

Abstract
Adenovirus is a well-established viral gene transfer model system that presents two major hurdles when being considered for cell specific targeting applications. First is the need to detarget the vector from inherent host binding mechanisms, and second is the need to establish a productive and stable method to retarget the vector to a desired cell receptor. In previous studies we had generated an adenovirus vector platform that lacks the normal targeting attributes derived from the fiber and penton capsid proteins. In the current study we characterized our detargeted Ad5-based vectors (Ad5.F7F41S and Ad5.F7F41SΔRGD) as platforms for novel retargeted viruses. The experimental strategy relied on incorporating small peptide ligands into several sites of the Ad 41 short fiber knob domain (AB, CD, HI, G and Cterm). Reengineering of Ad41 short fiber resulted either in a bypass to fiber 7 usage, or in a dominant negative packaging/production deficiency phenotype. Under specific growth conditions we could remedy some of the capsid deficiencies and generate high titer viruses. However when examined by Western blot analysis, the resulting viruses were still defective in capsid content. The tandem fiber F7F41S platform has revealed an unanticipated sensitivity of Adenovirus packaging to fiber 41 short structural modifications. These studies indicate fiber assembly into an intact virion or fiber influenced capsid stability as a bottleneck to efficient particle production. We also demonstrate that virus particles characterized as mature virions following CsCl banding can vary significantly in capsid protein content. Considering the complexity of virus entry into a target cell, modified “mature virions” may be compromised at the level of transduction not only through the intended modification, but also by virtue of secondary structural packaging conflicts.
Introduction
Adenovirus vectors (AdV) have been used in a variety of in vivo and in vitro gene transfer applications with varying degrees of success. These studies have revealed many technical difficulties when converting a pathogenic agent into a vehicle for long-term gene expression in vivo. Major hurdles to enhancing the use of AdV arise from vector immunogenicity and an inability to direct AdV transduction in cell/tissue specific manner. With standard adenovirus type 5 vectors (Ad5), the biological functions of the major capsid proteins have been shown to contribute significantly to these limitations. Thus, a goal in AdV biology has been to generate modified vectors that are targeted to specific cell types or tissues while blunting the host antiviral immune response.
The major AdV capsid proteins hexon, penton base, and fiber have been shown to contribute to a well-orchestrated gene delivery program in cell lines (reviewed in (Nemerow et al., 2009)). The canonical two-step entry process is initiated by high affinity attachment of the fiber homotrimeric knob domain to the cell-surface coxsackie-and-adenovirus receptor (CAR) (Bergelson et al., 1997). After fiber-CAR binding, virus internalization is triggered by engagement of the RGD motif of penton base with cell surface αv integrins (Wickham et al., 1993). In cells lacking CAR, binding through penton integrin can serve as both primary binding and virus internalization signals (Schoggins and Falck-Pedersen, 2006). Several strategies, both genetic and non-genetic, have been developed to alter AdV targeting (reviewed in (Campos and Barry, 2007)). The most common approach has been to genetically modifying capsid proteins, primarily fiber, to confer novel binding properties to the virus. Two strategies of genetic modification of fiber have been employed. The first is based on fiber pseudotyping, where fiber from a nonCAR-binding virus (Ad7) was used to replace the CAR binding Ad5 fiber (Gall et al., 1996; Nakamura, Sato, and Hamada, 2003). This strategy combines elimination of normal CAR binding with retargeting to the receptor for Ad7. The second strategy is more complex and relies on direct sequence manipulation of Ad5 fiber. Early examples of this approach demonstrated that a C terminal addition of a polylysine motif (Wickham et al., 1996) or an RGD motif (Wickham et al., 1997) could enhance Ad5 binding to heparin sulfate or integrin receptors, respectively. Atomic resolution of the Ad5 fiber knob revealed that the HI loop adopted a flexible conformation that may serve as a suitable scaffold for insertion of small targeting peptides. Inserting the targeting peptide RGD-4C (CDCRGDCFC) into the HI loop and demonstrating enhanced transduction in ovarian cancer cells confirmed this strategy(Dmitriev et al., 1998). Additional studies have demonstrated that this vector efficiently targets integrins and transduces cells types that are typically refractory to Ad5, such as glioma, pancreatic cancer cells, dendritic cells, endothelial cells, and smooth muscle cells (reviewed in (Barnett, Crews, and Douglas, 2002)). Other work has demonstrated that peptides targeting the transferrin receptor (Xia et al., 2000) or peptides identified by phage display to selectively bind endothelial cells (Nicklin et al., 2001; Nicklin et al., 2000) can be utilized to retarget Ad5 vectors to cells that were largely refractory to Ad5 transduction. Importantly, the early HI-targeted vectors were not ablated in fiber-CAR binding, and thus they enhanced cell tropism when used in vitro, but did not eliminate normal Ad5 targeting.
Accumulating evidence has shown that the processes of Ad5 binding and internalization differ greatly in tissue culture cell lines when compared to in vivo model systems. When administered systemically, Ad5-based vectors are primarily sequestered to hepatic tissue. Hepatic localization involves a variety of nonCAR binding interactions (Alemany and Curiel, 2001; Martin et al., 2003; Nicklin et al., 2001), and recent data demonstrate that a binding interaction between the hypervariable region of hexon and the vitamin K dependent coagulation factor X contributes to vector uptake into hepatocytes (Kalyuzhniy et al., 2008; Waddington et al., 2008). The different binding functions of Ad5 capsid proteins in these model infection systems present one of the major difficulties in generating targeted AdV, the need to detarget AdV5.
Two main strategies have been used to detarget Ad5-based vectors in vivo: incorporation of mutations that ablate capsid-receptor binding, and generation of capsid chimeras that reduce native Ad5 tropism. With respect to the former, the most successfully detargeted vectors have: 1) triple deletions that eliminate capsid interactions with CAR, integrins, and a putative interaction with heparin sulfate glycosaminoglycans via the fiber KKTK motif (Nicol et al., 2004; Smith et al., 2003), or 2) hexon modifications that prevent binding to blood factor X (Waddington et al., 2008). The simplest fiber chimeric vectors that confer a detargeted phenotype in a murine model are those that incorporate non-Ad5 fiber into the capsid (Gall et al., 1996; Nakamura, Sato, and Hamada, 2003), thus eliminating native fiber 5 binding functions.
We have extended the capsid chimera strategy to generate several fiber-modified vectors with desirable detargeting properties. By replacing the Ad5 fiber terminal exon with two genes encoding short-shafted fibers from Ad7 (fiber 7) and Ad41 (fiber 41S), we generated the Ad5-based F7F41S vector (Schoggins, Gall, and Falck-Pedersen, 2003). This virus expressed both fiber genes in infected cells; however using Western blot analysis we demonstrated that all detectable fiber incorporated into the F7F41S virion capsid was the medium shaft length fiber 41S. Since no receptor binding function for fiber 41S has been identified (Roelvink, Kovesdi, and Wickham, 1996), the F7F41S vector was highly refractory to transduction in vitro and in vivo (Schoggins et al., 2005). Subsequent mutation of the penton base RGD motif (F7F41SΔ vector) further compromised vector transduction and significantly reduced the ability of this virus to activate the host inflammatory response (Schoggins and Falck-Pedersen, 2006).
Based on these detargeting properties, we chose F7F41S and F7F41SΔ as platforms for retargeting and initiated a mutagenesis strategy to incorporate small peptide ligands into the fiber 41S knob domain. While these studies were being completed, Hesse et al (Hesse et al., 2007) carried out a strategy for modifying vector tropism by inserting the RGD-4C (CDCRGDCFC) motif into several domains of the fiber 41S knob region in an Ad5/41 chimeric fiber construct using a two step transfection/superinfection strategy. In our studies with the single step tandem fiber growth strategy, we found that viruses containing peptide insertions into fiber 41S exhibited severe growth defects and aberrant capsid protein composition. Mutant fiber 41S trimers formed, but were predominantly detected in empty capsids. For mature virions that were properly packaged, fiber incorporation was biased toward fiber 7, with little to no detectable mutant fiber 41S displayed. Upon removal of fiber 7 from the genome, capsid protein composition was partially restored and mutant fiber 41S molecules were incorporated into mature virions. However these virions still had demonstrable capsid defects, and were defective for transduction in a model ligand/receptor system.
