselectivity at the nascent stages of RCM with S-1 or R-1 (Tables 1-2). In reactions that commence with S-1, little or no ethylene is initially present; thus, RCM likely proceeds via R-9 to afford a significant amount of R-6 (minor product enantiomer). When the catalytic cycle is initiated by R-1, the faster reacting S-9 is formed and the major product isomer S-6 can be generated. If ethylene is available only at low concentration, however, maximum enantioselectivity cannot be achieved (~96:4 er), since ethylene can convert R-1 to S-11, which reacts with 5 to form R-9, leading to the minor product enantiomer (R-6). Only when sufficient ethylene is present, such that inversion at Mo occurs at an appropriately rapid rate, can the fast reacting S-9 become easily accessible and high enantioselectivity be obtained. The stereochemical outcome of the RCM reactions is thus independent of the identity of the initiating alkylidene S-1 or R-1 (Curtin-Hammett kinetics).10
Scheme 5.

Equilibria Promoted by Ethylene Critical to Efficiency and Enantioselectivity
An important feature of metal-catalyzed olefin metathesis promoted by stereogenic-at-metal complexes is that, with each reaction, the metal center is inverted. We demonstrate that, at steady state, such inversions are faster than product formation. The absence of multidentate ligands, which can raise the barrier to inversion at the metal and reduce catalyst activity, is therefore a significant attribute of the present class of catalysts. Our study highlights the principle that diastereomeric – not enantiomeric – chiral catalysts might be preferable to those that contain a C2-symmetric bidentate ligand2a (and thus a non-stereogenic metal center). In diastereomeric complexes that undergo rapid interconversion of metal center configuration by degenerate metathesis, stereomutation at the metal becomes inconsequential and, as a result, stereoselective synthesis of a chiral catalyst candidate is not required.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (Grant GM-59426) and AstraZeneca (graduate fellowship to S. Malcolmson). We are grateful to Professor K. Tan and A. Zhugralin for helpful discussions, and thank Dr. B. Bailey and K. Wampler for determination of X-ray structure of S-1.
Footnotes
Supporting Information Available: Experimental procedures and spectral, analytical data for all reaction products (PDF). This material is available on the web: http://www.pubs.acs.org.
References
- 1.For recent reviews on catalytic olefin metathesis, see: Grubbs RH, editor. Handbook of Metathesis. Wiley–VCH; Weinheim, Germany: 2003. Hoveyda AH, Zhugralin AR. Nature. 450. 2007:243–251. doi: 10.1038/nature06351.
- 2.For reviews on high oxidation state complexes used in catalytic olefin metathesis, see: Schrock RR, Hoveyda AH. Angew. Chem., Int. Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576.Schrock RR. Chem. Rev. 2009;109:3211–3226. doi: 10.1021/cr800502p.
- 3.a Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008;456:933–937. doi: 10.1038/nature07594. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sattely ES, Meek SJ, Malcolmson SJ, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:943–953. doi: 10.1021/ja8084934. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ibrahem I, Yu M, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3844–3845. doi: 10.1021/ja900097n. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lee Y-J, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:10652–10661. doi: 10.1021/ja904098h. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Marinescu SC, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2009;131:10840–10841. doi: 10.1021/ja904786y. [DOI] [PubMed] [Google Scholar]
- 4.a Solans-Monfort X, Clot E, Copéret C, Eisenstein O. J. Am. Chem. Soc. 2005;127:14015–14025. doi: 10.1021/ja053528i. [DOI] [PubMed] [Google Scholar]; b Poater A, Solans-Monfort X, Clot E, Copéret C, Eisenstein O. J. Am. Chem. Soc. 2007;129:8207–8216. doi: 10.1021/ja070625y. [DOI] [PubMed] [Google Scholar]
- 5.For applications of stereogenic-at-Ru complexes to enantio- or product-selective olefin metathesis reactions, see: Van Veldhuizen JJ, Gillingham DG, Garber SB, Kataoka O, Hoveyda AH. J. Am. Chem. Soc. 2003;125:12502–12508. doi: 10.1021/ja0302228.Gillingham DG, Kataoka O, Garber SB. J. Am. Chem. Soc. 2004;126:12288–12290. doi: 10.1021/ja0458672.Bornand M, Chen P. Angew. Chem., Int. Ed. 2005;44:7909–7911. doi: 10.1002/anie.200502606.
- 6.See the Supporting Information for details of crystal structure of R-1.
- 7.For crystallographic evidence that a Lewis basic PMe3 associates trans to the pyrrolide, see: Marinescu SC, Schrock RR, Li B, Hoveyda AH. J. Am. Chem. Soc. 2009;131:58–59. doi: 10.1021/ja808308e.
- 8.The observed Mo–Br distance (3.04 Å) is significantly less than the sum of the van der Waals radii for Mo and Br (1.85 Å and 2.00 Å, respectively).
- 9.For additional examples, see the Supporting Information.
- 10.For selected instances where Curtin-Hammett conditions have been illustrated for metal-catalyzed enantioselective reactions, see: Halpern J. Science. 1982;217:401–407. doi: 10.1126/science.217.4558.401.Hughes DL, Lloyd-Jones GC, Krska SW, Gouriou L, Bonnet VD, Jack K, Sun Y, Mathre DJ, Reamer RA. Proc. Nat. Acad. Sci. 2004;101:5379–5384. doi: 10.1073/pnas.0306918101. For additional cases, see the SI.
- 11.For an X-ray structure of monoaryloxide-monopyrrolide W-based methylidene complexes, see: Jiang AJ, Simpson JH, Müller P, Schrock RR. J. Am. Chem. Soc. 2009;131:7770–7780. doi: 10.1021/ja9012694.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.