Results
Characterization of F7F41S-based viruses expressing peptides in fiber 41S CD and HI loops
Previous work from a host of laboratories has demonstrated that the HI loop of Ad5 fiber is amenable to peptide insertions. Based on sequence alignment data and the success of HI insertions into Ad5 fiber, our first set of virus constructs focused on the CD and HI loops of fiber 41S as potential sites for ligand insertion. A PCR mutagenesis strategy was used to clone peptides of varying lengths (6aa, 14aa, 17aa for HI and 6aa, 37aa for CD) into the fiber 41S knob domain of the F7F41S vector as described in Materials and Methods (Figure 1 and 2 schematic). Viruses were generated by overlap homologous recombination in 293 cells followed by characterization and large-scale production. During plaque purification and virus propagation, we noted that viruses containing peptide insertions into fiber 41S formed larger plaques, grew more rapidly, and generated higher viral titers than the parental F7F41S construct (data not shown).
Figure 1. Peptide insertions into fiber 41 short CD and HI loops induce an F7 bypass event.

(A) Depiction of predicted fiber content in the Ad5-based constructs F7F41S and F7F41S.HI, with fiber 41S dominating fiber 7. (B) 1 × 1010 viral particles of vectors containing inserts of various lengths into the HI or CD loops were analyzed by Western blot using anti-fiber 4D2 antibody. The relative positions of fiber 41S and fiber 7 are demonstrated with control F7 and F7F41S vectors. (C) Total cell lysates from 293 cells infected with the indicated mutant viruses were analyzed by Western blot using the anti-fiber R72 antiserum. * denotes cells grown at 32°C. In all panels, numbers in parentheses indicate the length of amino acid insertion.
Figure 2. Fiber 41S knob and structure-based sequence alignment.

(A) Graphical representation of the fiber 41S homotrimeric knob domain with target insertion loops denoted by arrows. (B) Sequence alignment of fiber knobs from Ad2, Ad5, and Ad41 (fiber 41S). Alignments were generated by the 3DCOFFEE/EXPRESSO server using sequence and structure data obtained from atomic coordinates in the Protein Data Bank. Coloration from red → orange → yellow → green is indicative of crystal structure B values from low to high (A) and sequence conservation from high to low (B) Loop domains are shown and specific location of peptide insertions are indicated by arrows.
To assess the fiber content of viruses with CD and HI loop insertions, cesium-banded virions were analyzed by SDS-PAGE followed by Western blotting with a fiber-specific antibody (Figure 1B). A striking result was revealed with respect to capsid fiber incorporation. Compared to the F7F41S parental virus, all mutant viruses incorporated predominantly fiber 7 into the virions, with little or no detectable fiber 41S. With respect to fiber content, these viruses were indistinguishable from the single fiber-pseudotyped construct F7, which expresses only fiber 7 and exhibits a marked growth advantage over F7F41S. These results are consistent with our observations that mutant viruses grew faster and to higher titers than parental F7F41S.
The Western blot data suggest that mutant fiber 41S proteins were either not expressed or were expressed but not incorporated into capsids. To resolve this question, we measured mutant fiber 41S.HI protein levels in total infected cell lysates at 24hr and 48hr post-infection by Western blot on denaturing gels. Mutant fiber 41S proteins were detected in all of the infected cell lysates (Figure 1C), indicating that fiber monomers were expressed. Two possibilities may explain the absence of mutant fiber 41S in purified virions: 1) mutant fiber 41S monomers fail to trimerize and therefore are not incorporated, or 2) a packaging conflict prevents production of mature virions displaying the fiber 41S mutants. Importantly relatively minor peptide insertions into fiber 41 short completely changed the fiber bias toward fiber 7-only virions. Hence the switch phenotype serves as an indicator of an abnormal fiber 41 short fiber.
Unique structural features of the fiber 41S knob domain
During the course of these studies, x-ray crystallographic data for fiber 41S knob was published and provided insight into important structural features of the protein. Resolution of the fiber 41S knob revealed a structure that was similar to other fiber knobs (Seiradake and Cusack, 2005) (Figure 2A). The homotrimeric knob is formed by three monomers consisting of eight anti-parallel beta barrels connected by loop regions. Several features distinguish fiber 41S from other knob domains. Unlike fiber 5, the fiber 41S knob is more compact and has shorter loop domains connecting each of the beta barrels. Additionally, superimposition of AB loops from fiber 41S with other CAR-binding knobs revealed that the fiber 41S AB loop has a unique conformation, which may contribute to its inability to bind CAR (Seiradake and Cusack, 2005). The most notable feature with respect to our studies is the presence of a highly disordered region (the G region) that has partial beta sheet characteristics (Figure 2A). This region was only partially resolved in crystals prepared at pH5, while no density was obtained for the G region at pH8. Using the Expresso/3D-coffee server (Armougom et al., 2006), fiber knobs from Ad2, Ad5, and Ad41 were aligned based on structural information obtained from atomic coordinates in the Protein Data Bank. This structure-based sequence alignment confirmed that the G region of fiber 41S differs dramatically from homologous regions in other fiber knobs (Figure 2B).
Generating mutant fiber 41S proteins targeted to the melanocortin-1 receptor
Based on sequence alignment and structural analysis using the Protein Dossier 2.0 server (Neshich et al., 2005), we shifted the focus of our targeting studies from the CD/HI loops and chose to insert peptides into the AB loop, the G region, or the C terminus. To specifically target vectors in a model ligand/receptor system, we chose to insert sequences encoding the melanocyte stimulating hormone alpha (MSH), which binds with high affinity to the melanocortin-1 receptor (MC1R) on the surface of melanocytes and melanoma cells (Mountjoy et al., 1992; Yang et al., 1997). Due to its enhanced detargeted phenotype, the F7F41SΔ vector was selected as the platform for these mutagenesis studies.
To generate fiber 41S MSH mutants, a GS-rich flexible linker was inserted into 3 regions of fiber 41S (AB, G, and Cterm) (Figure 2B and 3A). Before generating novel MSH containing viruses, we sought to determine whether fiber 41S containing AB, G, or Cterm insertions were able to trimerize. Trimers were assayed by transient transfection of the following plasmids: pT.F41S.AB.Rec, pT.F41S.G.Rec, and pT.F41S.Cterm.Rec, each of which express a CMV-driven fiber 41S containing the linker region but no targeting MSH peptide. Lysates were generated from transfected 293 cells and analyzed by Western blot. Because fiber trimers remain stable in the presence of SDS at low temperatures (Novelli and Boulanger, 1991), boiled and unboiled samples were run on SDS-polyacrylamide gels, transferred to nitrocellulose, and blots were probed with the 4D2 anti-fiber antibody. Fibers containing linker insertions into the G and C terminal domains, but not the AB loop formed trimers (data not shown). Levels of F41S.Cterm trimer were diminished compared to F41S.G (data not shown), indicating a possible defect in trimer formation.
Figure 3. Incorporation of MSH peptide into fiber41S.

(A) Peptide insertion sites into the G region of fiber 41S. An oligo insertion strategy was used to clone 2 restriction sites, NotI and SanDI, flanked by flexible linkers into the G region. These restriction sites were used to insert oligos encoding two MSH peptides. (B) Trimerization of fiber 41S mutants. Expression vectors carrying each of the fiber 41S mutants were transfected into 293 cells, and cell lysates were analyzed by Western blot. To resolve trimer formation, samples remained unboiled before loading the gel. Blots were probed with 4D2 anti-fiber antibody.
Because of the expression/trimerization defect with the AB and Cterm insertions, we did not pursue these constructs further. Plasmids expressing fiber 41S with MSH peptides in the G region were cloned as described in Materials and Methods. Previous studies have mapped the MC1R binding sites of the MSH peptide to a minimal 4-aa core binding sequence (HFRW). MSH peptide truncations that include amino acids flanking the core region have been shown to bind the MC1R with higher affinity than the core alone (Hruby et al., 1987; Sawyer et al., 1993). Based on these data, two peptides were inserted into the G mutant: the HFRW core alone (MSH1), and the core plus several flanking amino acids (MSH2) (Figure 3B). Western blot trimerization assays revealed that G mutants were capable of producing monomers and trimers in 293 cells; expression of MSH1 and MSH2 variants was slightly lower than wild type or G versions (Figure 3C lanes 3,5,7). On some blots, we also detected a band of intermediate size that may represent a fiber dimer or a non-specific cellular protein.
Construction and characterization of F7F41SΔ-based MSH targeting viruses
Based on the trimerization assay results, we moved forward with construction of recombinant Ad5 vectors based on peptide insertions into F41S. A strategy was developed to clone our panel of fiber 41S mutants into the F7F41SΔ background using a well-characterized bacterial recombination scheme (Renaut, Bernard, and D’Halluin, 2002). This strategy takes advantage of the RecA+ BJ5183 bacterial cell line, which facilitates a double-overlapping recombination event between DNA fragments from two plasmids: 1) a parental plasmid containing the full-length F7F41SΔ genome with a truncation in the fiber 41S knob domain (pvAdCiG.F7tF41SΔ), and 2) a shuttle plasmid containing a mutant fiber 41S knob cassette flanked by recombination arms (e.g. pT.F41S.G.Rec) (Figure 4). DNA fragments were electroporated into BJ5183 cells and recombinant viral genomes verified by restriction digestion. Confirmed successful recombinants corresponding to F7F41SΔ.G, F7F41SΔ.G. MSH1, and F7F41SΔ.G.MSH2 were transfected into 293 cells to generate infectious virus.
Figure 4. Recombination scheme used to generate viral plasmids containing mutated fiber 41S proteins.
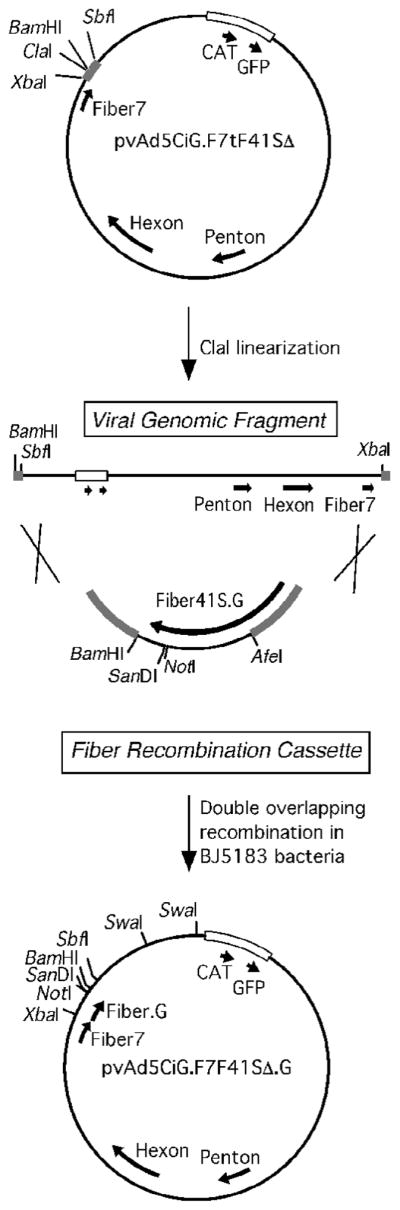
pvAd5CiG.F7tF41SΔ (top) has a truncated form of fiber 41S and a unique ClaI site flanked by two recombination arms (gray bars), which are also present in the pT.F41SRec.G plasmid. A double overlapping recombination event between these two arms permits rescue of full-length mutant viral plasmids. The viral genome is released from the plasmid backbone by SwaI digest and transfected into 293 cells to produce viral particles.
Initial characterization of the F7F41SΔ-based mutants revealed a striking growth defect. When viral preps were purified by cesium banding, we noted that compared to a control vector (F7F41SΔ), viruses expressing mutant fiber 41S exhibited a decrease in mature virions and an increase in the ratio of empty capsids: mature virions (Figure 5A). Calculation of total viral yield by spectrophotometric quantitation of particles at OD260 demonstrated a 30–50-fold decrease in mutant particle numbers compared to parental F7F41SΔ (Table I).
Figure 5. Growth characteristics and protein content of F7F41S-based mutant viruses.

Large scale preps of F7F41SΔ and F7F41SΔ.G.MSH1 were harvested and purified over two rounds of cesium chloride gradient centrifugation (A). Purified viral particles were separated by SDS-PAGE, transferred to nitrocellulose and probed with antibodies against fiber (B), IVa2 (C lower panel), or total Ad protein (C upper panel). Viral capsid protein assignments are indicated on the right and the position of molecular weight markers on the left. For lanes 3, 5, and 7, # marks putative preprocessed pVI/pVIII.
Table I.
Titers of fiber 41S-mutated vectors
| Vector | Titer (p/ml)a |
|---|---|
| F7F41SΔ | 1.0 ×1013 |
| F7F41SΔ.G | 9.2 × 1011 |
| F7F41SΔ.G.MSH1 | 5.6 × 1011 |
| F7F41SΔ.G.MSH2 | 6.0 × 1011 |
| F41SΔb | 1.8 × 1012 |
| F41SΔ.Gb | 5.2 ×1011 |
| F41SΔ.G.MSH1b | 1.4 ×1012 |
| F41SΔ.G.MSH2b | 2.0 ×1012 |
Viral prep volumes are typically 0.8 to 1.0 ml.
Fiber 41S-only viruses were generated under concentrating conditions in a fiber 5-complementing 293 cell line.
To determine whether empty capsids from F7F41SΔ.G, F7F41SΔ.G.MSH1, and F7F41SΔ.G2.MSH2 differ from mature virions with respect to fiber content, aliquots from both bands were analyzed by Western blot. Mature virions and empty capsids from F7F41SΔ.G incorporated high levels of mutant fiber 41S.G and low levels of fiber 7 (Figures 5B, lanes 3 and 4). This virus resembles the F7F41S in fiber content and suggests that the G peptide insertion does not significantly impair fiber incorporation into the virion. In contrast, the F7F41SΔ.G.MSH1 virus displayed predominantly fiber 7 in mature virions and a mix of both fibers in empty capsids (Figure 5B, lanes 5 and 6). Unexpectedly, the empty capsids from the F7F41SΔ.G.MSH2 prep contained almost exclusively fiber 41S.G.MSH2, while the mature virion band incorporated only fiber 7 (Figure 5B, lanes 7 and 8). The dominance of fiber 7 over mutant fiber 41S in the MSH1 and MSH2 mature virions resembles fiber levels in the CD and HI mutants of F7F41S (compare Figures 1B and 5B). These data suggest that MSH peptide insertions into the fiber 41S G region negatively impact the virus maturation process.
To assess the content of other capsid proteins, the same blots were probed with an anti-Ad rabbit antiserum (Figure 5C, upper panel). Previous work has established that mature virions and empty capsids have distinct banding patterns on denaturing gels (Edvardsson et al., 1976). For example, protein V (core minor) is present only in mature virions, while precursor polypeptides pVI and pVIII are detected in empty capsids. As expected, empty capsids had low levels of proteins V, VI, and VII, while bands corresponding to L1 52/55K, pVI, and pVII were present (Figure 5C, upper panel, lanes 3, 5, 7). Compared to mature virions, empty capsids also exhibited a higher hexon: fiber ratio. Interestingly, mature virion bands from the G mutants differ from the control F7F41S virus because they incorporate very low levels of proteins 100K, V, and VII (Figure 5C, upper panel, lanes 4, 6, 8). These data demonstrate that mature virions produced during infection with F7F41SΔ.G, F7F41SΔ.G.MSH1, and F7F41SΔ.G.MSH2 are highly defective with respect to total capsid protein content, and suggest that mutant fiber 41S trimers bearing peptides in the G region confer a dominant negative viral growth phenotype in an F7F41SΔ background.
The G region mutant fibers may be associated with a potential defect in Ad packaging. Adenovirus encapsidation is thought to occur via a process in which viral DNA is inserted into a pre-formed virion (i.e., empty capsid intermediate) (Ostapchuk and Hearing, 2005). Two virally encoded proteins are known to have important roles in packaging. The IVa2 protein is essential for virus production; mutant viruses that fail to express IVa2 do not assemble any virus particles (Zhang and Imperiale, 2003). L1 52/55K binds IVa2 and also plays a role in packaging. Viruses that do not express L1 52/55K are able to form empty capsids but do not encapsidate viral DNA (Gustin and Imperiale, 1998).
The IVa2 protein can be detected by Western blot in both mature virions and empty capsids (Winter and D’Halluin, 1991), while L1 52/55K is only present in empty capsids (Hasson, Ornelles, and Shenk, 1992). IVa2 levels in the F7F41SΔ mutant viruses were assessed by Western blot with a IVa2 rabbit polyclonal antibody. IVa2 was detected in both control vectors F7 and F7F41S (Figure 5C, lower panel, lanes 1 and 2). Low levels of IVa2 were detected in empty capsids and mature virions from F7F41SΔ.G, while IVa2 was not observed in empty capsids or mature virions from F7F41SΔ.G.MSH1 or F7F41SΔ.G.MSH2 (Figure 5C, lower panel, lanes 3–8). Taken together, these data suggest that incorporation of fiber 41S MSH mutants into virion capsid structures may compromise virion maturation. Alternatively, the incorporation of the mutant fiber may influence overall capsid stability by destabilizing the fiber trimer/penton base pentamer complex. Importantly, even the mature products that were obtained (which contain predominantly fiber 7 homotrimer) exhibit characteristics of defective particles (compromised levels of IVa2 and VII).
Impact of fiber 7 deletion on virus growth and virion maturation
In the current strategy, inclusion of fiber 7 enables F7F41SΔ vector production in 293 cells. In the case of fiber 41S CD, HI, and G mutants, we have shown that relatively modest peptide manipulations confer an overwhelming bias toward inclusion of fiber 7 into mature virions. We interpret this bypass phenotype as an indicator of defective fiber 41S homotrimer formation or incorporation into virion particles. Furthermore, the G region fiber 41S mutations confer a dominant negative viral packaging phenotype, which supersedes the fiber 7-bypass pathway. To determine if the presence of the fiber 7 exacerbates the mutant phenotype, a panel of fiber 41S-only (e.g. F41SΔ.G) viruses was constructed using the bacterial recombination scheme (Figure 4). We first attempted to propagate fiber 41S-only viruses in 293 cells under standard conditions, but the F41SΔ-based constructs failed to yield high titer lysates, presumably because these viruses are devoid of high affinity binding functions and or are defective in packaging functions. To overcome the severe growth defect in the fiber 41S-only background, viral DNAs were transfected into a fiber 5-complementing 293 cell-line (293-f5). Highly concentrated lysates from the 293-f5 cells were used to infect standard 293 cells for large-scale virus production. Under these conditions we were able to obtain viral yields that were comparable to the F7F41SΔ-based mutants (Table 1). Because these viruses could be rescued in the 293-f5 cells, we conclude that wt Ad5 fiber could compensate for the mutant fiber 41S deficiency.
To assess the impact fiber 7 elimination has on capsid content, equal particle numbers (approximated by protein assay) of each F41SΔ-based construct were characterized by Western blot using anti-fiber antibody. As expected, the control F41SΔ vector incorporated fiber 41S at levels comparable to F7F41S (Figure 6A, lanes 2 and 6) and F7F41SΔ (data not shown). Similar to F7F41SΔ.G, the F41SΔ.G virus displayed mutant fiber 41S, albeit at lower levels when compared to the parental F41SΔ virus (Figure 6A, compare lanes 3, 6, 7). When fiber 7 was deleted from the MSH variants, we observed incorporation of the fiber 41S.G.MSH1 and fiber. 41S.G.MSH2 into mature virions (Figure 6A, lanes 8 and 9). Levels of fiber 41S.MSH1 were markedly lower than fiber levels in the parental F41SΔ, where as elimination of fiber 7 restored fiber 41S in the MSH2 mutant to levels that were comparable to the parental vector.
Figure 6. Protein content of F41S-based mutant viruses.

Viral particles were separated by SDS-PAGE, transferred to nitrocellulose and probed with antibodies against fiber (A), IVa2 (B lower panel), or total Ad protein (B upper panel). Viral capsid protein assignments are indicated on the right and the position of molecular weight markers on the left. For lanes 7 and 8, # marks pVI/pVIII.
When Western blot analysis was carried out using anti-Ad serum or the anti-IVa2 antibody, IVa2 was quantitatively recovered in constructs lacking fiber 7 (Figure 6B lower panel). However, F41SΔ.G and F41SΔ.G.MSH1 differed from parental F41SΔ by the presence of a high hexon:fiber ratio, very high levels of pVI/pVIII proteins (Figure 6B, upper panel, lanes 8–9), and low levels of VII. These characteristics resemble the empty capsid phenotype observed in the F7F41SΔ mutants (compare Figure 5C, lanes 3,5,7). Of the three mutant constructs that were grown in the absence of fiber 7 from high concentration stocks, F41SΔ.G.MSH2 most closely resembled the protein composition pattern found in F41SΔ. The only major defect detectable by Western was diminished VII (Figure 6B upper panel compare lanes 6 and 9).
Influence of MSH2 peptide insertion on gene transduction
F41SΔ.G.MSH2 represents a candidate vector for retargeting to cells expressing the melanocortin-1 receptor (MC1R). A transduction assay was established using two murine cell lines, FL83B hepatocytes which express CAR and B16 melanoma cells which are poorly transduced by CAR binding vectors (JS unpublished observation) but express high levels of MC1R (Eberle et al., 1993). Infections in FL83B cells (5,000p/cell) demonstrated high levels of GFP by flow cytometry from a CAR targeting vector F41TΔ, which expresses both F41L (CAR binding) and F41S (Schoggins and Falck-Pedersen, 2006), very low levels of expression from the F41SΔ vector and even lower levels of GFP from the F41SΔ.G.MSH2 construct (Figure 7A). We next assessed whether this vector could transduce MC1R-expressing B16 cells. B16 cells were infected with 50,000 p/cell F41SΔ, F41SΔ.G.MSH2, and F41TΔ. In B16 cells, transduction levels for F41SΔ and F41TΔ were similar and about 10-fold over background. In contrast to our expectations, we did not observe enhancement of transduction by F41SΔ.G.MSH2 in B16 cells (Figure 7B). In fact, as found in the transduction assay of FL83 cells, transduction by F41SΔ.G.MSH2 was less than that found with F41SΔ and approached levels found in mock infected cells.
Figure 7. Transduction of FL83B and B16 cells by F41SΔ.G.MSH2.

FL83B (A) or B16 (B) cell lines were infected with the indicated virus at 5000 or 50000 p/cell, respectively. Transduction levels were measured by FACS-based quantitation of GFP. (C) B16 cell-associated viral genomes were measured by qPCR using hexon-specific primers. DNA from uninfected controls was spiked with 50,000 particles of each virus as a control.
The F41SΔ.G.MSH2 revealed a defect in VII content, which may impact on transduction but not viral DNA uptake. To characterize vector uptake into B16 cells, total DNA was harvested from cells 24 hours postinfection and viral genome copy number was determined by quantitative real-time PCR using hexon-specific primers (Figure 7C). A virus spike control for each vector established that the DNA content of each virus was consistent with particle estimation, and no unexpected harvesting artifacts were revealed. The total amount of infection recovered viral genomes was consistent with GFP transduction data (compare Figures 7B and 7C). Taken together, our results indicate that despite incorporation of MSH-bearing fibers into F41SΔ, targeted entry into B16 cells is not occurring under these conditions.
Discussion
The objective of the current study was to develop Ad5 pseudotyped F7F41S and F7F41SΔ vectors as experimental platforms for receptor specific retargeting through peptide insertions into fiber 41S. As an experimental model, the F7F41S platform provides several attractive features with respect to retargeting through fiber genetic modification. These viruses grow to high titers with complete capsid integrity in standard 293 E1 complementing cell lines. The fiber homotrimer that is incorporated into mature virions is essentially F41S only; it is detargeted from canonical and noncanonical Ad5 fiber binding, and offers an intermediate spacing between viral capsid and the target ligand. Using several strategies and target sites to insert peptide ligands into the fiber 41S knob region, we found that seemingly modest fiber 41S structural alterations in the knob domain resulted in either a bypass fiber phenotype or dominant negative influence on Ad capsid assembly.
Based on earlier studies, we had empirically concluded that the preference for fiber tail interactions with Ad5 penton was F5>F41S>F41L>F7 (Gall et al., 1996; Schoggins, Gall, and Falck-Pedersen, 2003). The dominance of fiber 41S over fiber 7 in our F7F41S virus (Figure 3C) is a striking demonstration of this bias. In the first set of constructs, peptide insertions into the HI or CD loops of fiber 41S resulted in fiber 7 incorporation into mature virions (Figure 1 and data not shown). Because mutant 41S fibers were well represented in infected cellular lysates, the bypass to fiber 7 was interpreted as a defect in mt fiber 41S trimerization and or assembly into penton. The peptide insertions rendered the fiber 41S mutants as noncompetitive fibers for virus assembly. In contrast, F7F41SΔ viruses containing MSH peptide inserts into the flexible G region of fiber 41S revealed diminished virus yield and produced an increase in the empty capsid/mature virion ratio. These fiber 41S mutations generate a fiber that suppresses the fiber 7-bypass phenotype, and functions as a dominant negative with respect to virus yield.
Upon examination of MSH empty capsid (EC) and mature virion (MV) polypeptide composition, both particle types were found to be defective and the degree of defect increased with size of peptide insert (Figures 5 and 6). Consistent with the HI and CD insertion studies, MSH insertions were shown to induce a bypass to fiber 7 in mature virions; however we were surprised to find a more significant representation of MSH containing fiber 41S in empty capsids. Other abnormalities included a disproportionate hexon: fiber ratio in empty capsids, reduced levels of proteins specific to mature virions, and diminished levels of the IVa2 packaging protein in both empty capsids and mature virions. The empty capsids we observe may represent a population of virions that are true packaging intermediates, whose encapsidation process failed to complete due to a fiber-mediated impairment. However, we cannot rule out the possibility that these empty capsids represent an accumulation of dead-end products. Regardless, transmission of knob structural alterations into a dominant negative packaging defect was unexpected, but may be better understood by studies characterizing the involvement of fiber in the process of late stage virus production.
The growth and production defects of our fiber 41S mutant viruses are reminiscent of fiberless Ad vectors (Falgout and Ketner, 1988; Legrand et al., 1999; Von Seggern et al., 1999). In Falgout et al., Ad2 and Ad5 fiber-deleted constructs were able to grow in noncomplementing cells due to trace contamination of helper virus. High titers of fiber-deleted vectors were obtained which were compromised not only in fiber, but were severely compromised in penton base and had diminished levels of mature protein VI, VII and VIII with increased levels of precursor products. The authors concluded that the absence of fiber influenced the action of viral protease responsible for conversion of capsid precursor proteins to full-length product. These observations are consistent with fiber 41S mutant capsids (Figure 5C and 6B).
In the studies by Legrand et al and Von Seggern et al, fiberless viruses were first grown in fiber complementing cell lines followed by growth in 293 cells in the absence of fiber complementation (Legrand et al., 1999; Von Seggern et al., 1999). In both studies, initial growth in noncomplementing cell lines was unproductive. These observations are completely consistent with each of our defective mutant constructs. Although there are some differences between the two fiberless Ad5 (AdFb°) studies, there are several important and relevant insights shared between each study. In the study by Legrand, total yields of AdFb° particles are diminished by approximately 2.5 fold compared to control AdLacZ. However the infectious unit/particle ratio of AdFb° virions is severely compromised (4 logs when grown on 293 cells and 2 logs when grown on 293Fcells (Legrand et al., 1999)). Irrespective of the cell line used for virus growth, analysis of radiolabeled particles showed that AdFb° capsids contain elevated levels of preprocessed pVI and pVIII proteins, indicating compromised virion maturation consistent with the Falgout study and our observations. All AdFb° capsids containded diminished levels of the DNA binding protein VII and protein VI (based on data from both studies). Based on Western analysis, AdFb° maintained a normal levels of penton base in purified capsids, and cryoEM revealed a structural similarity between wt and fiberless capsids (with some alterations in the angle of the RGD protrusions of fiberless penton base) (Von Seggern et al., 1999). This is one aspect where several of our mutant constructs differ with fiberless constructs; levels of penton are diminished in virions that demonstrate the dominant negative fiber 41S phenotype.
Several contributing factors may account for the deficient virus yields. The first and most obvious is that, unlike most other published targeting systems, our vectors are mutated in the penton RGD motif. We specifically deleted the RGD motif to avoid non-targeted uptake and diminish immune activation, a prerequisite for truly targeted gene therapy vectors. The lack of penton-dependent interactions with cell integrins affects both primary binding functions and internalization mediated through penton/integrin signaling (Wickham et al., 1993). Although the penton RGD motif is not anticipated to contribute in a significant manner to virus packaging or stability, fiber tail/penton RGD interactions have been implicated in cryoEM reconstructions (Fuschiotti et al., 2006) and may influence the stability of chimeric penton with fiber 7 or fiber 41S tail domains.
The second contributing factor is the method of virus production. In our experience with chimeric viruses, establishing a primary infective high titer stock is essential to producing a critical mass for efficient virus production. This point is illustrated with AdFb° 293 growth curves (Legrand et al., 1999). Infection at an MOI of 1 resulted in a viral yield that was diminished by 3 logs when compared to wt Ad5LacZ. With fiber 41S-only constructs, we were unable to grow high titer infectious virus in 293 cells; however, by using 293-f5 cells to establish a high titer infectious stock, sufficient virus was generated to produce high yields of these defective vectors. Our tandem two-fiber strategy was carried out in 293 cells, and the perturbation of fiber 41S structure resulted in defective virus production that was comparable to that seen with a fiberless vector grown in 293 cells (Legrand et al., 1999; Von Seggern et al., 1999). We would predict that virus production would be enhanced if the tandem constructs were first grown in a 293-f5 background.
Finally, how do fiber 41S knob mutations have a dominant negative effect on virus formation? In virus infection or when transiently coexpressed, penton base capsomers and fiber homotrimers form penton structures spontaneously (Karayan et al., 1994). During late stage adenovirus infection, large amounts of penton capsomers accumulate in the nucleus as “crystalline” structures (Franqueville et al., 2008). In fiberless mutant constructs, the nuclear organization of penton capsomer and hexon proteins are altered and shifted into PML containing inclusion bodies (Puvion-Dutilleul et al., 1999). Franqueville et al characterized the influence of fiber modifications on formation of the nuclear crystalline structures. They found that in specific instances, Ad5 fiber knob and shaft influenced the formation of the crystalline structures and that disruption of these structures correlated with diminished virus yield (Franqueville et al., 2008). These observations led to the hypothesis “that penton crystals represent a privileged assembly platform” (Franqueville et al., 2008) which facilitates assembly of a major limiting factor (penton capsomers) in virus production. This model fits very well with the observations made in our study. If our mutant fiber constructs disrupt normal formation of penton capsomers, formation of crystalline penton structures, or compete in an unproductive manner with the bypass fiber 7 in the tandem fiber vectors, then the expected outcome would be a severe compromise in virus production and capsid integrity.
When we eliminated the fiber 7-bypass route and forced high titer virus by the two-step 293-f5 virus production strategy, we were able to increase particle yields. However viruses produced in this manner were still clearly defective (Figure 6B). The F41SΔ.G and F41SΔ.G.MSH1 virions were compromised in penton, fiber and had high levels of unprocessed capsid proteins. The F41SΔ.G.MSH2 virus incorporated the mutant fiber and displayed an overall capsid content approaching the control F41SΔ (with previously noted exceptions). However, this vector did not exhibit enhanced transduction in the targeting cell line. Although many reasons can account for the lack of enhanced transduction by the F41SΔ.G.MSH2 virus (including a lack of proper presentation of ligand to receptor), the need to force virus production and the clear lack of protein VII in the purified virions argues that these viruses are structurally compromised which may impact on the efficiency of gene expression.
In the context of our current study, the recent work of Hesse et al provides additional insight into the use of fiber 41S as a targeting vector, particularly with respect to fiber 41S stability and virus production methods (Hesse et al., 2007). Hesse et al generated fiber 41S-targeted Ad5 vectors by transcomplementing a fiberless Ad5 backbone via transient transfection of plasmids expressing mutant fiber 41S. In their study, the 9 amino CDCRGDCFC targeting peptide, previously used in Ad5 fiber HI insertions (Dmitriev et al., 1998), was inserted into the EG, HI, and IJ loops of fiber 41S knob without loss of trimerization. SV-ori self-replicating plasmids expressing fiber 41S genes were transfected into 293T cells, then infected with Ad5.βGal.ΔF/F+ (Ad5.βGal.ΔF grown in 633 cells (a 293-f5 cell line)). Infectious virus generated in this manner was compared to virus generated by transfection of unmodified F5/41s.
Several features distinguish our study from that of Hesse et al. First, their base fiber F5/41s was a chimeric fiber containing Ad5 tail, Ad41s shaft and Ad41s knob. When expressing a virus based on wtF41S (F41S tail) with the CDCRGDCFC motif added to the C terminus, they observed a low level of trimerization product, whereas a construct that contained the Ad5 tail showed stable trimer product with the same Cterm addition. We found low level of trimerization with our Cterm fiber 41S construct as well, supporting the notion that the fiber tail domain influences trimer stability. Furthermore, although the tail domains of different serotypes are very highly conserved, we have previously described a fiber bias with respect to incorporation into an Ad5 capsid (Gall et al., 1996; Schoggins, Gall, and Falck-Pedersen, 2003). Since all of our constructs were based on authentic fiber 41S tail domains, and our HI loop insert was constructed in a manner similar to that used by Hesse et al, the data from both studies support the notion that use of an Ad5 fiber tail may enhance formation of a stable fiber/penton capsomer.
Another important difference in the two studies is our use of a two-fiber system. The transfection-based one-fiber system of Hesse et al provides an efficient method of driving high-level mutant fiber expression and incorporation into fiberless capsids. When we eliminated fiber 7 and generated a one-fiber system (using 293T-f5 cells) more closely resembling that of Hesse et al, we also observed enhanced virus production and an improved, but not completely normal, complement of capsid proteins. Hesse et al examined capsid integrity by Western blots using anti-hexon or anti-fiber antibodies, and revealed no distinguishable differences in the hexon/fiber ratio for the mutant constructs compared to the parental F5/41s. It is not clear whether CDCRGDCFC insertions into fiber 41S impact other capsid proteins. In our Western blot analysis of single fiber containing viruses (generated by the two-step production method) two clear conclusions can be made; first, fiber trimerization alone is not a true indicator of how a mutation will impact overall capsid integrity and second, CsCl purified mutant fiber viruses can demonstrate a spectrum of capsid anomalies (Figure 6B compare lanes 6–9). Simple analysis of the major capsid constituents (hexon, fiber, and penton) may not reveal subtle but important capsid deficiencies.
Overall, ligand insertion into fiber 41S is highly prone to defects in fiber trimerization and subsequent capsid integrity. Further experimentation will be needed to define the parameters needed to generate targeted vectors that exhibit high-level transduction (several log-fold) in physiologically relevant target cells. We believe that the F7F41S two-fiber strategy will serve as a useful and exquisitely sensitive platform to identify viable fiber 41S alterations that support capsid integrity, virion production, and cell-specific targeting.
Materials and Methods
Cell lines
The following monolayer cultures and corresponding media were used: HEK-293 (DMEM+5% cosmic calf serum), FL83B murine hepatocyte (F12K + 10% FBS), B16-F10 murine melanoma cells (DMEM+5% cosmic calf serum). The 293 fiber-5 cell line was generously provided by Transgene.
Cloning and recombinant virus production
Generation of fiber 41S mutant viruses containing CD and HI loop insertions
To generate mutant fiber 41S constructs expressing peptides in the CD and HI loops, the XbaI/BamHI fiber 41S fragment from pAd70-100.dlE3.F7F41S was subcloned into pSL301 to generate pSL301.F41S. A step-wise PCR mutagenesis strategy was used to insert two restriction sites, RsrII and PaeR7I, at the CD and HI insertion sites, resulting in two constructs: pSL301.F41S.CD and pSL301.F41S.HI. Oligos encoding peptides of varying lengths were inserted into these sites by directional cloning (details available upon request). Full-length fibers were excised from pSL301 plasmids by XbaI/BamHI digest and subcloned into pAd70-100.dlE3.F7F41S. Viruses were generated by recombination with left-end large fragments as previously described (Schoggins, Gall, and Falck-Pedersen, 2003).
Construction of recombination-competent fiber 41S expression shuttle plasmids
To generate mutated fiber 41S expressing MSH peptides, an XbaI/BamHI fiber 41S fragment from pAd70-100.dlE3.F7F41S was subcloned into the pTarget eukaryotic expression vector to generate pT.F41S. This plasmid was modified to generate pT.F41S.Rec by inserting a genomic fragment corresponding to the E4 region downstream of fiber (BamHI/SbfI fragment) that would serve as a homology arm for recombination. The QuikChange II site-directed mutagenesis kit (Stratagene) was used to insert oligos encoding two unique restriction sites (NotI and SanDI) flanked by a flexible linker. Oligos were inserted into the AB loop, the G region (G), and the C terminus to generate pT.F41S.Rec.AB, pT. F41S.Rec.G, and pT.F41S.Rec.Cterm. These plasmids were modified to express MSH peptides by ligating annealed oligos into NotI/SanDI sites. The resulting MSH targeting plasmids were: pT.F41S.Rec.AB.MSH1, pT.F41S.Rec.AB.MSH2, pT.F41S.Rec.G.MSH1, pT.F41S.Rec.G.MSH2, pT.F41S.Rec.Cterm.MSH1, pT.F41S.Rec.Cterm.MSH2.
Construction of Ad5-based viral plasmids expressing mutated fiber 41S
To generate viral plasmids containing mutant fiber 41S sequences, a ClaI/BstBI fragment from plasmid pTG15684 (kindly provided by Transgene) was recombined in BJ5183 cells with full-length viral DNA isolated from purified AdCMV.CATiresGFP (AdCiG) virus to generate the parental pvAdCiG plasmid. This construct bears the entire AdCiG viral genome along with a kanamycin resistance cassette and the Col E1 origin of replication. The pvAdCiG plasmid was digested with PsiI to release fiber 5 sequences, and the remaining large fragment was recombined with the AscI/SalI fragment of pAd70-100.dlE3.F7F41S to generate pvAdCiG.F7F41S. To create the RGD-deleted version of this plasmid, purified viral DNA from AdCiG.F7F41SΔ was digested with BstZ171 and Pac to release left and right ends of the genome flanking the penton RGD region. pvAdCiG.F7F41S was digested with RsrII and AscI, and the resulting fragment was recombined in BJ5183 cells with the BstZ171/PacI fragment from AdCiG.F7F41SΔ to generate pvAdCiG.F7F41SΔ.
Recombination-competent viral plasmids based on F7F41SΔ and F41SΔ and containing a truncation in the fiber 41S knob domain were developed as follows. The pT.F41S.Rec plasmid was digested with AfeI/BamHI to release the knob domain of fiber. A ClaI linker was inserted at the deletion site to give pT.F41S.delknob. The resulting plasmid contained a unique ClaI site flanked by two homology arms corresponding to the fiber 41S 5′ region and the E4 downstream domain. The truncated fiber 41S plus flanking homology arms were excised by XbaI/SbfI digest and subcloned into pAd70-100.dlE3.F7F41S to generate pAd70-100.dlE3.F7tF41S (“t” denotes truncated form of fiber 41S). The cassette containing truncated fiber and flanking homology arms was released by AscI/SacI digest and recombined in BJ5183 cells with the large fragment from PsiI-digested pvAdCiG.F7F41SΔ to generate pvAdCiG.F7tF41S Δ. A similar constructed was created in the fiber 41S-only background. To generate a fiber 41S-only expressing construct, the pAd70-100.dlE3.F7F41S was digested with PacI/XbaI, followed by a Klenow fill-in to remove the fiber 7 gene. This plasmid was recombined with a PsiI-digested pvAdCiG.F7F41SΔ to generate pvAdCiG.F41SΔ. The AscI/SacI fragment from pA70-100.dlE3.tF41SΔ was recombined with PsiI-digested pvACiG.F41SΔ to generate pvAdCiG.tF41SΔ. These constructs were recombined with the pTarget-based fiber plasmids described above to generate vectors expressing mutant fiber 41S.
Western blot of purified Ad capsids and transfected/infected 293 lysates
Purified Ad capsids (1010 particles) were separated by SDS-PAGE, transferred to nitrocellulose, and probed with either R72 or 4D2 anti-fiber antibodies, rabbit anti-Ad antisera, or polyclonal anti-IVa2 antibody (kindly provided by M. Imperiale) without stripping in between antibody treatments. Fiber from infected cell lysates was characterized by harvesting infected 293 cells with RIPA buffer (1x PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS), and analyzing by Western blot as described above. For analysis of fiber trimerization, pT.F41S-based plasmids were transfected into 293 cells and harvested 48 hours post-transfection. Cells were harvested in cold SDS-free lysis buffer (1% NP-40, 50 mM Tris pH 7.5, 150mM NaCl, 1mM EDTA) and analyzed by Western blot in either denatured (boiled) or non-denatured (unboiled) form.
In vitro transduction assays
FL83B and B16-F10 cells were seeded into 24-well plates and infected with 5000 (FL83B) or 50000 (B16-F10) p/cell in media without serum for 30 min at 37°C. After infection, virus was aspirated and fresh media was added back. Infected cells were harvested 24 hours post-infection and assayed for GFP expression by flow cytometry.
SYBR Green qPCR
For quantitative real-time PCR (qPCR), DNA was isolated from infected cells grown in 60mm dishes using Trizol reagent (Invitrogen) as instructed by the manufacturer. Amplifications of hexon or β-actin as a control were carried out in a total volume of 20 μl by using a one-step QuantiTect SYBR green kit (Qiagen) in an Applied Biosystems Prism 7900H sequence detection system with SDS 2.1 software. Cycles consisted of an initial incubation at 95°C for 15 min followed by 35 cycles at 94°C for 15 s, 60°C for 30 s, and 72°C for 20 s, with a final incubation at 72°C for 7 min. All determinations were performed in triplicate. Sequences of primers are available on request.
Acknowledgments
This research was supported by RO1AI06342 to EFP. GenVec and PaxVax have licensing agreements with Cornell Research Foundation for technology developed by E. Falck-Pedersen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alemany R, Curiel DT. CAR-binding ablation does not change biodistribution and toxicity of adenoviral vectors. Gene Ther. 2001;8(17):1347–53. doi: 10.1038/sj.gt.3301515. [DOI] [PubMed] [Google Scholar]
- Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, Keduas V, Notredame C. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006;34(Web Server issue):W604–8. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett BG, Crews CJ, Douglas JT. Targeted adenoviral vectors. Biochim Biophys Acta. 2002;1575(1–3):1–14. doi: 10.1016/s0167-4781(02)00249-x. [DOI] [PubMed] [Google Scholar]
- Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275(5304):1320–3. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- Campos SK, Barry MA. Current advances and future challenges in Adenoviral vector biology and targeting. Curr Gene Ther. 2007;7(3):189–204. doi: 10.2174/156652307780859062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev I, Krasnykh V, Miller CR, Wang M, Kashentseva E, Mikheeva G, Belousova N, Curiel DT. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J Virol. 1998;72(12):9706–13. doi: 10.1128/jvi.72.12.9706-9713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AN, Siegrist W, Bagutti C, Chluba-De Tapia J, Solca F, Wikberg JE, Chhajlani V. Receptors for melanocyte-stimulating hormone on melanoma cells. Ann N Y Acad Sci. 1993;680:320–41. doi: 10.1111/j.1749-6632.1993.tb19693.x. [DOI] [PubMed] [Google Scholar]
- Edvardsson B, Everitt E, Jornvall H, Prage L, Philipson L. Intermediates in adenovirus assembly. J Virol. 1976;19(2):533–47. doi: 10.1128/jvi.19.2.533-547.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falgout B, Ketner G. Characterization of adenovirus particles made by deletion mutants lacking the fiber gene. J Virol. 1988;62(2):622–5. doi: 10.1128/jvi.62.2.622-625.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franqueville L, Henning P, Magnusson M, Vigne E, Schoehn G, Blair-Zajdel ME, Habib N, Lindholm L, Blair GE, Hong SS, Boulanger P. Protein crystals in Adenovirus type 5-infected cells: requirements for intranuclear crystallogenesis, structural and functional analysis. PLoS One. 2008;3(8):e2894. doi: 10.1371/journal.pone.0002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuschiotti P, Schoehn G, Fender P, Fabry CM, Hewat EA, Chroboczek J, Ruigrok RW, Conway JF. Structure of the dodecahedral penton particle from human adenovirus type 3. J Mol Biol. 2006;356(2):510–20. doi: 10.1016/j.jmb.2005.11.048. [DOI] [PubMed] [Google Scholar]
- Gall J, Kass-Eisler A, Leinwand L, Falck-Pedersen E. Adenovirus Type 5 and 7 Capsid Chimera: Fiber Replacement Alters Receptor tropism without affecting primary immune neutralization epitopes. J Virol. 1996;70(4):2116–2123. doi: 10.1128/jvi.70.4.2116-2123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin KE, Imperiale MJ. Encapsidation of viral DNA requires the adenovirus L1 52/55-kilodalton protein. J Virol. 1998;72(10):7860–70. doi: 10.1128/jvi.72.10.7860-7870.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson TB, Ornelles DA, Shenk T. Adenovirus L1 52- and 55-kilodalton proteins are present within assembling virions and colocalize with nuclear structures distinct from replication centers. J Virol. 1992;66(10):6133–42. doi: 10.1128/jvi.66.10.6133-6142.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse A, Kosmides D, Kontermann RE, Nettelbeck DM. Tropism modification of adenovirus vectors by peptide ligand insertion into various positions of the adenovirus serotype 41 short-fiber knob domain. J Virol. 2007;81(6):2688–99. doi: 10.1128/JVI.02722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, Castrucci AM, Hintz MF, et al. alpha-Melanotropin: the minimal active sequence in the frog skin bioassay. J Med Chem. 1987;30(11):2126–30. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, Stewart PL, Shayakhmetov DM. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci U S A. 2008;105(14):5483–8. doi: 10.1073/pnas.0711757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayan L, Gay B, Gerfaux J, Boulanger PA. Oligomerization of recombinant penton base of adenovirus type 2 and its assembly with fiber in baculovirus-infected cells. Virology. 1994;202(2):782–95. doi: 10.1006/viro.1994.1400. [DOI] [PubMed] [Google Scholar]
- Legrand V, Spehner D, Schlesinger Y, Settelen N, Pavirani A, Mehtali M. Fiberless recombinant adenoviruses: virus maturation and infectivity in the absence of fiber. J Virol. 1999;73(2):907–19. doi: 10.1128/jvi.73.2.907-919.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, Brie A, Saulnier P, Perricaudet M, Yeh P, Vigne E. Simultaneous CAR- and alpha V integrin-binding ablation fails to reduce Ad5 liver tropism. Mol Ther. 2003;8(3):485–94. doi: 10.1016/s1525-0016(03)00182-5. [DOI] [PubMed] [Google Scholar]
- Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257(5074):1248–51. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Sato K, Hamada H. Reduction of natural adenovirus tropism to the liver by both ablation of fiber-coxsackievirus and adenovirus receptor interaction and use of replaceable short fiber. J Virol. 2003;77(4):2512–21. doi: 10.1128/JVI.77.4.2512-2521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemerow GR, Pache L, Reddy V, Stewart PL. Insights into adenovirus host cell interactions from structural studies. Virology. 2009;384(2):380–8. doi: 10.1016/j.virol.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neshich G, Borro LC, Higa RH, Kuser PR, Yamagishi ME, Franco EH, Krauchenco JN, Fileto R, Ribeiro AA, Bezerra GB, Velludo TM, Jimenez TS, Furukawa N, Teshima H, Kitajima K, Bava A, Sarai A, Togawa RC, Mancini AL. The Diamond STING server. Nucleic Acids Res. 2005;33(Web Server issue):W29– 35. doi: 10.1093/nar/gki397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin SA, Von Seggern DJ, Work LM, Pek DC, Dominiczak AF, Nemerow GR, Baker AH. Ablating adenovirus type 5 fiber-CAR binding and HI loop insertion of the SIGYPLP peptide generate an endothelial cell-selective adenovirus. Mol Ther. 2001;4(6):534–42. doi: 10.1006/mthe.2001.0489. [DOI] [PubMed] [Google Scholar]
- Nicklin SA, White SJ, Watkins SJ, Hawkins RE, Baker AH. Selective targeting of gene transfer to vascular endothelial cells by use of peptides isolated by phage display. Circulation. 2000;102(2):231–7. doi: 10.1161/01.cir.102.2.231. [DOI] [PubMed] [Google Scholar]
- Nicol CG, Graham D, Miller WH, White SJ, Smith TA, Nicklin SA, Stevenson SC, Baker AH. Effect of adenovirus serotype 5 fiber and penton modifications on in vivo tropism in rats. Mol Ther. 2004;10(2):344–54. doi: 10.1016/j.ymthe.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Novelli A, Boulanger PA. Deletion analysis of functional domains in baculovirus-expressed adenovirus type 2 fiber. Virology. 1991;185(1):365–76. doi: 10.1016/0042-6822(91)90784-9. [DOI] [PubMed] [Google Scholar]
- Ostapchuk P, Hearing P. Control of adenovirus packaging. J Cell Biochem. 2005;96(1):25–35. doi: 10.1002/jcb.20523. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F, Legrand V, Mehtali M, Chelbi-Alix MK, de The H, Puvion E. Deletion of the fiber gene induces the storage of hexon and penton base proteins in PML/Sp100-containing inclusions during adenovirus infection. Biol Cell. 1999;91(8):617–28. [PubMed] [Google Scholar]
- Renaut L, Bernard C, D’Halluin JC. A rapid and easy method for production and selection of recombinant adenovirus genomes. J Virol Methods. 2002;100(1–2):121– 31. doi: 10.1016/s0166-0934(01)00407-4. [DOI] [PubMed] [Google Scholar]
- Roelvink PW, Kovesdi I, Wickham TJ. Comparative analysis of adenovirus fiber-cell interaction: adenovirus type 2 (Ad2) and Ad9 utilize the same cellular fiber receptor but use different binding strategies for attachment. J Virol. 1996;70(11):7614–21. doi: 10.1128/jvi.70.11.7614-7621.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer TK, Castrucci AM, Staples DJ, Affholter JA, De Vaux A, Hruby VJ, Hadley ME. Structure-activity relationships of [Nle4, D-Phe7]alpha-MSH. Discovery of a tripeptidyl agonist exhibiting sustained bioactivity. Ann N Y Acad Sci. 1993;680:597–9. doi: 10.1111/j.1749-6632.1993.tb19749.x. [DOI] [PubMed] [Google Scholar]
- Schoggins JW, Falck-Pedersen E. Fiber and penton base capsid modifications yield diminished adenovirus type 5 transduction and proinflammatory gene expression with retention of antigen-specific humoral immunity. J Virol. 2006;80(21):10634–44. doi: 10.1128/JVI.01359-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Gall JG, Falck-Pedersen E. Subgroup B and F fiber chimeras eliminate normal adenovirus type 5 vector transduction in vitro and in vivo. J Virol. 2003;77(2):1039–48. doi: 10.1128/JVI.77.2.1039-1048.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Nociari M, Philpott N, Falck-Pedersen E. Influence of fiber detargeting on adenovirus-mediated innate and adaptive immune activation. J Virol. 2005;79(18):11627–37. doi: 10.1128/JVI.79.18.11627-11637.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiradake E, Cusack S. Crystal structure of enteric adenovirus serotype 41 short fiber head. J Virol. 2005;79(22):14088–94. doi: 10.1128/JVI.79.22.14088-14094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TA, Idamakanti N, Marshall-Neff J, Rollence ML, Wright P, Kaloss M, King L, Mech C, Dinges L, Iverson WO, Sherer AD, Markovits JE, Lyons RM, Kaleko M, Stevenson SC. Receptor interactions involved in adenoviral- mediated gene delivery after systemic administration in non-human primates. Hum Gene Ther. 2003;14(17):1595–604. doi: 10.1089/104303403322542248. [DOI] [PubMed] [Google Scholar]
- Von Seggern DJ, Chiu CY, Fleck SK, Stewart PL, Nemerow GR. A helper-independent adenovirus vector with E1, E3, and fiber deleted: structure and infectivity of fiberless particles. J Virol. 1999;73(2):1601–8. doi: 10.1128/jvi.73.2.1601-1608.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, Atoda H, Pink R, Buckley SM, Greig JA, Denby L, Custers J, Morita T, Francischetti IM, Monteiro RQ, Barouch DH, van Rooijen N, Napoli C, Havenga MJ, Nicklin SA, Baker AH. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132(3):397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73(2):309–19. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- Wickham TJ, Roelvink PW, Brough DE, Kovesdi I. Adenovirus targeted to heparan-containing receptors increases its gene delivery efficiency to multiple cell types. Nat Biotechnol. 1996;14(11):1570–3. doi: 10.1038/nbt1196-1570. [DOI] [PubMed] [Google Scholar]
- Wickham TJ, Tzeng E, Shears LL, 2nd, Roelvink PW, Li Y, Lee GM, Brough DE, Lizonova A, Kovesdi I. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. J Virol. 1997;71(11):8221–9. doi: 10.1128/jvi.71.11.8221-8229.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter N, D’Halluin JC. Regulation of the biosynthesis of subgroup C adenovirus protein IVa2. J Virol. 1991;65(10):5250–9. doi: 10.1128/jvi.65.10.5250-5259.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Anderson B, Mao Q, Davidson BL. Recombinant human adenovirus: targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J Virol. 2000;74(23):11359–66. doi: 10.1128/jvi.74.23.11359-11366.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Dickinson C, Haskell-Luevano C, Gantz I. Molecular basis for the interaction of [Nle4,D-Phe7]melanocyte stimulating hormone with the human melanocortin-1 receptor. J Biol Chem. 1997;272(37):23000–10. doi: 10.1074/jbc.272.37.23000. [DOI] [PubMed] [Google Scholar]
- Zhang W, Imperiale MJ. Requirement of the adenovirus IVa2 protein for virus assembly. J Virol. 2003;77(6):3586–94. doi: 10.1128/JVI.77.6.3586-3594.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]